
1. část: Technologie genomového sekvenování pro účely precizní onkologie
Datum publikace: 28. 5. 2019
prof. RNDr. Ondřej Slabý, Ph.D.
Masarykův onkologický ústav, Brno
Úvod – genom, nádorový genom a co v něm hledáme
Lidský genom se skládá z celkem 3,2 miliardy párů bází (bp – base pairs), ze kterých přibližně 1 % (30–40 milionů bp) tvoří tzv. exom neboli soubor všech exonů dohromady čítajících asi 23 tisíc genů. Tato kódující část genomu je přepisována do struktury proteinů a její sekvenční variabilita tak může mít přímý dopad na buněčné funkce. Odhaduje se, že přibližně 85 % mutací zodpovědných za vznik různých onemocnění leží právě v exomové části genomu. Také nádorová transformace je důsledek akumulace změn (mutací) v sekvenci DNA.
Z hlediska funkce v kancerogenezi dělíme tyto mutace do dvou základních skupin: doporovodné (passenger) mutace, které nejsou zapojené do patogeneze nádoru, a řídicí (driver) mutace, které jsou kauzálně zapojené do procesu maligní transformace. Počet doprovodných mutací v individuálním nádorovém genomu je odhadován na 10 tisíc, řídicích mutací je 5–10, a tzv. actionable neboli klinicky relevantní mutace umožňující léčebnou intervenci jsou pouze 1–2. Tyto mutace pak mají zásadní význam pro účely precizní onkologie.
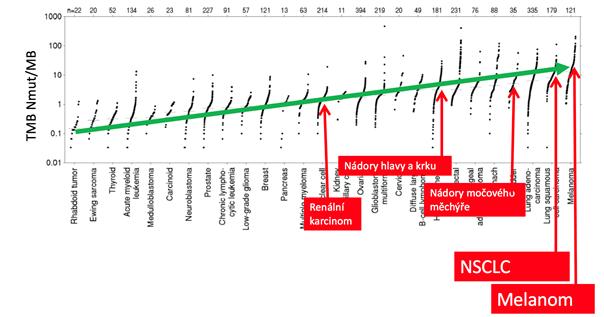
Sekvence nádorového genomu je díky jeho inherentně narušené stabilitě enormně dynamickou entitou, a je proto vždy pouze „momentkou“ charakterizující tumor v daném čase a stadiu nádorové choroby. Různé typy nádorů, ale i různé nádory v rámci jedné histopatologické jednotky mohou být charakteristické různou mírou genomové nestability či působením kancerogenů v rámci své etiologie (melanom – UV záření, karcinom plic – kouření) a počtem somatických mutací v nádorovém genomu, tzv. mutační náloží (TMB – tumor mutational burden, viz obr. 1). Protože TMB lze považovat za surogát neoantigenní nálože, tedy počtu neoantigenů na povrchu nádorové buňky, souvisí TMB také s odpovědí na protinádorovou imunoterapii. Typickým příkladem nádorů s extrémně vysokou TMB jsou nádory vykazující tzv. mikrosatelitvou nestabilitu (MSI high).
Cílem sekvenačních technologií používaných v současnosti je tedy především nalézt tzv. actionable genomové varianty a stanovit mutační nálož nádoru, což je soubor informací o individuálním nádorovém genomu, který se stává podkladem pro jednání molekulárně onkologické indikační komise (MTB – molecular tumor board).
Obr. 1 Mutační nálož (TMB) u různých typů nádorových onemocnění (upraveno dle: Lawrence et al., 2013)
Jaké typy genomových variant hledáme a jaké technologie lze použít k jejich identifikaci
Genomové varianty lze klasifikovat nejen podle jejich významu pro maligní transformaci, ale také na základě jejich chemismu. Jsou to nejčastěji:
- záměny jedné báze v sekvenci DNA neboli bodové mutace (SNV – single nucleotide variant, např. BRAFV600E: citlivost k inhibitorům BRAF);
- krátké genové inzerce a delece (1–40 párů bází; indels, např. delece v exonu 19 EGFR: citlivost k inhibitorům EGFR);
- variabilita v počtu kopií genů (CNV – copy number variation, např. amplifikace HER2: citlivost k trastuzumabu);
- chromosomové zlomy vedoucí ke ko-lokalizaci 2 genů nebo jejich částí, které normálně neležící blízko sebe, tzv. fúzní geny (např. BCR-ABL: imatinib; ALK-EML4: citlivost k inhibitoru ALK).
Kliničtí genetici pracují s genomem zárodečným (germinálním) a klasifikují varianty (nejčastěji inaktivační v nádorových supresorech s nízkou frekvencí v populaci) především s ohledem na jejich patogenicitu, resp. jejich potenciál způsobovat, případně zvyšovat riziko nádorové choroby. Pro tyto účely mají historicky etablovaný systém klasifikace těchto variant (IARC 1 benigní – 5 vysoce penetrantní a patogenní).
Pro účely precizní onkologie a terapeutické plánování pracujeme především s variantami v genomu nádorovém, tedy s mutacemi somatickými, nejčastěji aktivačními mutacemi v onkogenech, ideálně v genech pro terapeutické cíle případně v komponentách významných patogenních signálních drah, které lze terapeuticky zacílit. Klasifikace těchto variant potom vychází ze síly klinické evidence existující pro vztah dané varianty a léčebné odpovědi na konkrétní léčebný přípravek (např. klasifikace používaná v Memorial Sloan Kettering Cancer Center, viz obr. 2).
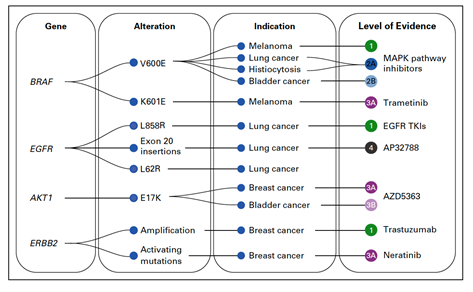
Příklady použití této klasifikace na konkrétní genomové varianty jsou potom znázorněny na obr. 3. Například mutace BRAFV600E dosahuje u melanomu nejvyššího stupně evidence (1), což znamená, že mutace je schválená americkým Úřadem pro kontrolu léků a potravin (FDA) jako prediktor pro lék schválený FDA u daného nádoru. U karcinomu močového měchýře dosahuje stejná mutace evidence 2B, což znamená, že se jedná o standard-of-care biomarker pro lék schválený FDA v jiné indikaci.
Výjimečně jsou i germinální mutace nositelkami tzv. actionable informace, např. zárodečná inaktivace genů pro mismatch repair způsobující mikrosatelitovou nestabilitu (MSI high) a následně vysokou TMB spojenou s dobrou léčebnou odpovědí na imunoterapii.
Obr. 2 Klasifikace genomových variant používaná v Memorial Sloan Kettering Cancer Center pro účely precizní onkologie (dle: Chakravarty et al., 2017)
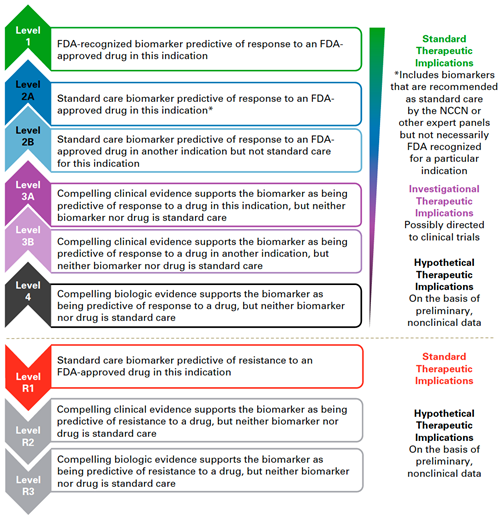
Obr. 3 Ukázka klasifikování konkrétních genomových variant dle schématu na obr. 2 (dle: Chakravarty et al., 2017)
Základní principy sekvenování nové generace jako optimální metody pro účely precizní onkologie
Sekvenování nové generace (NGS – next generation sequencing) vykazuje oproti tradičním metodám používaným pro detekci genomových variant řadu výhod. Tradiční technologie jako Sangerovo sekvenování, sekvenčně specifická PCR nebo fluorescenční in situ hybridizace (FISH) jsou vždy cílené pouze na konkrétní gen nebo variantu. V současné době, kdy je v onkologii často nezbytné paralelní vyšetřování většího množství genů / variant, kladou tyto konvenční metody vyšší nároky na množství biologického materiálu, často opakované biopsie a také delší čas do poskytnutí výsledků, protože jednotlivá vyšetření musejí být prováděna vždy nezávisle na sobě. Oproti tomu NGS umožňuje paralelní testování většího množství genů, případně otestovat celý exom či genom, a to i od většího množství pacientů v jednom experimentu. Tento přístup je spojen s nižšími nároky na množství vstupního biologického materiálu, a především s redukcí času do poskytnutí výsledků vyšetření, což má v onkologii zásadní význam.
NGS je založeno na masivním paralelním sekvenování mnoha fragmentů DNA. Tato technologie někdy také bývá označována jako vysokokapacitní sekvenování, hluboké sekvenování nebo sekvenování 2. generace. V současnosti již existuje řada technologií (nejčastěji používanými jsou Illumina a Ion Torrent) umožňujících diagnostickou aplikaci NGS. Napříč všemi platformami jsou zachovány základní principy NGS.
Technologie NGS umožňuje sekvenovat DNA, a i když tři základní typy genomových variant – jednonukleotidové záměny (SNV), variabilita v počtu kopií genů (CNV) a krátké inzerce a delece (indels) – lze identifikovat na úrovni nádorové DNA, neméně důležité fúzní geny detekujeme pomocí analýzy nádorové RNA. Na jednu kopii fúzního genu v nádorovém genomu totiž připadají řádově stovky až tisíce kopií jeho RNA transkriptu. Detekcí fúzních genů na úrovni RNA proto ve srovnání s DNA dosahujeme mnohonásobně vyšší citlivosti. Před vlastní sekvenační reakcí je ovšem nezbytné provést přepis (reverzní transkripci).
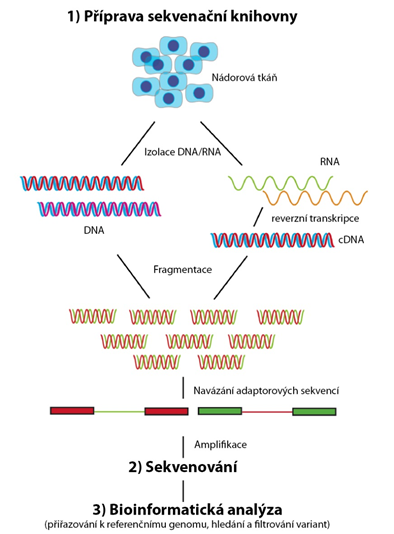
Prvním krokem NGS je příprava vzorku DNA pro účely sekvenace neboli příprava tzv. sekvenační knihovny. V tomto kroku je vždy zahrnuta izolace DNA ze vzorku nádoru (případně přepis nádorové RNA do cDNA), fragmentace DNA, navázaní tzv. adaptorových sekvencí na konce fragmentů DNA, což umožní jejich globální namnožení (amplifikaci) a dále jejich fixaci na solidní povrch. Následně, ve druhém kroku, dochází k vlastní sekvenaci syntézou. K imobilizovaným fragmentům DNA jsou komplementárně připojovány jednotlivé nukleotidy (ACTG) což je spojeno s emisí vždy jiného fyzikálního signálu zachycovaného detektorem. Takto získané sekvence jednotlivých fragmentů jsou v třetím kroku bioinformaticky analyzovány. V první fázi jsou získané sekvence přiřazovány (alignment) k referenčnímu genomu či sekvenci, považované za sekvenci referenční. Následně probíhá analýza jejíž cílem je nalézt varianty v sekvenovaném vzorku DNA (variant calling), poté anotace nalezených variant a dále přichází nejsložitější krok bioinformatické analýzy, kterým je filtrování nalezených variant. Zde jsou postupně eliminovány varianty podle různých kritérií, v první fázi automaticky (například dle schopnosti varianty měnit strukturu kódovaného proteinu, tzv. nesynonymní varianty), následně manuálně (například filtrování podle klinické evidence). Cílem je zúžit seznam nalezených variant na takové, které jsou biologicky relevantní v kontextu daného nádorového onemocnění (driver mutace) a optimálně také klinicky relevantní (actionable mutace) s jasným dopadem pro terapeutické plánování. Workflow technologie sekvenování nové generace je shrnuto na obr. 4.
Obr. 4 Využití technologie sekvenování nové generace pro studium nádorové DNA
Představíme-li si konkrétní pozici v lidském genomu, resp. genu, např. BRAFV600E (v sekvenci genu pro BRAF to odpovídá pozici a záměně c.1799T>A), pak počet fragmentů DNA získaných z nádorového vzorku topologicky pokrývajících tuto pozici určuje, kolikrát bude daná pozice genu v sekvenační reakci tzv. přečtena, tedy jaká bude v dané pozici hloubka čtení (sequencing depth), resp. jaké zde bude pokrytí (neboli coverage).
Představme si, že máme biologických vzorek tvořený tisícem buněk, ze kterých je 50 % nádorových. Z těchto 500 nádorových buněk vykazuje 400 heterozygotní stav pro aktivační mutaci BRAFV600E → 40 % všech buněk obsahuje mutaci v jedné alele, tzn. 20 % DNA ve vzorku nese mutaci. Pokud danou pozici při sekvenaci přečteme s hloubkou čtení 20X, teoreticky 4 z 20 čtení zachytí mutovanou alelu A, 16 z 20 čtení potom nemutovanou alelu T.
K charakterizaci sekvenační reakce daného vzorku se někdy uvádí průměrná hloubka čtení (pokytí) neboli tzv. average coverage (např. průměrné pokrytí sekvenační reakce bylo 30X). Ten parametr má ovšem omezenou výpovědní hodnotu, protože hloubka čtení / pokrytí není uniformní napříč celou sekvenovanou DNA. Počty fragmentů překrývajících různé pozice lidského genomu nejsou vždy identické, což je podmíněno řadou fyzikalně-chemických faktorů a také samotnou technologií přípravy sekvenačních knihoven.
Z hlediska kvality sekvenační reakce pro konkrétní vzorek DNA a pro odhad senzitivity záchytu genových variant má vyšší výpovědní hodnotu (než průměrné pokrytí) informace o tom, jak velké procento sekvenované DNA při sekvenaci dosáhlo předem definovaného pokrytí. Například 98 % sekvenované DNA mělo minimální pokrytí 10X.
Základní aplikace sekvenování nové generace
Za dvě základní aplikace NGS lze považovat:
- sekvenaci genomové DNA nebo jejích částí s cílem identifikovat SNV, CNV a indels;
- sekvenaci transkriptomu, tzv. RNAseq; technicky se jedná o sekvenaci DNA získané přepisem nádorové RNA s cílem identifikovat přítomnost fúzních genů.
Z hlediska genomové DNA lze potom sekvenovat zárodečnou DNA (germinální varianty) získanou obvykle z periferních leukocytů či bukálního stěru, zpravidla pro účely klinické genetiky, případně DNA nádorovou (somatické varianty) získanou izolací ze vzorku nádorové tkáně pro účely terapeutického plánování.
Z hlediska tzv. šíře pokrytí (breadth of coverage) neboli procenta genomu pokrytého sekvenační reakcí dělíme základní aplikace NGS na:
- celogenomové sekvenování (WGS – whole genome sequencing);
- celoexomové sekvenování (WES – whole exome sequencing);
- cílené (hotspot) sekvenování založení na použití tzv. sekvenančních panelů.
Tab. 1 Srovnání základních aplikací sekvenování nové generace
|
Aplikace NGS |
Popis |
Výhody |
Nevýhody |
|
Celogenomové sekvenování (WGS) |
Stanovení kompletní genomové DNA sekvence |
|
|
|
Celoexomové sekvenování (WES) |
Stanovení sekvence přibližně 1 % genomové DNA představující kódující oblasti |
|
|
|
Cílené (hotspot) sekvenování (sekvenační panely) |
Stanovení sekvence vybraných genů, případně genomových oblastí |
|
|
Celogenomové sekvenování umožňuje detekovat všechny typy variant napříč celým genomem, v kódujících (tzv. exom) i nekódujících oblastech, a tak umožňuje identifikovat i nové, dříve nepopsané nádorové varianty. Srovnání zárodečného genomu s genomem nádorovým poskytne skutečně komplexní pohled na specifické změny přítomné v nádorovém genomu. Nicméně množství dat získané pomocí WGS klade vysoké nároky na jejich interpretaci, a to především v případě náhodných potenciálně rizikových, ale těžce interpretovatelných nálezů.
Celoexomové sekvenování svojí šíří pokrývá pouze kódující oblasti genomu, tedy přibližně 1 % z jeho celkové sekvence. Nicméně přibližně 85 % patogenních variant se nachází právě v kódujících oblastech, což činní výstupy WES oproti WGS jednodušší z hlediska interpretace. Nižší šíře pokrytí WES umožňuje oproti WGS větší hloubku čtení, a tudíž i vyšší senzitivitu pro záchyt variant v heterogenním vzorku nádorové tkáně. Z hlediska klinicky významného informačního obsahu představuje WES velice dobrou alternativu k WGS.
V současnosti je pro účely precizní onkologie nejvíce používáno cílené (hotspot) sekvenování založené na tzv. sekvenačních panelech. V rámci tohoto přístupu je sekvenován předem definovaný panel řádově desítek až stovek genů významných v nádorové biologii, případně pro terapeutické plánování. Tento přístup má oproti WGS a WES řadu výhod především z praktického hlediska. Ještě menší šíře pokrytí než v případě WES umožňuje větší hloubku čtení, a zajišťuje tedy nejvyšší senzitivitu, což může mít svůj význam především u nádorových biopsií s nízkým zastoupením nádorových buněk. Sekvenační panely jsou méně finančně nákladné, jednodušší z hlediska interpretace a nevedou k náhodným nálezům. Z hlediska technologického panely umožňuji lepší optimalizaci sekvenačního protokolu a vyšší míru jeho standardizace. Mezi komerčně nabízenými sekvenačními panely se setkáme s panely obsahujícími jak desítky, tak i stovky genů, přičemž s narůstajícím počtem genů zařazených do panelu logicky narůstá také pravděpodobnost záchytu klinicky relevantní mutace a rovněž přesnost výpočtu tzv. mutační nálože nádoru (viz níže). Na druhou stranu cílené (hotspot) sekvenování, nikdy nepokrývá všechny potenciálně klinicky relevantní genomové varianty, takže pravděpodobnost jejich záchytu pomocí WGS a WES bude proto vždy vyšší. Informace o individuálních nádorových exomech či genomech má rovněž významnou hodnotu pro translační výzkum v onkologii.
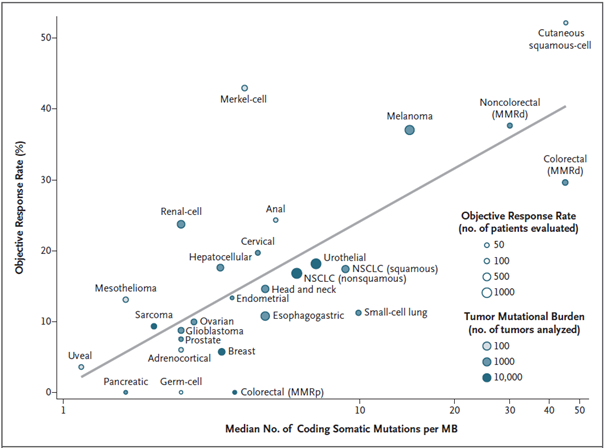
Ať již použijeme kteroukoliv z výše uvedených aplikací genomového sekvenování, je možné její výstupy použít pro výpočet mutační nálože nádoru neboli TMB (viz výše). V případě sekvenačních panelů lze výpočet TMB provést pouze v případě, že panel pokrývá 300 a více genů (odpovídá přibližně 1 milionu bází = 1 Mb). Teprve od tohoto limitního počtu genů začíná hodnota TMB získaná ze sekvenačního panelu korelovat s TMB získanou pomocí WGS a WES. TMB je definována jako počet nesynonymních (protein měnících) mutací na 1 Mb (na milion bází) DNA a jeho hodnota může sloužit jako citlivý ukazatel odpovědi na protinádorovou imunoterapii (viz obr. 5).
Obr. 5 Korelace mutační nálože a objektivní léčebné odpovědi u různých typů nádorů (dle: Yarchoan et al., 2017)
Závěr – role sekvenování nové generace ve fungování molekulárně onkologické indikační komise
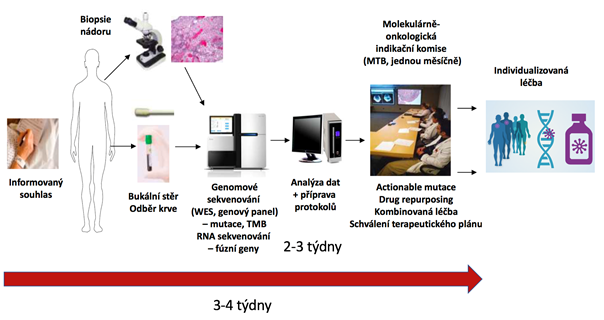
Použitím jakékoliv z výše uvedených aplikací NGS získáme více či méně komplexní informaci o individuálním nádorovém genomu. Ta je pak použita jako podklad pro přípravu detailních výsledkových protokolů obsahujících přehled klinicky relevantních variant a stupeň klinické evidence jejich vztahu ke konkrétnímu preparátu a léčebné odpovědi. Takto připravené protokoly jsou následně projednávány v kontextu celkového klinického stavu pacienta na multioborové molekulárně onkologické indikační komisi (MTB – molecular tumor board, viz obr. 6) s cílem vytvoření individuálního terapeutického plánu založeného na biologických vlastnostech daného tumoru.
Obr. 6 Typické schéma zapojení genomového sekvenování do fungování molekulárně onkologické indikační komise
Zdroje:
- Carter T. C., He M. M. Challenges of identifying clinically actionable genetic variants for precision medicine. J Healthc Eng 2016; 2016, doi: 10.1155/2016/3617572.
- Chakravarty D., Gao J., Phillips S. M. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017; 2017, doi: 10.1200/PO.17.00011.
- Lawrence M. S., Stojanov P., Polak P. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499 (7457): 214–218, doi: 10.1038/nature12213.
- Moghadasi S., Eccles D. M., Devilee P. et al. Classification and clinical management of variants of uncertain significance in high penetrance cancer predisposition genes. Hum Mutat 2016; 37 (4): 331–336, doi: 10.1002/humu.22956.
- Moorcraft S. Y., Gonzalez D., Walker B. A. Understanding next generation sequencing in oncology: a guide for oncologists. Crit Rev Oncol Hematol 2015; 96 (3): 463–474, doi: 10.1016/j.critrevonc.2015.06.007.
- Strom S. P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol Med 2016; 13 (1): 3–11, doi: 10.28092/j.issn.2095-3941.2016.0004.
- Yarchoan M., Hopkins A., Jaffee E. M. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017; 377 (25): 2500–2501, doi: 10.1056/NEJMc1713444.
Byl pro Vás kurz přínosný? Rádi byste se k němu vyjádřili? Napište nám − Vaše názory a postřehy nás zajímají. Zveřejňovat je nebudeme, ale rádi Vám na ně odpovíme.