
Radioaktivně značené peptidy v diagnostice a terapii nádorů
Radiolabelled peptides in the diagnosis and therapy of tumours
In the last two decades, radiolabelled receptor-specific peptides have profiled themselves as excellent tools that have expressively increased the diagnostic and therapeutical possibilities of tumours predominantly of neuroendocrine origin. The aim of this review paper is to transparently inform the Czech scientific fraternity about the significance and limitations of the use of this group of radiopharmaceuticals in clinical practice.
Key words:
radiopharmaceuticals – diagnosis – therapy – cancers – receptor-specific peptides
Autoři:
J. Cihlo; M. Lázníček
Působiště autorů:
Univerzita Karlova v Praze, Farmaceutická fakulta v Hradci Králové, katedra farmakologie a toxikologie
Vyšlo v časopise:
Čes. slov. Farm., 2008; 57, 70-77
Kategorie:
Přehledy a odborná sdělení
Souhrn
Radioaktivně značené receptorově specifické peptidy se v posledních dvaceti letech profilovaly jako významná léčiva výrazně rozšiřující diagnostické a terapeutické možnosti převážně neuroendokrinních nádorů. Zásadním cílem této přehledné práce je co nejpřehledněji informovat českou vědeckou veřejnost o významu a limetách použití této cenné skupiny radiofarmak v klinické praxi.
Klíčová slova:
radiofarmaka – diagnostika – terapie – nádory – receptorově specifické peptidy
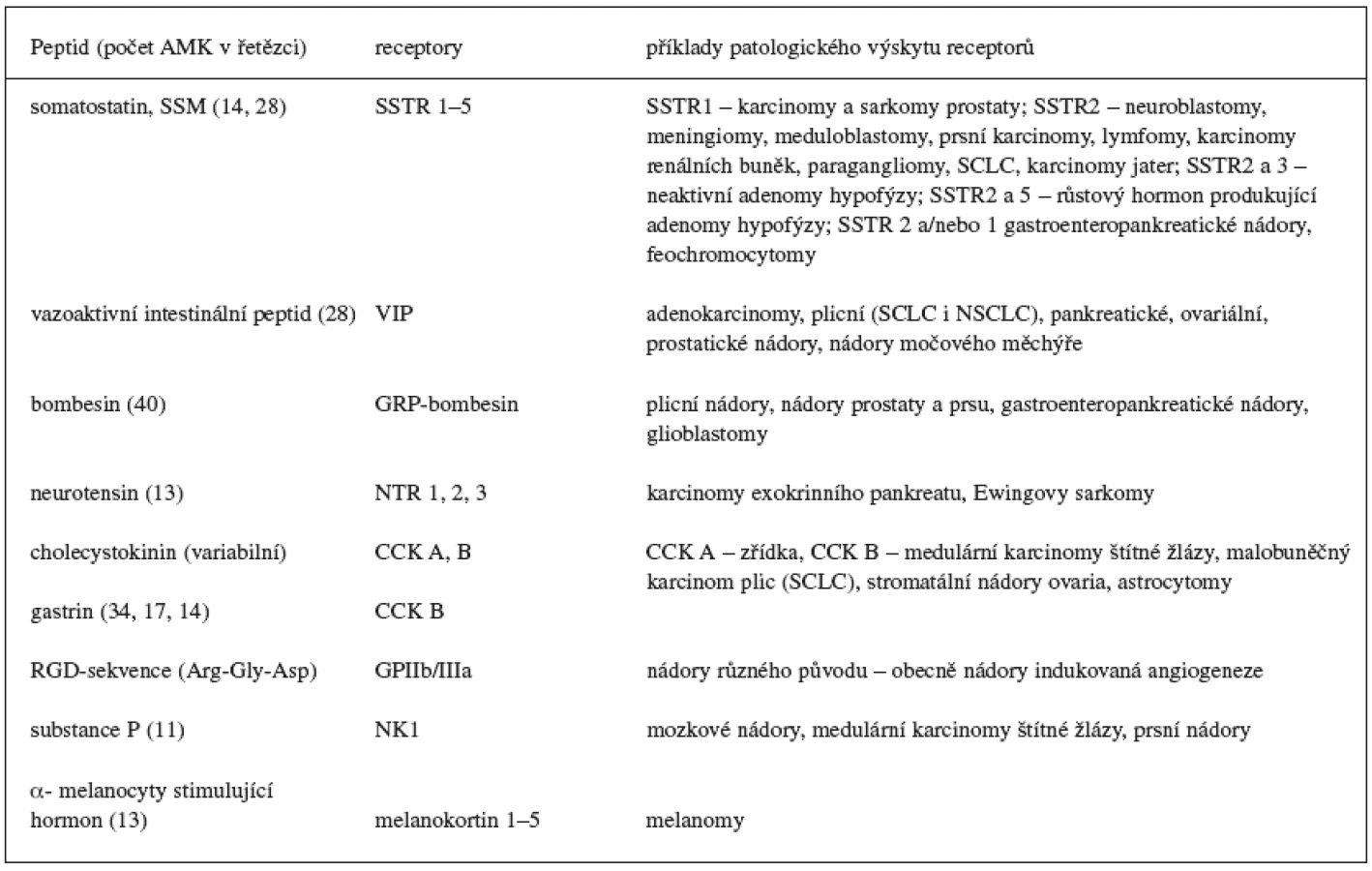
Radioaktivně značené peptidy tvoří významnou skupinu léčiv, které umožňují pomocí vysoce citlivých neinvazivních metod identifikaci, lokalizaci a terapii určitého typu nádorů. Podstatou jejich diagnosticko-terapeutického využití v nukleární medicíně je výskyt receptorů v nádorových tkáních schopných tyto látky specificky vázat 1). Mezi nejčastěji používané radioaktivně značené receptorově specifické peptidy patří analogy odvozené od fyziologického hormonu somatostatinu 2, 3). Spolu s dalšími přirozeně se vyskytujícími peptidy (tab. 1) patří somatostatin do skupiny regulačních peptidů projevujících se v organismu širokou škálou farmakologických účinků. Zatímco fyziologické působení somatostatinu je spíše inhibičního charakteru a zahrnuje například útlum sekrece růstového hormonu, insulinu, glukagonu, gastrinu, cholecystokininu a dalších hormonů, ostatní peptidy této skupiny naopak často sekreci bioaktivních látek stimulují, a vyvolávají tak odpovídající farmakologický efekt 9–11). Toto rozsáhlé fyziologické působení je na cílových orgánech zprostředkováno stejnými membránovými receptory, které se ve velké hustotě vyskytují také v nádorových tkáních. Například výskyt somatostatinových receptorů je typický pro neuroendokrinní typ nádorů, receptory pro bombesin (gastrin releasing peptid) lze identifikovat u prsních nádorů a nádorů prostaty, receptory cholecystokininu v medulárních karcinomech štítné žlázy a receptory neurotensinu v exokrinních nádorech pankreatu 1, 12). Specifické vychytávání radioaktivity na těchto receptorech je základním předpokladem využití radioaktivně značených peptidů v diagnostice a terapii tohoto typu nádorů.
Vývoj radioaktivně značených receptorově specifických radiofarmak
Jako specifické nosiče radioaktivity se využívají převážně syntetické analogy fyziologických peptidů. Za jejich prototypy lze bezesporu označit analogy odvozené od endogenního hormonu somatostatinu. Tohoto cyklického peptidu, který se skládá ze 14, respektive 28 aminokyselin (polypeptid z 28 aminokyselin se 14 aminokyselinovým somatostatinem na C-konci), nelze pro účely (nukleární) medicíny použít 8, 9). Podobně jako u většiny ostatních fyziologických peptidů je limitem jeho použití krátký biologický poločas zapříčiněný rychlým štěpením struktury nativního peptidu sérovými endo - a exopeptidasami. Obměnami v sekvenci aminokyselin přirozených peptidů (zkrácení peptidického řetězce, jeho amidace, acetylace, zavedení atypických D-aminokyselin, aminoalkoholů, neobvyklých aminokyselin nebo postranních řetězců) bylo dosaženo prodloužení biologického poločasu. Tyto stabilní peptidické struktury umožňují použití, ať už ve formě radioaktivně značených sloučenin (k diagnostice a terapii nádorů), nebo ve formě neznačených peptidů, například k inhibici symptomů souvisejících se zvýšenou hormonální sekrecí (analogy somatostatinu u adenomu hypofýzy produkujícího růstový hormon) 13–16). Příkladem stabilních peptidických derivátů s dostatečně dlouhým biologickým poločasem, které jsou používané/testované v souvislosti s nádory, mohou být analogy somatostatinu (oktreotid, lanreotid, vapreotid, oktreotát, 1-Nal3-oktreotid). U oktreotidu bylo například dosaženo prodloužení biologického poločasu až na 90 minut oproti původní 1 až 3 minutám nativního somatostatinu, a to při zachování většiny jeho biologických funkcí 16, 17).
Zatímco afinita přirozeně se vyskytujících peptidů je ke všem receptorovým subtypům přibližně stejná, tedy vysoká, může se afinita syntetických analogů k jednotlivým receptorům značně lišit. Obměna peptidického řetězce podílející se na vzniku stabilních analogů není jediným parametrem, který ovlivňuje afinitu peptidických sloučenin k receptorům. Připojení chelatační části (DTPA – diethylentriaminpentaoctová kyselina, DOTA – 1,4,7,10-tetraazacyklododekan-N,N’,N’’,N’’’-tetraoctová kyselina, HYNIC – hydrazinonikotinamid; TETA – 1,4,8,11-tetraazacyklotetradekan-N,N’,N’’,N’’’-tetraoctová kyselina) (obr. 1) k těmto stabilním strukturám, které umožňují radionuklidové značení a nakonec i chemické vlastnosti zvoleného radionuklidu (tab. 2), mohou mít podstatný vliv na výslednou receptorovou afinitu diagnostického nebo terapeutického radioaktivně značeného peptidu 32). Dalšími důležitými parametry, které se mohou významně spolupodílet na výsledném nukleárně medicínském potenciálu diskutovaných peptidů, jsou molekulová hmotnost a lipofilita peptidů, vazebnost na plasmatické proteiny a v neposlední řadě také náboj radioligandu 9, 33). Tyto parametry ovlivňující farmakokinetický profil peptidů mají významný vliv na biodistribuci a clearance, a významně tak předurčují typ exkrece nebo případnou míru kumulace radioaktivně značených receptorově specifických peptidů.


Diagnostické uplatnění radioaktivně značených peptidů (analogů somatostatinu)
Výzkumu receptorově specifických peptidů je věnována velká pozornost především v souvislosti s neuroendokrinními nádory. Právě v tomto typu nádorů byla nalezena velká hustota peptidických receptorů, z nichž exprese somatostatinových patří k nejčetnějším. Neuroendokrinní nádory představují heterogenní skupinu nádorů difuzního endokrinního systému, které se mohou lišit velikostí, histologií, biologickým a klinickým chováním 34). Většinou jde o benigní metastázující nádory s pouze výjimečným přechodem do malignity. V současné době jsou rozlišovány dvě hlavní kategorie neuroendokrinních nádorů: gastroenteropankreatické (GEP) (gastrinom, insulinom, glukagon, VIPom) a karcinoidní nádory 34). Jejich počáteční diagnóza může být založena na klinických symptomech (flush syndrom, průjem) způsobených nadprodukcí hormonů 8, 35). Typickým znakem neuroendokrinních nádorů je výskyt somatostatinových receptorů, kterých bylo dodnes popsáno 5 subtypů (SSTR1 – SSTR5). Jde často o malé, pomalu rostoucí nádory, což znesnadňuje jejich detekci jinými, pro tyto účely méně citlivými metodami (CT, magnetická rezonance, ultrazvuk) 8, 36). Zavedení syntetického somatostatinového analogu oktreotidu s navázaným chelátorem DTPA (obr. 2) stabilně vázajícím indium-111 (111In-pentetreotid, Octreoscan) umožnilo detekci právě tohoto typu nádorů. Afinita této sloučeniny je omezena převážně na somatostatinové receptorové subtypy 2 a 5, které se právě v neuroendokrinních nádorech vyskytují nejčastěji 34).

Jelikož se koncentrace receptorů a výskyt jednotlivých subtypů u různých nádorů může lišit, může se lišit i senzitivita diagnostického vyšetření. Příkladem může být diagnostické užití Octreoscanu u jiných než neuroendokrinních nádorů, kde byly somatostatinové receptory subtypu 2 také popsány (prsní nádory, malobuněčné nádory plic, lymfomy). Při těchto vyšetřeních ovšem nebylo dosaženo tak uspokojivých výsledků, což bývá zdůvodňováno nerovnoměrnou distribucí receptorů v těchto nádorových tkáních 8).
Za účelem zvýšení citlivosti a umožnění detekce také ostatních nádorů exprimujících jiné než zmíněné subtypy somatostatinových receptorů jsou prováděny variace v zavedených strukturách syntetických derivátů. Příkladem dnes již standardních peptidů vzniklých substitucí aminokyselin fenylalaninu za tyrozin v bioaktivním jádře oktreotidu může být DTPA(DOTA)-Tyr3-oktreotid a DTPA(DOTA)-Tyr3-oktreotát s vystupňovanou afinitou k somatostatinovému receptovému subtypu 2. Mezi novějšími analogy se širším afinitním spektrem je třeba analog DOTA-1-Nal3-oktreotid (substituce fenylalaninu 1-naftylovou funkční skupinou), který má navíc také afinitu k subtypům 3 a 5 17).
Jelikož je technecium-99m díky svým fyzikálním vlastnostem a snadné dostupnosti považováno za ideální diagnostický radionuklid, je snaha vyvíjet peptidy se schopností vázat tento gama zářič. Takovým peptidem je depreotid, komerčně vyráběný analog somatostatinu, jež našel uplatnění v diagnostice maligních plicních nádorů 8). Chelatační tetraaminová skupina připojená k analogu Tyr3-oktreotátu dala vznik dalšímu derivátu s názvem demotát, který je rovněž schopný vázat 99mTc a jehož vlastnosti se jeví příznivě pro diagnostické využití somatostatin-receptorově pozitivních nádorů 19, 20).
Terapeutické uplatnění radioaktivně značených peptidů (analogů somatostatinu)
Úspěšné použití receptorově specifických peptidů značených gama zářiči k vizualizaci nádorů a metastáz otevřelo fascinující možnost využít tyto peptidy značené terapeutickými radionuklidy k léčbě nádorů, a ovlivnit tak stav a progresi onemocnění. Základním faktorem tohoto způsobu terapie je cílená distribuce efektivní radiační dávky k nádorovým buňkám, respektive k jejich jaderné DNA. Tato forma terapie je především vhodná pro maligní, roztroušené nebo příliš malé nádory nevhodné pro odstranění klasickými chirurgickými postupy nebo pro využití externích ozařovacích technik. Výběr radiofarmaka/radionuklidu je kritickou částí plánování terapie, při které by měl být dbán zřetel na individuální vlastnosti nádorů (velikost a lokalizace) se zvláštním ohledem také na přiléhající tkáň 34, 37). Počáteční pokusy s vysokými dávkami Octreoscanu vystřídaly analogy značené 177Lu a 90Y, při kterých byla nejednou pozorována stabilizace onemocnění či úplná remise nádorů 12, 34, 38). Možnost využití Octreoscanu k terapii nádorů je dána spektrem emitovaného záření jeho radioizotopu (111In), který je kromě zdroje gama záření také emitorem Augerových elektronů s prokázaným antiproliferativním účinkem 39). Radiotoxicita přípravků značených 111In je však omezena krátkým dosahem těchto emitovaných částic 16). Jejich terapeutický efekt je přímo podmíněn těsné lokalizaci u DNA rakovinných buněk, respektive procesu internalizace (receptorově zprostředkovanému endocytickému transportu), který je za tuto lokalizaci zodpovědný 40). Naopak účinek radiopeptidů značených radionuklidy emitující beta částice s dostatečnou energií a dosahem není bezprostředně spojen s těsnou penetrací do nádorových struktur. Jejich značení si vyžádalo zavedení chelatačních činidel (DOTA, TETA) stabilně vázajících hlavní terapeuticky využívané beta zářiče 90Y, 177Lu, ale i jejich případné alternativy 153Sm, 64Cu 15, 41–43). Radiobiologické vlastnosti terapeutických izotopů naznačily, že každý radionuklid vlastní specifický potenciál pro terapii určitého typu nádorů. Částice 90Y mají v porovnání s 177Lu vyšší energii a vyšší maximální dosah v tkáních a jsou tedy vhodné pro větší nádory i s heterogenní distribucí receptorů. Naopak menší nádory nejsou schopné vzhledem ke své velikosti absorbovat celou emitovanou energii 90Y, která pak může toxicky působit na zdravou tkáň. Pro tyto menší nádory a metastázy se jeví příhodněji využití radioizotopu 177Lu s nižší energií a nižším tkáňovým dosahem, což bylo dokázáno ve výsledcích preklinických studií, ve kterých byly testovány peptidy 90Y-DOTA-oktreotid a 177Lu DOTA-oktreotát 39, 44). 188Re je radioizotopem emitujícím cytotoxické beta záření s energií a dosahem porovnatelným s 90Y ovšem s mnohem kratším fyzikálním poločasem 8, 29). Tento radionuklid má podobné dispozice ke značení jako 99mTc 45). Z tohoto důvodu je snaha převést diagnostické peptidické analogy (99mTc-depreotid) s chelatační částí vazající 99mTc také na terapeuticky využitelné peptidy radioaktivně značené 188Re 29).
Většina zmíněných radioizotopů emitujících terapeuticky prospěšné beta záření je také zdrojem gama záření (177Lu,188Re, 153Sm), případně zdrojem pozitronů (64Cu). Tohoto paralelního záření lze za pomoci scintigraficko-dozimetrického vyšetření využít k přibližné kvantifikaci retence absorbované dávky v cílových a limitních orgánech, a tudíž k vyhodnocení prospěšnosti/nevhodnosti zvolené strategie léčby. U analogů značených ytriem-90 (90Y je pouze čistý beta zářič) je k simulaci terapie možno využít peptidy značené 111In nebo k radioaktivnímu značení daného peptidu použít izotop 86Y, který je zdrojem pozitronového záření. Tento posledně jmenovaný přístup může být výhodný z hlediska odfiltrování zkreslení vazebné receptorové afinity způsobené rozdílnou chemickou povahou 90Y/111In. Nevýhodou je omezená dostupnost 86Y a poměrně krátký fyzikální poločas limitující získání pozdějších dat 37, 46, 47). 90Y-DOTA-oktreotid, 90Y-DOTA-lanreotid, 177Lu/90Y/153Sm-DOTA-Tyr3-oktreotát nebo peptid značený 188Re, prozatím pojmenovaný P2045, jsou příklady radioaktivně značených peptidů, které jsou klinicky testovány v souvislosti s využitím v terapii neuroendokrinních nádorů.
Nežádoucí účinky související s použitím radioaktivně značených peptidů a možnost jejich ovlivnění
Radioaktivně značené receptorově specifické peptidy našly prozatím své pevné místo především v diagnostice neuroendokrinních nádorů (Octreoscan). Peptidy značené terapeutickými radionuklidy procházejí v současné době řadou klinických studií, ve kterých jsou kromě pozitivních vlivů na progresi onemocnění sledovány nežádoucí účinky, které ve většině případů souvisejí právě s agresivním chováním emitovaného záření.
V porovnání s vedlejšími účinky chemoterapie jsou nežádoucí projevy peptidické radioterapie méně časté, a proto lze tento léčebný postup označit za relativně bezpečný 38, 48). Akutní následky aplikace radiopeptidu (nauzea, zvracení, bolest v místě nádoru) mohou být kontrolovány symptomatickou terapií, na ty, které se projevují až s časovou latencí a které jsou výsledkem absorpce radiofarmaka ve zdravé tkáni, je nutno dbát zřetel při samotném plánování terapie (volba dávky a typu radionuklidu, dozimetrická vyšetření s diagnostickými analogy atd.) 38).
Kumulace radioaktivně značených peptidů v jiných než nádorových tkáních může být významným klinickým problémem nejen z hlediska zkreslení interpretace případných patologických nálezů při scintigrafickém vyšetření, ale hlavně z důvodu možných radiotoxických účinků. Následky léčby radioaktivně značenými peptidy přímo souvisí s velikostí dávky (aktivitou), agresivitou záření a farmakokinetickým profilem dané radioaktivně značené sloučeniny. Pečlivé vyhodnocení dozimetrických parametrů u kritických orgánů diagnosticky aplikovaných peptidů může předejít radiotoxickému působení samotné radionuklidové terapie. Výsledky biodistribučních studií studovaných radioaktivně značených peptidů ukázaly, že k nejvyšší retenci radioaktivity dochází ve slezině, ledvinách, játrech, kostní dřeni a SSTR-pozitivních nádorech 34). Zatímco vliv absorbovaného záření na tkáň sleziny nebyl pozorován a jaterní toxicita, pokud se vyskytne, je většinou mírná a reverzibilní, kostní dřeň a hlavně ledviny byly v souvislosti s terapeuticky užívanými peptidy označeny za kritické, dávku limitující tkáně 47, 49). V ledvinách, hlavním exkrečním orgánu radioaktivně značených peptidů, dochází k částečné reabsorpci primárně zfiltrovaných radiopeptidů a vlivem jejich polárního charakteru také ke kumulování, které je spojené s radiotoxickými účinky na ledvinnou tkáň. Barone a spol. v roce 2005 publikovala závěry své práce, ze kterých plyne, že radiační nefrotoxita je závislá na velikosti dávky a farmakologickém profilu radiofarmaka, individuálním objemu ledvin a případně frakcionalizaci dávky 50). Ve většině popsaných klinických případů využívajících k léčbě radioaktivně značené receptorově specifické peptidy byla ovšem radiotoxicita nepředvídatelná, k čemuž přispívá také fakt, že není zcela přesně objasněn mechanismus retence radioaktivně značených peptidů v ledvinách 37). Z tohoto důvodu jsou studovány mechanismy reabsorpce a hledány látky s potencionálním inhibičním vlivem na tento renální uptake.
Mnoho vědeckých prací dokládá, že ledvinné vychytávání analogů somatostatinu může být sníženo současným podáním pozitivně nabitých aminokyselin lysinu (Lys) a argininu (Arg) nebo želatinového plazma expandéru gelofusinu 51–53).
Snížení renálního vychytávání 111In-D-Glu1-minigastrinu bylo dosaženo polyglutamovou kyselinou (5 a více glutamátových zbytků), zatímco efekt podání aminokyselin byl u této látky minimální 54).
Testováním látek s potenciálem inhibovat vychytávání v ledvinných buňkách bylo zjištěno, že se na procesu akumulace podílí patrně více typů transportních mechanismů 55). Za hlavní mechanismus vychytávání látek podobných oktreotidu je považován mechanismus endocytózy pomocí multiligandového megalinového receptoru, který je v proximálním tubulu hojně exprimován. Tato hypotéza byla potvrzena řadou studií jak in vitro na buňečných liniích, tak in vivo pokusech prováděných na myších se specifickými megalin-deficitními ledvinami 56–58). Megalin se v proximálním renálním tubulu společně s dalším glykoproteinem cubilinem podílí na udržování homeostázy organismu kontrolou reabsorpce řady fyziologických i syntetických látek (hormony, vitaminy vázající proteiny, enzymy, léčiva atd.) 59–62). Tyto látky mají nezřídka charakter kationaktivních sloučenin nebo charakter peptidů a proteinů bohatých na pozitivně nabité aminokyseliny. Tento fakt může vysvětlovat inhibiční schopnosti Arg a Lys na reabsorpci derivátů somatostatinu vlivem saturace o vazebná místa megalinového receptoru. Teorie inhibičního efektu pozitivně nabitých aminokyselin může být potvrzena i redukčními projevy plazma expandéru gelofusinu majícího ve své struktuře pozitivně nabité skupiny. Na druhou stranu mechanismus inhibice pomocí aniontových sloučenin, což je příkladem polyglutamátu u derivátu minigastrinu, naznačuje, že vychytávání radiopeptidů v ledvinách je velice komplexní děj a může být ovlivněn řadou vlastností radiopeptidů i potenciálních inhibitorů (velikost, struktura a náboj) 55). Dalším perspektivním postupem při snižování radiotoxického působení diskutovaných peptidů na ledvinnou tkáň je použití amifostinu 63). Tato látka neredukuje vychytávání radiopeptidů v renálních tubulech, ale mírní toxický efekt radiace vychytáváním volných radikálů. Kombinací amifostinu a lysinu bylo dosaženo signifikantního snížení renálního postižení u potkanů medikovaných177Lu-DOTA-Tyr3-oktreotátem 63).
Hematologická toxicita provázející léčbu radiopeptidy projevující se snížením počtu krevních elementů je častá, ale dá se obecně označit za mírnou a přechodnou 38). V průběhu léčby radiofarmaky bylo dále popsáno přechodné porušení spermatogeneze, zatímco vzhledem k fyziologickému výskytu somatostatinových receptorů pochopitelná porucha endokrinních funkcí adenohypofyzární osy ani diabetes mellitus nebyly vůbec pozorovány 34, 64, 65).
Nové strategie využití radioaktivně značených peptidů
Zvýšení efektivity scintigrafických a terapeutických metod využívajících radioaktivně značených somatostatinových analogů je možné dosáhnout kromě vývoje nových stabilních analogů s vyšší afinitou k receptorům aplikací dvou či více radioaktivně značených peptidů nebo jejich analogů. Výskyt receptorů v nádorech není totiž většinou omezen na jeden receptorový subtyp, respektive výčet potenciálních cílů pro metody nukleární medicíny je u mnoha nádorů velice široký. Například GEP nádory exprimují kromě somatostatinových receptorů GLIP-1 (glukagon-like peptid 1) receptory a receptory VIP (vazoaktivní intestinální peptid), cholecystokyninu a bombesinu 66). V prsních nádorech byl zase zjištěn současný výskyt receptorů somatostatinu, VIP, gastrin-releasing peptidu a NPY-Y1 (neuropeptid Y, subtyp Y1) receptorů 67). Kombinace peptidických analogů odvozených od jednonoho či více přirozených peptidů by mohla vést ke zvýšení akumulace radioaktivity nebo k optimalizaci jejího rozložení hlavně v nádorech s heterogenní strukturou. Kromě toho některé ze zmíněných peptidů mají potenciál inhibovat nádorový růst 1). Využití této strategie léčby využívající současné aplikace více analogů je ovšem v současné době limitováno chudšími poznatky o ostatních peptidech a jejich receptorech, jelikož je výzkum soustředěn převážně na vývoj derivátů somatostatinu. Existují ale už i výjimky potvrzující tuto skutečnost. Radioaktivně značené analogy minigastrinu byly úspěšně použity pro diagnostiku cholecystokinin B receptorově pozitivních nádorů. Cyklický analog RGD sekvence c(Arg-Gly-Asp-D-Tyr-Lys) s navázanou chelatační skupinou DTPA, který antagonizuje nádorovou angiogenezi navázáním na receptory avb3,je dalším peptidickým analogem, který může být použit ke scintigrafii nebo radionuklidové terapii nádorového onemocnění 68).
Problém nízké specifické aktivity u některých DOTA-peptidů limitující terapeutické použití například u ligandů DOTA-substance P a DOTA-bombesinu (pro nukleárně medicínské účely by se aplikované množství peptidu mělo pohybovat hluboko pod hladinou dávek, které způsobují farmakologický efekt) může být částečně vyřešen rozdělením dávky nebo jejím infuzním, intratumorálním nebo intraarteriálním podáním 9, 69). Zavedení necyklických chelátorů a vyvinutí peptidů s větším počtem navázaných chelatačních činidel patří mezi další možné principy, které by měly zvýšit specifickou aktivitu diskutovaných radiofarmak, a tím i jejich využitelnost pro potřeby nukleární medicíny 9).
Závěr
Radioaktivně značené receptorově specifické peptidy našly své uplatnění v diagnostice neuroendokrinních nádorů (Octreoscan). Také jejich další využití v terapii tohoto typu nádorů se jeví velice perspektivně.
I když byla výzkumu těchto látek věnována pozornost mnoha vědeckých skupin, je zde ještě velký prostor především pro vývoj nových radioaktivně značených peptidických analogů s vysokou afinitou ke specifickým nádorovým receptorům. Optimalizace postupů využití těchto peptidů v nukleární medicíně a ostatních zainteresovaných lékařských disciplínách je jedním ze základních předpokladů vedoucích ke zlepšení diagnostických a terapeutických možností léčby nádorového onemocnění neuroendokrinního původu.
Tato práce byla podpořena grantem č. 305/07/0535 Grantové agentury České republiky.
Došlo: 10. ledna 2007
Přijato: 31. ledna 2008
Adresa pro korespondenci:
prof. PharmDr. Ing. Milan Lázníček, CSc.
Katedra farmakologie a toxikologie FaF UK
Heyrovského 1203, Hradec Králové 500 05
e-mail: milan.laznicek@faf.cuni.cz
Zdroje
1. Reubi, J. C.: Endocr. Rev., 2003; 24, 389–427.
2. de Herder, W. W., Lamberts, S. W.: Curr. Opin. Oncol., 2002; 14, 53–57.
3. Lamberts, S. W., de Herder, W. W., Hofland, L. J.: Trends Endocrinol. Metab., 2002; 13, 451–457.
4. Behr, T. M., Behe, M., Becker, W.: Q. J. Nucl. Med., 1999; 43, 268–280.
5. de Visser, M., Janssen, P. J., Srinivasan, A. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 1134–1139.
6. Koikov, L. N., Ebetino, F. H., Solinsky, M. G. et al.: Bioorg. Med. Chem. Lett., 2003; 13, 2647–2650.
7. Reubi, J. C., Waser, B., Schaer, J. C., Laissue, J. A.: Eur. J. Nucl. Med., 2001; 28, 836–846.
8. Weiner, R. E.,Thakur, M. L.: BioDrugs, 2005; 19, 145–163.
9. Breeman, W. A., Kwekkeboom, D. J., de Blois, E. et al.: Anticancer Agents Med. Chem., 2007; 7, 345–357.
10. Patel, Y. C., Greenwood, M. T., Panetta, R. et al.: Life Sci., 1995; 57, 1249–1265.
11. Scherubl, H., Hescheler, J.,Riecken, E. O.: Horm. Metab. Res. Suppl., 1993; 27, 1–4.
12. Reubi, J. C.: Neuroendocrinology, 2004; 80 (Suppl. 1), 51–56.
13. Blok, D., Feitsma, R. I., Vermeij, P., Pauwels, E. J.: Eur. J. Nucl. Med., 1999; 26, 1511–1519.
14. Hejna, M., Schmidinger, M., Raderer, M.: Ann. Oncol., 2002; 13, 653–668.
15. Gotthardt, M., Boermann, O. C., Behr, T. M. et al.: Curr. Pharm. Des., 2004; 10, 2951–2963.
16. Breeman, W. A., de Jong, M., Kwekkeboom, D. J. et al.: Eur. J. Nucl. Med., 2001; 28, 1421–1429.
17. Wild, D., Schmitt, J. S., Ginj, M. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 1338–1347.
18. Virgolini, I., Traub, T., Novotny, C. et al.: Curr. Pharm. Des., 2002; 8, 1781–1807.
19. Decristoforo, C., Maina, T., Nock, B. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 1211–1219.
20. Kopecky, M., Trejtnar, F., Laznicek, M. et al.: Nucl. Med. Commun., 2005; 26, 549–554.
21. Maina, T., Nock, B., Nikolopoulou, A. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2002; 29, 742–753.
22. Maina, T., Nock, B. A., Cordopatis, P. et al.: Eur. J. Nucl. Med. Mo. Imaging, 2006; 33, 831–840.
23. Decristoforo, C., Mather, S. J., Cholewinski, W. et al.: Eur. J. Nucl. Med., 2000; 27, 1318–1325.
24. Decristoforo, C., Melendez-Alafort, L., Sosabowski, J. K., Mather, S. J.: J. Nucl. Med., 2000; 41, 1114–1119.
25. Cremonesi, M., Ferrari, M., Bodei, L. et al.: J. Nucl. Med., 2006; 47, 1467–1475.
26. de Jong, M., Breeman, W. A., Bernard, B. F. et al.: Int. J. Cancer, 2001; 92, 628–633.
27. Lewis, J. S., Wang, M., Laforest, R. et al.: Int. J. Cancer, 2001; 94, 873–877.
28. Bugaj, J. E., Erion, J. L., Johnson, M. A. et al.: Nucl. Med. Biol., 2001; 28, 327–334.
29. Cyr, J. E., Pearson, D. A., Wilson, D. M. et al.: J. Med. Chem., 2007; 50, 1354–1364.
30. Lewis, J. S., Lewis, M. R., Cutler, P. D. et al.: Clin. Cancer Res., 1999; 5, 3608–3616.
31. Lewis, J. S., Srinivasan, A., Schmidt, M. A., Anderson, C. J.: Nucl. Med. Biol., 1999; 26, 267–273.
32. Blower, P.: Dalton Trans, 2006; 1705–1711.
33. Lister-James, J., Moyer, B. R.,Dean, R. T.: Q. J. Nucl. Med., 1997; 41, 111–118.
34. Kaltsas, G. A., Papadogias, D., Makras, P., Grossman, A. B.: Endocr. Relat. Cancer, 2005; 12, 683–699.
35. Caplin, M. E., Buscombe, J. R., Hilson, A. J. et al.: Lancet, 1998; 352, 799–805.
36. Gibril, F., Reynolds, J. C., Doppman, J. L. et al.: Ann. Intern. Med., 1996; 125, 26–34.
37. Cremonesi, M., Ferrari, M., Bodei, L. et al.: Q. J. Nucl. Med. Mol. Imaging, 2006; 50, 288–295.
38. Forrer, F., Valkema, R., Kwekkeboom, D. J. et al.: Best. Pract. Res. Clin. Endocrinol. Metab, 2007; 21, 111–129.
39. Teunissen, J. J., Kwekkeboom, D. J., de Jong, M. et al.: Best. Pract. Res. Clin. Gastroenterol., 2005; 19, 595–616.
40. Capello, A., Krenning, E. P., Breeman, W. A. et al.: J. Nucl. Med., 2003; 44, 98–104.
41. de Jong, M., Bakker, W. H., Krenning, E. P. et al.: Eur. J. Nucl. Med., 1997; 24, 368–371.
42. Krenning, E. P., de Jong, M., Kooij, P. P. et al.: Ann. Oncol., 1999; 10 (Suppl. 2), S23–S29.
43. Anderson, C. J., Dehdashti, F., Cutler, P. D. et al.: J. Nucl. Med., 2001; 42, 213–221.
44. Kwekkeboom, D. J., Bakker, W. H., Kooij, P. P. et al.: Eur. J. Nucl. Med., 2001; 28, 1319–1325.
45. Deutsch, E., Libson, K., Vanderheyden, J. L. et al.: Int. J. Rad. Appl. Instrum. B, 1986; 13, 465–477.
46. Cremonesi, M., Ferrari, M., Zoboli, S. et al.: Eur. J. Nucl. Med., 1999; 26, 877–886.
47. Pauwels, S., Barone, R., Walrand, S. et al.: J. Nucl. Med., 2005; 46 (Suppl. 1), 92S–98S.
48. Engstrom, P. F., Lavin, P. T., Moertel, C. G. et al.: J. Clin. Oncol., 1984; 2, 1255–1259.
49. Bodei, L., Cremonesi, M., Zoboli, S. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 207–216.
50. Barone, R., Borson-Chazot, F., Valkema, R. et al.: J. Nucl. Med., 2005; 46 (Suppl. 1), 99S–106S.
51. Rolleman, E. J., Valkema, R., de Jong, M. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 9–15.
52. van Eerd, J. E., Vegt, E., Wetzels, J. F. et al.: J. Nucl. Med., 2006; 47, 528–533.
53. Vegt, E., Wetzels, J. F., Russel, F. G. et al.: J. Nucl. Med., 2006; 47, 432–436.
54. Behe, M., Kluge, G., Becker, W. et al.: J. Nucl. Med., 2005; 46, 1012–1015.
55. Gotthardt, M., van Eerd-Vismale, J., Oyen, W. J. et al.: J. Nucl. Med., 2007; 48, 596–601.
56. Barone, R., Van Der Smissen, P., Devuyst, O. et al.: Kidney Int., 2005; 67, 969–976.
57. de Jong, M., Barone, R., Krenning, E. et al.: J. Nucl. Med., 2005; 46, 1696–1700.
58. Melis, M., Krenning, E. P., Bernard, B. F. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2005; 32, 1136–1143.
59. Christensen, E. I., Nielsen, R.: Rev. Physiol. Biochem. Pharmacol., 2007; 158, 1–22.
60. Christensen, E. I.,Verroust, P. J.: Pediatr. Nephrol., 2002; 17, 993–999.
61. Verroust, P. J.,Christensen, E. I.: Nephrol. Dial. Transplant., 2002; 17, 1867–1871.
62. Moestrup, S. K., Cui, S., Vorum, H. et al.: J. Clin. Invest., 1995; 96, 1404–1413.
63. Rolleman, E. J., Forrer, F., Bernard, B. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2007; 34, 763–771.
64. Bodei, L., Cremonesi, M., Grana, C. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2004; 31, 1038–1046.
65. de Jong, M., Valkema, R., Jamar, F. et al.: Semin. Nucl. Med., 2002; 32, 133–140.
66. Reubi, J. C.,Waser, B.: Eur. J. Nucl. Med. Mol. Imaging, 2003; 30, 781–793.
67. Reubi, C., Gugger, M., Waser, B.: Eur. J. Nucl. Med. Mol. Imaging, 2002; 29, 855–862.
68. van Hagen, P. M., Breeman, W. A., Bernard, H. F. et al.: Int. J. Cancer, 2000; 90, 186–198.
69. Schumacher, T., Hofer, S., Eichhorn, K. et al.: Eur. J. Nucl. Med. Mol. Imaging, 2002; 29, 486–493.
Štítky
Farmacie FarmakologieČlánek vyšel v časopise
Česká a slovenská farmacie

2008 Číslo 2
- Využití diklofenaku ve formě gelu v terapii seboroické keratózy
- Aceklofenak v léčbě muskuloskeletálních onemocnění – srovnání s dalšími NSAIDs z hlediska účinnosti a bezpečnosti
- Jaké zdravotní benefity může mít popíjení kávy nebo čaje?
- Jak a kdy u celiakie začíná reakce na lepek? Možnou odpověď poodkryla čerstvá kanadská studie
Nejčtenější v tomto čísle
- Polosyntetické deriváty celulosy jako základ hydrofilních gelových systémů
- Laktobacily a ich probiotické vlastnosti
- Standardní receptura pro přípravu léčivých přípravků v lékárnách I. Suspenze k aplikaci na kůži
- Léčivé rostliny a diabetes mellitus