
Biochemical Properties of Highly Neuroinvasive Prion Strains
Infectious prions propagate from peripheral entry sites into the central nervous system (CNS), where they cause progressive neurodegeneration that ultimately leads to death. Yet the pathogenesis of prion disease can vary dramatically depending on the strain, or conformational variant of the aberrantly folded and aggregated protein, PrPSc. Although most prion strains invade the CNS, some prion strains cannot gain entry and do not cause clinical signs of disease. The conformational basis for this remarkable variation in the pathogenesis among strains is unclear. Using mouse-adapted prion strains, here we show that highly neuroinvasive prion strains primarily form diffuse aggregates in brain and are noncongophilic, conformationally unstable in denaturing conditions, and lead to rapidly lethal disease. These neuroinvasive strains efficiently generate PrPSc over short incubation periods. In contrast, the weakly neuroinvasive prion strains form large fibrillary plaques and are stable, congophilic, and inefficiently generate PrPSc over long incubation periods. Overall, these results indicate that the most neuroinvasive prion strains are also the least stable, and support the concept that the efficient replication and unstable nature of the most rapidly converting prions may be a feature linked to their efficient spread into the CNS.
Published in the journal:
. PLoS Pathog 8(2): e32767. doi:10.1371/journal.ppat.1002522
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002522
Summary
Infectious prions propagate from peripheral entry sites into the central nervous system (CNS), where they cause progressive neurodegeneration that ultimately leads to death. Yet the pathogenesis of prion disease can vary dramatically depending on the strain, or conformational variant of the aberrantly folded and aggregated protein, PrPSc. Although most prion strains invade the CNS, some prion strains cannot gain entry and do not cause clinical signs of disease. The conformational basis for this remarkable variation in the pathogenesis among strains is unclear. Using mouse-adapted prion strains, here we show that highly neuroinvasive prion strains primarily form diffuse aggregates in brain and are noncongophilic, conformationally unstable in denaturing conditions, and lead to rapidly lethal disease. These neuroinvasive strains efficiently generate PrPSc over short incubation periods. In contrast, the weakly neuroinvasive prion strains form large fibrillary plaques and are stable, congophilic, and inefficiently generate PrPSc over long incubation periods. Overall, these results indicate that the most neuroinvasive prion strains are also the least stable, and support the concept that the efficient replication and unstable nature of the most rapidly converting prions may be a feature linked to their efficient spread into the CNS.
Introduction
Prion diseases are fatal neurodegenerative disorders that include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in cervids (reviewed in [1]). These disorders are caused by misfolding of the cellular prion protein, PrPC, into a β-sheet-rich, aggregated isoform known as PrPSc [2]–[5]. PrPSc can exist as distinct conformational variants or strains, which show strikingly different disease phenotypes even when PrPC sequences are identical [6]–[10]. Prions strains may vary in their aggregate size, stability in chaotropes, PrP epitopes exposed, glycosylation profile, and core of shielded hydrogen atoms as assessed by H/D exchange and mass spectrometry [11]–[15]. Nonetheless, the critical conformational features of PrPSc that drive rapidly lethal disease remain unclear.
Conformational determinants of PrPSc that impact the key events in prion pathogenesis are emerging. Incubation periods from time of exposure to terminal disease vary widely among prion strains, sometimes by more than two-fold [16]. Prion stability, or resistance to denaturation, has been assessed in chaotrope-based assays and has revealed that short incubation period strains correlated with unstable PrPSc in mouse [17], yet with stable PrPSc in hamster [18]. Together these findings suggest that other factors such as differences in prion structure or cellular processing influence survival times.
PrPSc particle size varies among prion aggregates, from oligomers to long fibrils, with the most highly infectious PrPSc size identified as small oligomers [19], [20]. Prion strains also show differences in the amount of proteinase K (PK)-sensitive versus PK-resistant aggregates [21], [22], with some of the most virulent strains having an estimated 80% of aggregates being PK-sensitive [13], [21], [23]. Additionally, the electrophoretic mobility of the PK-resistant core can differ between strains [11]. Thus, the conformational variability among distinct prion strains is pronounced and correlates with some features of disease.
Following entry of prions, many prion strains accumulate in lymphoid tissue during the early stages of disease. Prions spread to the CNS through peripheral nerves in a process known as neuroinvasion [24], [25]. Although it has not yet been directly demonstrated how PrPSc transits via peripheral nerves, a wealth of indirect evidence exists that prion transport by nerves is a major entry route into the brain [26]–[30]. Manipulating the density of nerves or the distance between nerves and the PrPSc peripheral reservoirs in lymphoid tissue has a robust impact on neuroinvasion [26], [27], [31]. Additionally, gastric, ocular, or lingual exposure to prions induces prion replication initially at CNS sites with direct neural connections to the entry site, also suggestive of neural transport of prions into the CNS [30], [32]–[34]. Yet little is known about how strains differ in their capacity for neuroinvasion.
Neuroinvasion is dependent upon (1) host and (2) the specific strain. Certain critical host factors that enhance neuroinvasion, such as CD21/35 complement receptor or C1q, have been identified through prion infection studies in which receptors or complement were depleted [35]–[37]. Yet within the same host species, distinct strains show varying abilities to invade the CNS. For example, the mouse-adapted sheep scrapie strain 87 V is poorly neuroinvasive whereas strain 22A is highly neuroinvasive [38]. Yet, the structural determinants that underlie strain differences in CNS entry are unknown. Here we investigated the biochemical properties of prion strains that efficiently or poorly invade the CNS. We demonstrate that the most rapidly lethal strains are highly neuroinvasive, physically unstable, and form primarily diffuse aggregates in the brain.
Results
The capacity of prions to neuroinvade is strain dependent
To compare the pathogenesis of prion strains, we inoculated WT mice (VM/Dk background) with mouse-adapted strains 22 L or 87 V by intracerebral (IC) or intraperitoneal (IP) routes. Prion infection in brain was determined by three independent assays: western blot, histoblots, and immunohistochemistry. Following IC inoculation, both 22 L and 87 V led to terminal prion disease in all mice, although with a significantly longer incubation period in 87 V-exposed mice (22 L: 200±2 days; 87 V: 302±2 days) (Figure 1A). Yet after IP inoculation, only 22 L was neuroinvasive and led to the consistent development of clinical prion disease and PrPSc in the brain (5/5, 100% attack rate, Figure 2A, left panel). No prions were detected in the brains of mice inoculated IP with strain 87 V (0/6), consistent with previous reports indicating that CNS invasion was inefficient for this strain (Figure 2A, right panel).


To determine how additional strains vary in their ability to neuroinvade, we inoculated mice expressing WT mouse PrPC (Tga20 mice) with mouse-adapted CWD (mCWD) and mouse-adapted scrapie strains RML, 22 L, and ME7 by the IC route. Tga20 mice overexpress a WT PrP sequence that varies from VM/DK mice by two dimorphisms (108L/F and 189T/V) [39]. Here we again found that following IC inoculation, the strains led to disease with 100% attack rate but showed significant differences in the time to terminal prion disease. The mean incubation period for strains RML, 22 L, and ME7 was 71–85 days, whereas the mean incubation period for strain mCWD was significantly longer at 164 days (Figure 1A). Upon IP inoculation of RML and mCWD, the rapid strain RML developed prion disease with an incubation period that was extended 40 days beyond that of the IC route and had an attack rate of 100% (n = 10). In contrast, the slow mCWD strain developed prion disease with an incubation period of approximately 460 days and a very low attack rate of 20% (3/15) (Figure 2B). ME7 and 22 L are well-established to be strongly neuroinvasive strains following IP inoculation [35], [40]–[43]. Taken together with the WT mouse infections, we found that the rapid strains (22 L, RML) tended to be strongly neuroinvasive with all mice developing prion disease following a peripheral exposure, whereas the slower strains (87 V, mCWD) tended to be weakly or non-neuroinvasive.
Morphologic properties of strongly and weakly neuroinvasive (NI) prions
We found dramatic differences in the PrPSc aggregate morphology in brains of mice that were IC inoculated with strongly and weakly NI strains. In the WT brain, the strongly NI strain, 22 L, led to diffuse PrPSc deposits that did not stain with the amyloid binding dye Congo red, hence deposits are referred to as noncongophilic. In contrast, the weakly NI strain, 87 V, led to dense, large aggregates that bound CR, and are referred to as congophilic (Figure 1B). Similarly, in the Tga20 brain, RML, ME7, 22 L, all strongly NI strains [31], [35], [40]–[43], led to diffuse, fine 2–5 µm aggregates and occasionally small 10–15 µm plaques that were noncongophilic, whereas the weakly NI strain, mCWD, led to focal, dense, large 20–50 µm plaques that were congophilic (Figure 1B). Ultrastructurally, the strongly NI strains had no visible fibrils consistent with previous reports for ME7 and RML [44], [45], whereas the weakly NI prion mCWD consistently had long fibrils visible within the plaques (Figure 1C and Figure S1). Plaques of fibrils have been previously observed for 87 V [45].
We then compared the prion aggregate morphology of the same strain entering the brain following either an IC or IP exposure. The strongly NI strains showed identical aggregate morphologies in the brains of mice exposed either by the IC or IP routes (Figure 2A,B). However in the weakly NI strains, there were surprising differences in the PrPSc aggregate morphology. After IP inoculation, the weakly NI strains showed none of the large congophilic aggregates in the brain that were seen after the IC inoculation, although scattered diffuse aggregates were seen in the brain of three Tga20 mice IP exposed to mCWD (data not shown).
Peripheral prion replication varies among the strongly and weakly NI prion strains
We next assessed whether PrPSc accrued in the lymphoid tissues after IP inoculation with the different strains. Here we found that PrPSc was detectable in spleens of all mice inoculated with the strongly NI strains RML and 22 L, but in none of the weakly NI 87 V-inoculated and only approximately 20% of mCWD-inoculated mice (Figure 2). Thus, the strongly NI strains also developed a more robust accumulation of prions in the lymphoid tissues.
Electrophoretic mobility and glycoform profiles of the strains
Having found that the less neuroinvasive prions form large, dense congophilic core plaques in histologic sections suggested that there were structural differences among the strains that may alter their ability to spread to the brain. To test this hypothesis, we first investigated the structural and biochemical differences underlying the biological properties of the strongly and weakly NI prions. We compared the mobility of the PK resistant core fragment and the ratios of di-, mono - and unglycosylated PrPSc glycoforms. In assessing the PK-resistant core fragment, we found no differences in the electrophoretic mobility among the strains. The glycoform ratios varied among the strains, however neither the PK-resistant PrP core size nor the glycoform profile correlated with neuroinvasion ability (data not shown).
Strongly NI strains degrade with limited proteolysis at low chaotrope concentrations
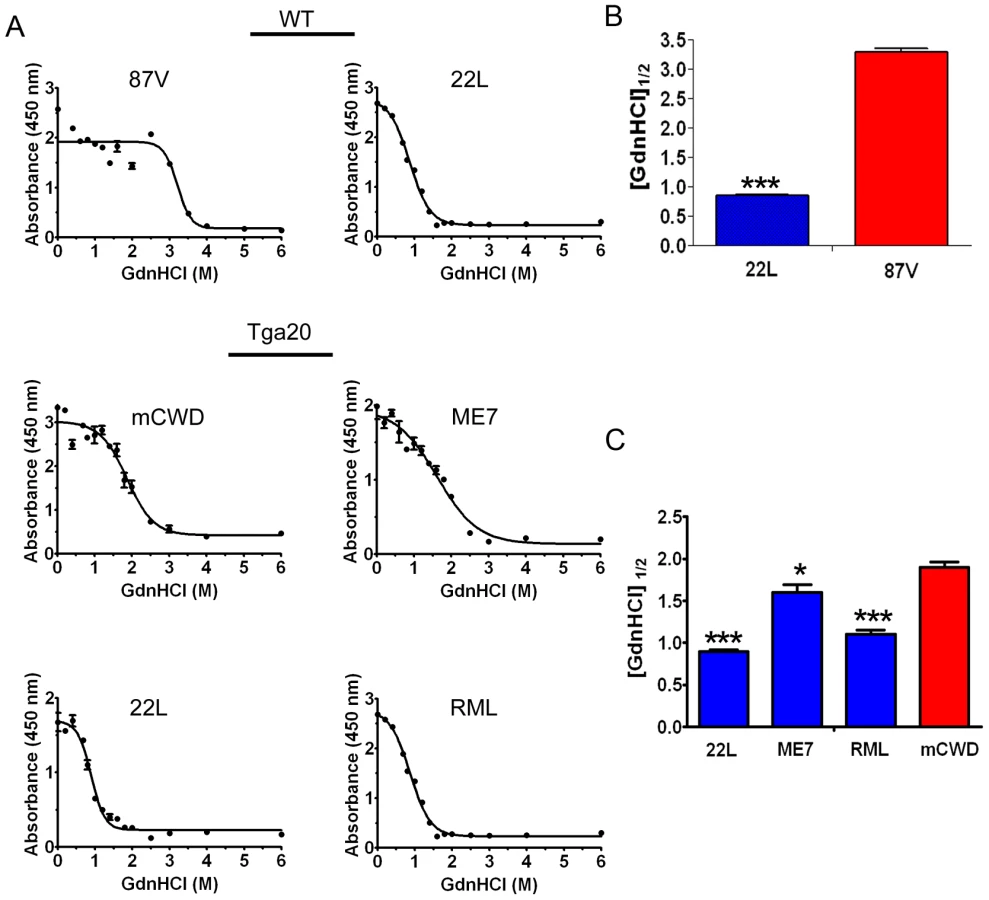
Prion strains have been shown to vary dramatically in their conformational stability. To next determine how the strongly and weakly NI prions varied in their stability in chaotropes, we exposed brain homogenates to 14 concentrations of guanidine hydrochloride (GdnHCl) ranging from 0 M to 6 M. We then diluted the samples to 0.15 M GdnHCl, digested with PK, denatured and measured the PrPSc remaining by ELISA, and calculated the [GdnHCl]1/2. ELISA was used to quantify PrPSc since western blots of murine prion strains showed no visible mobility shift in PK-resistant PrPSc at the higher GdnHCl concentrations (data not shown). This suggests global PrPSc denaturation and complete PrP digestion with PK occurs, consistent with the results of Peretz and colleagues [46]. Interestingly, the strongly NI, nonfibrillar strains showed the lowest [GdnHCl]1/2, which was 0.9±0.2, 1.1±0.1 and 1.6±0.1 for the 22 L, RML and ME7 strains, respectively. The [GdnHCl]1/2 was significantly higher for the weakly neuroinvasive strains mCWD at 1.9±0.1 and 87 V at 3.2±0.1. (Figure 3A–C). These results indicate that the strongly NI, nonfibrillar, noncongophilic strains 22 L, RML, and ME7 were less stable in chaotropes as compared to the weakly NI, fibrillar, congophilic strains mCWD and 87 V.
Strongly NI strains readily disassemble at low temperatures
To further confirm the stability differences among strains, we also assessed the thermostability of the prion aggregates. Here we digested brain homogenate with PK and exposed PrPSc to a thermal gradient from 25–99°C in the presence of 1.6% SDS for 6 minutes, then performed one dimensional denaturing gel electrophoresis and quantified levels of monomeric PrP, as has been previously shown with yeast prions [47]. These measurements indicate how readily the aggregates disassemble into monomers at increasing temperatures. We determined the temperature at which half of the total PrPSc, measured at 99°C, appeared as monomers (T1/2). The thermostability and chaotrope stability measurements showed remarkable agreement. Whereas the strongly NI strains showed T1/2 from 61–70°C, the weakly NI, fibrillar strains disassembled at higher temperatures, with a T1/2 from 86–90°C (Figure 4). These results again indicated that the strongly NI, noncongophilic strains formed less stable aggregates.

Strongly neuroinvasive prion strain has high levels of insoluble PrPSc
To assess the relative efficiencies of PrP conversion, we measured the amount of soluble and insoluble PrP in brain at terminal disease for each WT strain. The insoluble PrP fraction was significantly higher in the strongly NI 22 L versus the weakly NI 87 V, at 90%±1 versus 53%±7 insoluble PrP, respectively (Figure 5). The 22 L strain notably accumulated 11±4-fold more total PrPSc at terminal disease than 87 V, which occurred over a shorter incubation period. These data suggest that the noncongophilic 22 L is more efficient at rapidly converting PrPC in vivo as compared to the congophilic 87 V strain. The Tga20 strains showed highly variable insoluble∶soluble PrP ratios, likely due to the high PrPC expression.

Since the relative level of PK-sensitive PrPSc may influence the neuroinvasive ability of a strain, we quantified the total insoluble and PK-resistant PrPSc of strains 22 L and 87 V. PK-digested and non-digested aliquots of brain homogenate were ultracentrifuged, and PrPSc was measured in the pellet fractions. Interestingly the levels of PK-resistant PrPSc were similar and consisted of 40±3% and 46±6% of the total insoluble PrP for 22 L and 87 V, respectively, indicating that >50% of the total insoluble PrPSc was PK-sensitive (Figure S2).
To determine whether the total insoluble PrPSc more readily dissembled into monomers at lower temperatures as compared to the PK-resistant fraction, the thermostability measurements were repeated on total insoluble PrPSc in the absence of PK. The T1/2 was similar to the values determined for the PK-resistant PrPSc for both the 22 L and 87 V (T1/2: 22 L = 65°C±5; 87 V = 92°C±3) (Figure S3).
Discussion
Infectious prions invade the body through peripheral sites such as the gastrointestinal tract, and following amplification in lymphoid tissues for some strains, spread mainly via peripheral nerves into the CNS [48]. Experimentally, certain fibril-forming prion strains replicate peripherally and do not enter the CNS, leading to a persistence of prions in extraneural tissues [24], [38], [49]. GPI-anchorless prion strains are also fibrillar and also show infrequent neuroinvasion after inoculation by tongue, ocular, intravenous, or intraperitoneal routes [50]. Natural infection with similar weakly or non-neuroinvasive strains may yield asymptomatic, long-term carriers of infectious prions, thus could pose a risk for transmission to other humans or animals. Here we investigated the pathologic phenotype and the biochemical properties of strongly and weakly neuroinvasive prion strains using a range of assays. Our results establish that the strongly neuroinvasive murine strains are less stable and efficiently accumulate PrPSc over a short incubation period.
The strongly and weakly neuroinvasive prions form distinct aggregate morphologies after intracerebral challenge
The strongly and weakly neuroinvasive mouse strains showed profound differences in the PrP aggregate morphology and incubation period to terminal disease. After intracerebral inoculation, strongly neuroinvasive strains form diffuse, nonfibrillar PrP aggregates and mice rapidly progressed to terminal disease. In contrast, weakly neuroinvasive strains form dense, congophilic, fibrillar plaques and mice slowly progressed to terminal disease. These findings suggest that the congophilic, fibrillar PrPSc accumulates slowly or is less toxic, the latter consistent with recent evidence that fibrils in general are less toxic than oligomers [51]. Interestingly, there are new reports of strains that form large, dense plaques and show prolonged incubation periods, such as vCJD prions in transgenic mice expressing human 129VV PrPC [52] and Gerstmann–Straussler-Scheinker (GSS) prions in TgPrP101LL mice [53]. Similar to our mCWD strain, the vCJD and GSS mouse strains show a predilection for the corpus callosum region [52], [53].
PrPSc spread to the CNS
We found that the pathogenesis diverged among the strains inoculated IP, in that only the diffuse, noncongophilic strains were strongly neuroinvasive. Since several murine prion strains inoculated IP require amplification in the lymphoid tissues for efficient neuroinvasion to occur [54], [55], it is possible that the strongly neuroinvasive prion strains replicate readily to higher levels in lymphoid tissue, which may facilitate spread through peripheral nerves. Here the strongly neuroinvasive strains had more abundant PrPSc aggregates in the spleen, and had a short incubation period after IC inoculation. Together these findings suggest that the strongly neuroinvasive strains could propagate efficiently in lymphoid tissues, which may enhance neuroinvasion.
What is the biochemical basis for a strain's efficient spread to the CNS? Marked differences were detected in the stability assays where the strongly neuroinvasive strains were the most unstable. The stability could account for the differences in the disease pathogenesis. First, it is possible that the unstable strains have small PrPSc aggregates that efficiently spread via peripheral nerves to the CNS. Consistent with this hypothesis, previous studies have shown that small or low density PrP aggregates were unstable and rapidly lethal after IC inoculation [20]. Whether these strains also spread efficiently after IP inoculation is unknown. Second, the higher stability of the congophilic strains may lead to such slow fragmentation and PrPC recruitment that incubation times exceeding the mouse lifespan would be required for prion replication in spleen and spread to the CNS. Although we did not detect splenic PrPSc in 87 V-infected mice, others have shown with sensitive infectivity assays that 87 V accumulates in the spleen at early timepoints post-inoculation [38]. This suggests that the failure of 87 V to spread to the CNS was not due to a delayed replication in the spleen [38]. A third possibility is that instability of a strain generates high levels of infectious particles necessary to initiate peripheral nerve spread. Indeed, a report of 87 V indicates that very high doses of infectious prions can lead to brain entry [56]. The lymphoid tissues may serve to amplify PrPSc to high levels near nerve terminals, and thus efficient lymphoid PrPSc amplification may be crucial for neuroinvasion after IP inoculation. Alternatively, the strain may change after replication in the spleen. Lastly, it is possible that the instability does not underlie the efficient spread of prions, but that there is another yet undiscovered factor, such as a PrPSc interacting protein, that is required for spread. These possibilities are not mutually exclusive.
PK-sensitive PrPSc
Previous studies indicate that for many strains, a major fraction of PrPSc consists of PK-sensitive conformers [57], consistent with our findings of 87 V and 22 L. In human CJD cases, higher levels of stable PK-sensitive conformers were associated with extended disease duration [58]. Similarly, in hamster strains, high levels of PK-sensitive PrPSc correlate with longer incubation periods [13]. In mice, the level of PK-sensitive forms was found to be higher in RML as compared to ME7 [59]. Although a full characterization of the role of the PK-sensitive and resistant conformers in neuroinvasion is beyond the scope of this study, it will be interesting in future studies to determine which fraction is more involved in neuroinvasion.
Congophilic prions
Is congophilia predictive of biological behavior? CR staining has been a gold-standard for the histologic diagnosis of amyloid fibrils, in which tightly interdigitated β-sheets are aligned perpendicular to the fibril axis. Recently, CR was shown to bind parallel to the fibril axis in a surface groove of yeast Het-s fibrils, and to require electrostatic interactions for binding [60]. One key residue exchange in Het-s abrogated the binding of CR, even though fibrils were present, thus CR-negative status of aggregates does not necessarily indicate a lack of fibrillar structure. Nevertheless, the noncongophilic prion strains RML and ME7 used here previously have not revealed fibrils after direct examination of the brain ultrastructure [44], [45]. In studies of partially purified hamster prions, fibrils were found only when PrPSc was truncated by digestion with PK [61], [62]. To our knowledge, fibrils of full length PrP have not been observed for noncongophilic prion strains [62]. In contrast, fibrils are readily detected for strains mCWD [63] and 87 V [45]. Taken together, these studies suggest that for the mCWD and 87 V strains, congophilia correlates with plaques of PrP fibrils.
Prion stability in mice and yeast
Notably, the unstable and stable prion strains reported here share properties with the yeast Sup35 prion strains, Sc4 and SCS. Sc4 prions are thermally unstable, readily fragment to generate short fibrils, and have low levels of soluble monomers, whereas the SCS prions are highly thermally stable, rarely fragment, develop long fibrils, and have high levels of soluble monomers [64]. By comparison, strain 22 L, like Sc4, is thermally unstable and has low levels of soluble monomers, whereas 87 V, like SCS, is highly stable with long fibrils and high levels of soluble monomers. Although we did not directly measure fibril breakage or the propensity to shear, in yeast strains, low thermal stability correlates with high fragmentation rate [64]. Whether this is also true for PrP remains to be determined.
Recent studies in yeast have shown that transmission of a prion aggregate to the daughter cell selects for a certain aggregate size [65], which is governed in part by fragmentation properties. In mice and yeast, the prion stability appears to greatly influence prion amplification, and lends support to the proposed mathematical model that fibril breakage is a dominating factor in the kinetics of prion propagation [66].
This study shows that the more unstable, noncongophilic and nonfibrillar murine prion strains efficiently spread from extraneural sites to the central nervous system. Future studies on the relationship between the biochemical properties of misfolded proteins and the disease phenotype are essential for deciphering how aggregates spread in prion and other protein misfolding diseases.
Materials and Methods
Ethics statement
All procedures involving animals were performed to minimize suffering and were approved by the Institutional Animal Care and Use Committee at UC San Diego. Protocols were performed in strict accordance with good animal practices, as described in the Guide for the Use and Care of Laboratory Animals published by the National Institutes of Health.
Prion inoculations
WT (VM/Dk inbred mice, kindly provided by Dr. Byron Caughey) or Tga20 transgenic mice [67] (groups of n = 4–15 mice) were intracerebrally inoculated into the left parietal cortex with 30 µl of a 0.1% or 1% (w/v) prion-infected brain homogenate prepared from terminally diseased mice. Strain 87 V was a gift from Dr. Thomas Wisniewski. Uninfected brain homogenate served as a negative control. Intraperitoneal inoculations were performed using 100 µl of a 0.1% or 1% prion-infected or uninfected brain homogenate. Mice were monitored three times weekly, and TSE was diagnosed according to clinical criteria including ataxia, kyphosis, stiff tail, hind leg clasp, and hind leg paresis. Mice were sacrificed at the onset of terminal disease and incubation period was calculated from the day of inoculation to the day of terminal clinical disease. Mice were maintained under specific pathogen-free conditions.
Histoblots
Histoblots were performed on tissue cryosections as reported in Taraboulos et al. [68], using up to 100 µg/ml of PK for the digestion of PrPC. Histoblots were developed using the anti-PrP POM1 antibody (epitope in the globular domain, amino acids 121–231, a kind gift from Dr. Adriano Aguzzi) [69].
Histopathology and immunohistochemical stains
Two-µm thick sections were cut onto positively charged silanized glass slides and stained with hematoxylin and eosin, or immunostained using antibodies for PrP (SAF84) or GFAP for astrocytes. For PrP staining, sections were deparaffinized and incubated for 5 min in 88% formic acid, then washed in water for 5 min, treated with 5 µg/ml of proteinase-K, and washed in water for 5 min. Sections were then autoclaved in citrate buffer (pH 6), cooled for 3 min, and washed in distilled water for 2 min. Immunohistochemical stains were performed using the TSA Plus DNP kit (PerkinElmer). Sections were blocked and incubated with anti-PrP SAF-84 (SPI bio; 1∶400) for 45 min followed by anti-mouse HRP (Jackson Immunolabs; 1∶500) for 30 min. Slides were then incubated with anti-DNP-HRP (PerkinElmer, 1∶100) for 30 min, followed by 6 min incubation with DAB. Sections were counterstained with hematoxylin.
Western blotting for PrPSc in brain and spleen
Samples were electrophoresed in 10% Bis-Tris SDS-PAGE gels (Invitrogen), transferred onto a nitrocellulose membrane, and PrP detected using the anti-PrP primary antibody POM1 and an HRP-conjugated anti-mouse IgG secondary antibody. The blots were developed using a chemiluminescent substrate (ECL detection Kit, Pierce) and visualized on a Fuji LAS 4000 imager. Quantification of PrPSc glycoforms was performed using Multigauge V3 software (Fujifilm). Prior to western blotting, PrPSc was concentrated from the brain homogenates of IP inoculated mice by sodium phosphotungstic acid precipitation as previously described [70].
Electron microscopy
Tissues were post-fixed in osmium tetroxide, embedded in epon araldite, sectioned with the ultramicrotome, then collected on grids and post-stained using saturated uranyl acetate solution and bismuth subnitrate. Grids were analyzed with a Zeiss EM10 electron microscope.
Thermostability assay
Brain homogenate in Tris lysis buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 2% sarcosyl) was digested with 50 µg/ml PK for 30 min at 37°C and then treated with phenylmethylsulfonyl fluoride (PMSF) (2 mM final concentration) and Complete TM protease inhibitor (Roche). Individual aliquots were incubated in 1.6% SDS (final) and subjected to temperatures ranging from 25°C to 99°C in 10° intervals for 6 min with shaking at 1000 rpm. Western blotting was performed on the samples by electrophoresis in a 10% Bis-Tris SDS-PAGE gel as described above. PrP signals from monomers were captured and quantified using a Fujifilm LAS-4000 imager and Multigage software. Each strain was analyzed in at least 3 separate experiments using 3–5 mice.
Conformation stability assay
Prion strain stability in guanidine hydrochloride (GdnHCl) was performed as previously described [14] with minor modifications. Briefly, brain homogenates in Tris lysis buffer were incubated in GdnHCl for 1 hr and then diluted with lysis buffer to a final concentration of 0.15 M GdnHCl. Samples were digested with PK at a ratio of 1∶500 (1 µg PK : 500 µg total protein) for 1 hr at 37°C, treated with protease inhibitors, and centrifuged at 18,000 g for 1 hr at 4°C. The pellets were washed with 500 µl of 0.1 M NaHCO3, pH 9.6 and centrifuged for 20 min at 18,000 g. Pellets were denatured in 6 M GdnSCN for 20 min, diluted 2× with 0.1 M NaHCO3 and coated passively onto an ELISA plate. PrP was detected with anti-PrP biotinylated-POM1 antibody and a streptavidin HRP-conjugated anti-mouse IgG secondary antibody. The signals were detected with a chemiluminescent substrate (1-Step Ultra TMB-ELISA, Thermo-Scientific). Each strain was analyzed in at least 3 separate experiments using 3–5 mice. Statistical analysis was performed using a Student's t test.
Quantification of soluble and insoluble PrP
Brain homogenate in Tris lysis buffer was centrifuged at 150,000 g for 1 hr at 4°C. The supernatant and pellet were separately collected. Proteins in the supernatant were precipitated using cold methanol. Supernatant and pellet proteins were then analyzed and quantified by western blotting for PrP. Each strain was analyzed in at least 3 separate experiments using 3–5 mice.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AguzziAPolymenidouM 2004 Mammalian prion biology. One century of evolving concepts. Cell 116 313 327
2. PrusinerSB 1982 Novel proteinaceous infectious particles cause scrapie. Science 216 136 144
3. DeleaultNRHarrisBTReesJRSupattaponeS 2007 Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A 104 9741 9746
4. WangFWangXYuanCGMaJ 2010 Generating a prion with bacterially expressed recombinant prion protein. Science 327 1132 1135
5. MakaravaNKovacsGGBocharovaOSavtchenkoRAlexeevaI 2010 Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 119 177 187
6. FraserHDickinsonAG 1968 The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78 301 311
7. FraserHDickinsonAG 1973 Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J Comp Pathol 83 29 40
8. BruceMEMcBridePAFarquharCF 1989 Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett 102 1 6
9. CollingeJClarkeAR 2007 A general model of prion strains and their pathogenicity. Science 318 930 936
10. TellingGC 2011 Transgenic mouse models and prion strains. Top Curr Chem 305 79 99
11. BessenRAMarshRF 1992 Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66 2096 2101
12. HillAFJoinerSBeckJACampbellTADickinsonA 2006 Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 129 676 685
13. SafarJWilleHItriVGrothDSerbanH 1998 Eight prion strains have PrPSc molecules with different conformations. Nat Med 4 1157 1165
14. PeretzDWilliamsonRALegnameGMatsunagaYVergaraJ 2002 A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34 921 932
15. SmirnovasVBaronGSOfferdahlDKRaymondGJCaugheyB 2011 Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 18 504 506
16. BruceMEMcConnellIFraserHDickinsonAG 1991 The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72 595 603
17. LegnameGNguyenHOPeretzDCohenFEDeArmondSJ 2006 Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A 103 19105 19110
18. AyersJISchuttCRShikiyaRAAguzziAKincaidAE 2011 The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7 e1001317
19. SilveiraJRRaymondGJHughsonAGRaceRESimVL 2005 The most infectious prion protein particles. Nature 437 257 261
20. TixadorPHerzogLReineFJaumainEChapuisJ 2010 The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog 6 e1000859
21. TzabanSFriedlanderGSchonbergerOHoronchikLYedidiaY 2002 Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 41 12868 12875
22. PastranaMASajnaniGOniskoBCastillaJMoralesR 2006 Isolation and Characterization of a Proteinase K-Sensitive PrP(Sc) Fraction. Biochemistry 45 15710 15717
23. SafarJGGeschwindMDDeeringCDidorenkoSSattavatM 2005 Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102 3501 3506
24. KimberlinRHWalkerCA 1988 Incubation periods in six models of intraperitoneally injected scrapie depend mainly on the dynamics of agent replication within the nervous system and not the lymphoreticular system. J Gen Virol 69 2953 2960
25. KleinMAFriggRRaeberAJFlechsigEHegyiI 1998 PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4 1429 1433
26. GlatzelMAguzziA 2000 PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol 81 2813 2821
27. GlatzelMHeppnerFLAlbersKMAguzziA 2001 Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 31 25 34
28. HaikSFaucheuxBASazdovitchVPrivatNKemenyJL 2003 The sympathetic nervous system is involved in variant Creutzfeldt-Jakob disease. Nat Med 9 1121 1122
29. BartzJCKincaidAEBessenRA 2002 Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J Virol 76 5759 5768
30. BartzJCKincaidAEBessenRA 2003 Rapid prion neuroinvasion following tongue infection. J Virol 77 583 591
31. PrinzMHeikenwalderMJuntTSchwarzPGlatzelM 2003 Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425 957 962
32. KimberlinRHWalkerCA 1989 Pathogenesis of scrapie in mice after intragastric infection. Virus Res 12 213 220
33. BeekesMMcBridePABaldaufE 1998 Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol 79 Part 3 601 607
34. KimberlinRHWalkerCA 1986 Pathogenesis of scrapie (strain 263 K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol 67 255 263
35. MabbottNABruceMEBottoMWalportMJPepysMB 2001 Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat Med 7 485 487
36. KleinMAKaeserPSSchwarzPWeydHXenariosI 2001 Complement facilitates early prion pathogenesis. Nat Med 7 488 492
37. ZabelMDHeikenwalderMPrinzMArrighiISchwarzP 2007 Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J Immunol 179 6144 6152
38. CollisSCKimberlinRH 1985 Long-term persistence of scrapie infection in mouse spleens in the absence of clinical disease. FEMS Microbiol Lett 29 111 114
39. MooreRCHopeJMcBridePAMcConnellISelfridgeJ 1998 Mice with gene targetted prion protein alterations show that Prnp, Sinc and Prni are congruent. Nat Genet 18 118 125
40. MabbottNAMcGovernGJeffreyMBruceME 2002 Temporary blockade of the tumor necrosis factor receptor signaling pathway impedes the spread of scrapie to the brain. J Virol 76 5131 5139
41. MabbottNAWilliamsAFarquharCFPasparakisMKolliasG 2000 Tumor necrosis factor alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J Virol 74 3338 3344
42. MabbottNAYoungJMcConnellIBruceME 2003 Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J Virol 77 6845 6854
43. SadowskiMJPankiewiczJPrelliFScholtzovaHSpinnerDS 2009 Anti-PrP Mab 6D11 suppresses PrP(Sc) replication in prion infected myeloid precursor line FDC-P1/22 L and in the lymphoreticular system in vivo. Neurobiol Dis 34 267 278
44. GodsaveSFWilleHKujalaPLatawiecDDeArmondSJ 2008 Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci 28 12489 12499
45. JeffreyMGoodsirCMBruceMEMcBridePAFraserJR 1997 In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol Appl Neurobiol 23 93 101
46. PeretzDScottMRGrothDWilliamsonRABurtonDR 2001 Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci 10 854 863
47. TanakaMChienPNaberNCookeRWeissmanJS 2004 Conformational variations in an infectious protein determine prion strain differences. Nature 428 323 328
48. MabbottNAMacPhersonGG 2006 Prions and their lethal journey to the brain. Nat Rev Microbiol 4 201 211
49. BeringueVLe DurATixadorPReineFLepourryL 2008 Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE 3 e1419
50. KlingebornMRaceBMeade-WhiteKDRosenkeRStriebelJF 2011 Crucial role for prion protein membrane anchoring in the neuroinvasion and neural spread of prion infection. J Virol 85 1484 1494
51. CaugheyBLansburyPTJr 2003 Protofibrils, Pores, Fibrils, and Neurodegeneration: Separating the Responsible Protein Aggregates from the Innocent Bystanders. Annu Rev Neurosci 26 267 98
52. WadsworthJDAsanteEADesbruslaisMLinehanJMJoinerS 2004 Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 306 1793 1796
53. PiccardoPMansonJCKingDGhettiBBarronRM 2007 Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A 104 4712 4717
54. MontrasioFFriggRGlatzelMKleinMAMackayF 2000 Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 288 1257 1259
55. MabbottNAMackayFMinnsFBruceME 2000 Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie [letter]. Nat Med 6 719 720
56. BruceME 1985 Agent replication dynamics in a long incubation period model of mouse scrapie. J Gen Virol 66 2517 2522
57. CronierSGrosNTattumMHJacksonGSClarkeAR 2008 Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416 297 305
58. KimCHaldimanTCohenYChenWBlevinsJ 2011 Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog 7 e1002242
59. ThackrayAMHopkinsLKleinMABujdosoR 2007 Mouse-adapted ovine scrapie prion strains are characterized by different conformers of PrPSc. J Virol 81 12119 12127
60. SchutzAKSoragniAHornemannSAguzziAErnstM 2011 The amyloid-Congo red interface at atomic resolution. Angew Chem Int Ed Engl 50 5956 5960
61. PrusinerSBMcKinleyMPBowmanKABoltonDCBendheimPE 1983 Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35 349 358
62. McKinleyMPMeyerRKKenagaLRahbarFCotterR 1991 Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J Virol 65 1340 1351
63. SigurdsonCJMancoGSchwarzPLiberskiPHooverEA 2006 Strain fidelity of chronic wasting disease upon murine adaptation. J Virol 80 12303 12311
64. TanakaMCollinsSRToyamaBHWeissmanJS 2006 The physical basis of how prion conformations determine strain phenotypes. Nature 442 585 589
65. DerdowskiASindiSSKlaipsCLDiSalvoSSerioTR 2010 A size threshold limits prion transmission and establishes phenotypic diversity. Science 330 680 683
66. KnowlesTPWaudbyCADevlinGLCohenSIAguzziA 2009 An analytical solution to the kinetics of breakable filament assembly. Science 326 1533 1537
67. SigurdsonCJNilssonKPHornemannSHeikenwalderMMancoG 2009 De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proc Natl Acad Sci U S A 106 304 309
68. TaraboulosAJendroskaKSerbanDYangSLDeArmondSJ 1992 Regional mapping of prion proteins in brain. Proc Natl Acad Sci U S A 89 7620 7624
69. PolymenidouMMoosRScottMSigurdsonCShiYZ 2008 The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS ONE 3 e3872
70. WadsworthJDFJoinerSHillAFCampbellTADesbruslaisM 2001 Tissue distribution of protease resistant prion protein in variant CJD using a highly sensitive immuno-blotting assay. Lancet 358 171 180
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Discrete Cyclic di-GMP-Dependent Control of Bacterial Predation versus Axenic Growth in
- Characterising the Mucosal and Systemic Immune Responses to Experimental Human Hookworm Infection
- How Do Microbial Pathogens Make s?
- Substance P Causes Seizures in Neurocysticercosis