
Distinct APC Subtypes Drive Spatially Segregated CD4 and CD8 T-Cell Effector Activity during Skin Infection with HSV-1
HSV-1 is a widely distributed pathogen causing a life-long latent infection associated with periodic bouts of reactivation and severe clinical complications. Adaptive immune responses encompassing CD4+ and CD8+ T-cell activities are key to both the clearance of infectious virus and the control of latent infection. However, precisely how such T-cell responses are regulated, particularly within acutely infected peripheral tissues, remains poorly understood. Using a mouse model of HSV-1 skin infection, we describe a complex regulation of T-cell responses at the site of acute infection. These responses were subset-specific and anatomically distinct, with CD4+ and CD8+ T-cell activities being directed to distinct anatomical compartments within the skin. While IFN-γ-producing CD4+ T cells were broadly distributed, including skin regions a considerable distance away from infected cells, CD8+ T-cell activity was strictly confined to directly infected epithelial compartments. This unexpected spatial segregation was a direct consequence of the involvement of largely non-overlapping types of antigen-presenting cells in driving CD4+ and CD8+ T-cell effector activity. Our results provide novel insights into the cellular regulation of T-cell immunity within peripheral tissues and have the potential to guide the development of T-cell subset-specific approaches for therapeutic and prophylactic intervention in antimicrobial immunity and autoimmunity.
Published in the journal:
. PLoS Pathog 10(8): e32767. doi:10.1371/journal.ppat.1004303
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004303
Summary
HSV-1 is a widely distributed pathogen causing a life-long latent infection associated with periodic bouts of reactivation and severe clinical complications. Adaptive immune responses encompassing CD4+ and CD8+ T-cell activities are key to both the clearance of infectious virus and the control of latent infection. However, precisely how such T-cell responses are regulated, particularly within acutely infected peripheral tissues, remains poorly understood. Using a mouse model of HSV-1 skin infection, we describe a complex regulation of T-cell responses at the site of acute infection. These responses were subset-specific and anatomically distinct, with CD4+ and CD8+ T-cell activities being directed to distinct anatomical compartments within the skin. While IFN-γ-producing CD4+ T cells were broadly distributed, including skin regions a considerable distance away from infected cells, CD8+ T-cell activity was strictly confined to directly infected epithelial compartments. This unexpected spatial segregation was a direct consequence of the involvement of largely non-overlapping types of antigen-presenting cells in driving CD4+ and CD8+ T-cell effector activity. Our results provide novel insights into the cellular regulation of T-cell immunity within peripheral tissues and have the potential to guide the development of T-cell subset-specific approaches for therapeutic and prophylactic intervention in antimicrobial immunity and autoimmunity.
Introduction
Infection results in the priming of pathogen-specific T-cell responses in LNs draining the site of infection. Depending on the nature of the pathogen, this critical step in generating adaptive immunity involves the interaction of naive T cells with various types of migrating and LN-resident DCs [1], [2]. During skin infection with herpes simplex virus (HSV)-1, LN-resident CD8α+ DCs and skin-derived CD103+ DCs can activate naïve CD8+ T-cells, presumably through the cross-presentation pathway involving the acquisition of noninfectious antigen [1]–[4]. By contrast, all subsets of skin-derived migratory DCs, including epidermal Langerhans cells, dermal CD11b+ and dermal CD103+ DCs, in addition to LN-resident CD8α+ DCs acquire the ability to stimulate naive HSV-specific CD4+ T cells [1], [2], [4]. Following appropriate activation by DCs, T cells undergo a program of clonal expansion, which is accompanied by the acquisition of effector functions and the induction of migration molecules that facilitate their infiltration of infected tissues.
While CD4+ helper T cells support the generation of antibody and CD8+ T-cell responses in lymphoid tissues, both CD4+ and CD8+ T-cells also contribute directly to pathogen control at sites of infection [5], [6]. The latter is achieved through two principle effector functions: the contact-dependent elimination of infected tissue cells and the local production of inflammatory and antimicrobial cytokines [5], [6]. The extent to which these T-cell activities contribute to immunity depends on the nature of the infection. For instance, control of non-cytopathic viruses, such as lymphocytic choriomeningitis virus, strictly requires cytolytic T-cell activity [7]. By contrast, immunity against cytolytic viruses, such as vaccinia and vesicular stomatitis virus, does not rely on target cell elimination by T cells [8]. Instead, under circumstances where infection will ultimately result in lytic cell death regardless of T-cell killing, pathogen containment and clearance is dependent on the production of cytokines by effector CD4+ and CD8+ T cells [9]–[11]. Together these diverse effector T-cell (TEFF) activities are essential for efficient immune protection, however, they may also cause the destruction of uninfected tissues, as seen in the context of immunopathology, autoimmunity or transplant rejection. Therefore, a detailed understanding of T-cell-mediated immunity in peripheral tissues forms an essential basis for therapeutic interventions to modulate T-cell responses against both harmful and innocuous antigens. Nevertheless, the cellular and molecular mechanisms controlling T-cell effector activities in nonlymphoid organs remain poorly defined [2], [10].
At its simplest, T-cell effector functions are regulated by T-cell receptor (TCR) stimulation through peptide-MHC complexes on APCs. Importantly in this respect, disengagement of the TCR from antigen-MHC complexes results in the immediate cessation of T-cell cytokine production [10], [12]. This “on-off cycling” of effector activity provides a sophisticated level of antigen specificity and places important temporal and spatial constraints on TEFF-cell responses [10]. As a consequence, effector T cells circulating through the blood or uninfected tissues are thought to shutdown cytokine production and to regain this effector function only upon reencounter with antigen in infected tissues [10]. In addition, noncognate signals delivered through inflammatory mediators and costimulatory molecules, such as interleukin (IL)-18, IL-12, type I IFNs or CD80 and CD86, may also trigger or further modulate T-cell cytokine production and cytotoxic activity [13]–[16]. Thus, the presence of appropriate APCs providing antigen stimulation together with accessory signals is critical in regulating T-cell immunity in situ and targeting effector activities to pathogen-containing tissues [2], [17]. Indeed, various types of professional and nonprofessional APCs, including monocyte-derived inflammatory DCs, B cells, neutrophils and parenchymal cells, have been suggested to elicit T-cell effector functions within nonlymphoid tissues [15], [18]–[21]. Key aspects in this regulation, however, particularly those pertaining to the infection status of APCs and the role of distinct APC subtypes in driving CD4+ versus CD8+ TEFF-cell responses, remain poorly understood.
Here, we define the cellular interactions that control TEFF-cell activity during the course of skin infection with HSV-1. We focus our analysis on the production of IFN-γ, a central component of adaptive immune responses. IFN-γexerts proinflammatory and regulatory effects on a variety of target cells, including the stimulation of antimicrobial activity and the induction of MHC molecules and inflammatory chemokines [5], [6]. Protection from HSV infection strictly requires TEFF-cell activities, with both CD4+ and CD8+ T cells contributing to virus control in skin, mucosa and sensory ganglia [22]–[26]. Moreover, efficient immunity against HSV infection requires IFN-γ [27] and, interestingly, it has been proposed that production of this cytokine rather than cytolytic activity is the major CD8+ T-cell mechanism for virus control in neuronal tissues [28], [29] as well as during lytic infection in genital mucosa [26]. The shared role of IFN-γ as a key effector molecule produced by both CD4+ and CD8+ T cells allowed us to directly compare the regulation of these TEFF-cell subsets side by side. We further took advantage of the tropism of HSV-1 for epithelial tissues [30] to document a distinct anatomical distribution of IFN-producing TEFF-cell subsets in relation to the presence or absence of infectious virus in different microanatomical compartments. Importantly, this unexpected spatial segregation of TEFF-cell effector activity was a direct result of the involvement of largely non-overlapping subsets of professional and nonprofessional APCs in driving CD4+ and CD8+ TEFF-cell responses.
Results
Distribution of IFN-γ-producing TEFF cells
To determine population kinetics and cytokine production by TEFF cells in lymphoid and peripheral tissues, we utilized a skin infection with HSV-1 in combination with adoptive transfer of TCR-transgenic T cells specific for determinants derived from the HSV glycoproteins gB (CD8+ gBT-I cells) [31] and gD (CD4+ gDT-II cells) [4], respectively. Consistent with the tropism of HSV for epithelial tissues, immunofluorescence microscopy (IFM) of skin revealed that infection was largely confined to the epidermal layer and hair follicle epithelium (Fig. S1A). Separation of epidermal and dermal tissue (Fig. S1B,C) revealed that HSV-specific TEFF cells began to infiltrate infected skin around 5 days post-infection, albeit with fewer cells in the smaller epidermal compartment (Fig. 1A). TEFF-cell numbers peaked around 8 days after inoculation and declined thereafter (Fig. 1A).
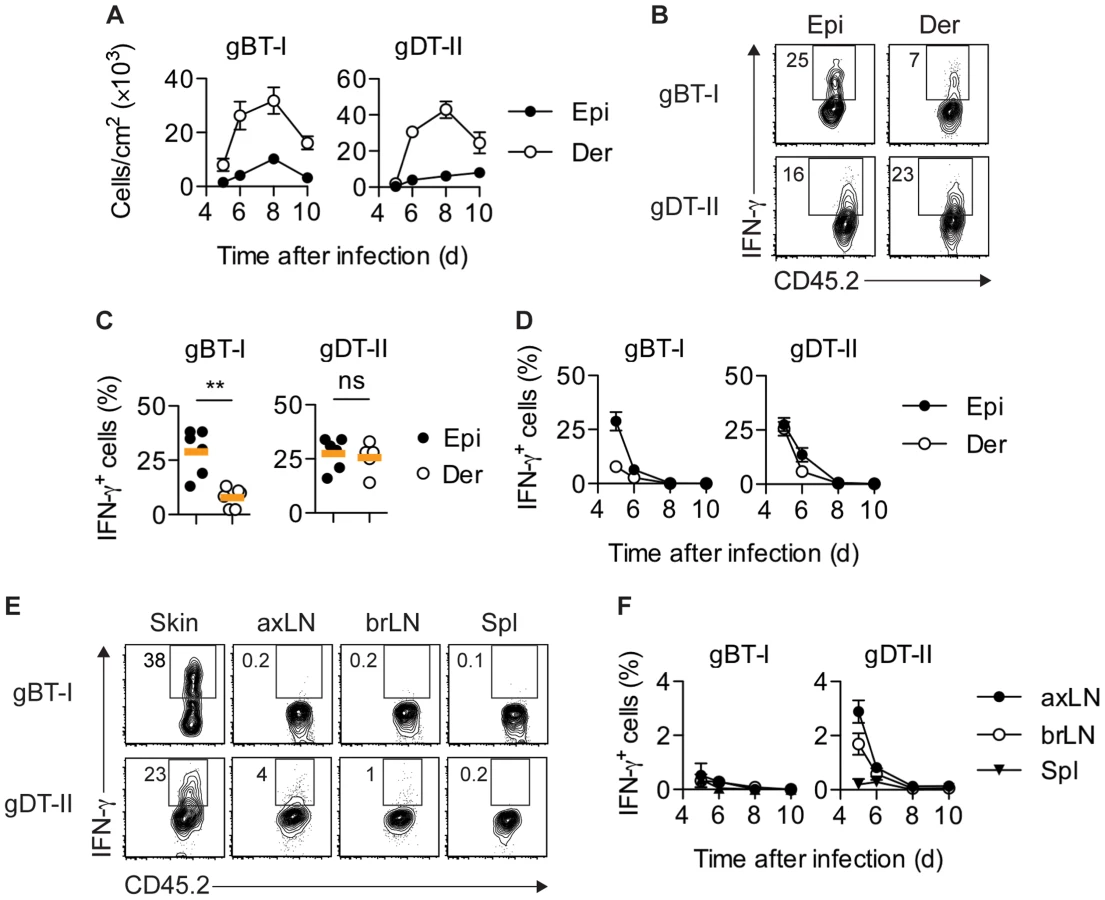
To analyze the production of IFN-γ by TEFF cells in situ, we adopted protocols that facilitate intracellular cytokine staining following exposure to the Golgi inhibitor brefeldin A (BFA), either in vivo after intravenous injection [15], [32] and/or ex vivo immediately after tissue harvest and during enzymatic digestion [21]. Of note, in order to focus our analysis on IFN-γ production in situ, neither of these approaches involved overt restimulation with high concentrations of peptide antigen ex vivo. A considerable portion of gBT-I and gDT-II TEFF cells in the epidermis and dermis of infected skin produced IFN-γ 5–6 days post-infection (Fig. 1B–D). Concomitant with clearance of infectious virus from skin [25], IFN-γ production by TEFF cells ceased around day 7, with virtually no IFN-γ+ TEFF cells present 8 days post-infection (Fig. 1D). Similar kinetics of IFN-γ production were also observed for endogenous CD8+ and CD4+ TEFF cells (Fig. S2A,B). While roughly equal portions of gDT-II cells produced IFN-γ in the epidermal and dermal layers of skin 5 days after infection, the fraction of IFN-γ+ gBT-I cells was approximately 3-fold higher in the epidermis as compared to the dermis (Fig. 1C,D). Note, that dermal preparations contained hair follicles of epithelial origin and therefore also harbored some replicating virus (Fig. S1A–C). The broader distribution of IFN-γ+ gDT-II cells in the skin also extended to lymphoid tissues, with skin-draining axillary and brachial LNs, but not spleen, containing an appreciable fraction of IFN-γ-producing gDT-II cells (Fig. 1E,F). By contrast, IFN-γ+ gBT-I cells were virtually absent from all lymphoid tissues. Together, these results suggested a distinct anatomical distribution of IFN-γ-producing CD4+ and CD8+ TEFF cells in both peripheral and lymphoid tissues.
Confinement of IFN-γ+ CD8+ TEFF cells to skin epithelium
To gain further insight into the microanatomical localization of IFN-γ-producing TEFF-cell subsets, we obtained skin tissue for IFM analysis. Staining of skin sections with anti-IFN-γ antibodies confirmed the presence of IFN-γ-producing cells during the acute phase of infection (Fig. 2A,B), with both endogenous CD4+ and CD8+ (Fig. S2C,D), as well as transgenic gBT-I and gDT-II TEFF cells (Fig. 2C,D) contributing to this response. Interestingly, although gBT-I TEFF cells were broadly distributed throughout the skin, IFN-γ+ gBT-I cells were strictly confined to the epidermis and hair follicle epithelium (Fig. 2C). By contrast, the majority of IFN-γ+ gDT-II cells localized to the dermal layer, where they were found either in association with hair follicles or in considerable distance to the epithelium (Fig. 2D). Thus, in contrast to the strict confinement of IFN-γ+ CD8+ TEFF cells to the epithelium, the dermal layer was the predominant site of the CD4+ T-cell IFN-γ response.

Antigen requirements for IFN-γ production by TEFF cells
The kinetics of IFN-γ production by TEFF cells suggested that the presence of infectious virus was likely to play a role in the induction of cytokine production. This was indeed the case, as gBT-I and gDT-II TEFF cells primed by HSV infection did not produce IFN-γ in non-specifically inflamed skin after treatment with 1-fluoro-2,4-dinitrobenzene (DNFB) (Figs. 3A and S3A). Likewise, in vitro activated gBT-I TEFF cells transferred into HSV-infected mice lacking H-2Kb molecules (H-2Kb−/−) did not produce significant amounts of IFN-γ in the skin (Figs. 3B,C and S3B). Transfer of activated TEFF cells was necessary as H-2Kb−/− mice cannot prime naïve gBT-I cells due to lack of the relevant MHC-I restriction element. IFN-γ production by transferred gBT-I TEFF cells was completely restored in similar experiments following HSV-1 infection of bone marrow chimeric mice in which H-2Kb molecules were expressed exclusively in radioresistant cells, but were absent from the radiosensitive hematopoietic compartment (Figs. 3D and S3C). By contrast, we observed a significant reduction in the frequency of in vivo primed IFN-γ+ gBT-I cells in chimeric mice in which H-2Kb molecules were selectively missing from radioresistant cells, when compared with fully MHC-I-sufficient control chimeras (Figs. 3E and S3D). Interestingly, the overall frequencies of IFN-γ+ gBT-I TEFF cells appeared to be increased in this particular experimental set-up, possibly related to altered immune activation thresholds in previously irradiated recipient mice. Regardless, these results indicated that presentation of viral antigens by radioresistant epithelial cells, such as keratinocytes, Langerhans cells [33] and dendritic epidermal T cells (DETC) [34], was necessary and sufficient for optimal IFN-γ responses by gBT-I TEFF cells.

IFN-γ production by gDT-II cells was also a consequence of antigen recognition, as in vitro activated gDT-II TEFF cells transferred into infected MHC-II-deficient mice (I-A/E−/−) failed to produce IFN-γ (Figs. 3F,G and S3E). Transfer of activated gDT-II cells was necessary to overcome the inability of I-A/E−/− mice to support CD4+ T-cell priming. By contrast, gDT-II TEFF cells primed in vivo produced IFN-γ in chimeric mice in which only radiosensitive, but not radioresistant cells expressed MHC-II molecules, although we observed a moderate, yet significant reduction in the frequency of IFN-γ+ gDT-II cells in this situation (Figs. 3H and S3F). These results implied that bone marrow-derived MHC-II+ APCs were largely responsible for presentation of viral antigens and eliciting cytokine production by CD4+ TEFF cells.
DCs trigger IFN-γ production by CD4+ TEFF cells
Consistent with an involvement of MHC-II-expressing APCs in driving CD4+ TEFF-cell responses, MHC-IIhi cells accumulated in the skin during the first week post-infection [35] (Fig. S4A,B). The majority of these cells had a CD11cintCD11b+ phenotype and further expressed CD64 and MAR-1, identifying them as monocyte-derived inflammatory DCs [36], [37] (Fig. S4C). IFM of infected skin revealed a broad distribution of MHC-II+ cells, with the vast majority localizing to the dermal layer, where they were found in close proximity to IFN-γ+ gDT-II cells (Fig. 4A). Indeed, partial depletion of CD11c+ DCs upon diphtheria toxin (DT) treatment of CD11c.DTR mice resulted in a significant reduction in the frequency of IFN-γ+ gDT-II cells in infected skin (Figs. 4B,C and S5A). Furthermore, treatment of mice with antibodies blocking the costimulatory molecules CD80 and CD86, typically expressed by professional APCs such as DCs, also abrogated the CD4+ TEFF-cell IFN-γ in skin and draining LNs (Figs. 4D and S5B). In stark contrast, costimulation blockade had no bearing on IFN-γ production by gBT-I TEFF cells (Fig. 4D). These experiments suggested that MHC-II+ DCs were the main drivers of cytokine production by CD4+ TEFF cells. Nevertheless, in Ccr2−/− mice, an absence of monocyte-derived DCs [20], [38], which numerically dominated the cutaneous DC network during acute infection, rather increased than decreased the frequency of IFN-γ+ gDT-II and gBT-I cells (Fig. S5C,D), potentially related to impaired virus control in these mice [20]. Furthermore, we observed normal IFN-γ production by gDT-II and gBT-I TEFF cells in the absence of Langerhans cells, CD103+ dermal DC and CD8+ LN-resident DCs upon DT treatment of Langerin.DTR mice [39], [40], and similarly, also in B-cell-deficient μMT mice [41] (Fig. S5E–G). These results suggested a level of redundancy regarding the involvement of different types of professional APCs in regulating CD4+ TEFF activities in infected skin.

Multiple DC subsets activate CD4+ TEFF cells
To more directly establish a role for DCs in CD4+ T-cell responses, we utilized an ex vivo stimulation assay in which APCs purified from infected mice were cocultured with in vitro generated TEFF cells. Initial experiments revealed that maximal IFN-γ production occurred 18 hours after antigen-dependent restimulation for gDT-II and 5 hours for gBT-I TEFF cells (Fig. S6A,B). We purified CD11chi DCs from HSV-infected skin and divided them into CD11bhi and CD11blo subsets (Fig. 5A), with the former expected to contain monocyte-derived and dermal DCs and the latter expected to contain Langerhans cells and CD103+ dermal DCs [4], [37], [40]. Notably, both subsets induced robust IFN-γ production by gDT-II TEFF cells, whereas monocytes (CD11b+CD11c−Ly6Chi) and neutrophils (CD11b+CD11c−Ly6Cint) failed to do so (Fig. 5B,C). Unexpectedly, none of these APCs induced IFN-γ production by gBT-I TEFF cells (Fig. 5B,C). This was not related to potentially compromised expression of H-2Kb molecules after APC isolation, since all subtypes triggered IFN-γ production by gBT-I TEFF cells when pulsed with high doses of gB-peptide prior to cell sorting (Fig. S6C,D). In separate experiments, we specifically sorted CD103+ dermal DCs, as this DC subset is capable of cross-presenting viral antigens to CD8+ T cells during skin infection [4]. Once again, while both CD103+ and CD103− CD11chi DCs triggered IFN-γ production by gDT-II TEFF cells, neither of the two subsets activated gBT-I TEFF cells (Fig. 5D). Remarkably, this disparate response also extended to DCs isolated from LNs draining the site of infection with CD103+, CD11b+, CD8α+ and Langerhans cell-containing CD103−CD11b−CD8α− subsets inducing IFN-γ production by gDT-II, but not gBT-I TEFF cells (Fig. 5E). Of note, gBT-I TEFF cell unresponsiveness towards DC stimulation was observed irrespective of the culture period for 5–18 hours (not shown). Together, these results demonstrated that various skin-derived and LN-resident DC subsets acquired and presented viral antigen for the activation of CD4+ TEFF cells. By contrast, none of these DCs elicited IFN-γ-production by CD8+ TEFF cells, even though at least some of them, namely the CD8α+ and CD103+ subsets, have the ability to present viral antigens for the activation of naïve CD8+ T cells [3], [4].

Epithelial APCs activate CD8+ TEFF cells
Given that IFN-γ+ gBT-I cells were found exclusively in skin epithelium (Figs. 1B–D and 2B), we reasoned that this compartment contained APCs capable of stimulating CD8+ TEFF cells. Therefore, we purified CD45.2+ hematopoetic and CD45.2− parenchymal and stromal cells from epidermal sheets of infected mice by cell sorting and tested their ability to activate CD8+ TEFF cells. Note that here we used expression of CD45.2 to distinguish between hematopoeitc and non-hematopoeitc epidermal cells, whereas in other analyses we used this molecule as a marker for CD45.1+CD45.2+ gBT-I and gDT-II cells. Both fractions induced IFN-γ production by gBT-I TEFF cells, although the keratinocyte-containing CD45.2− subset appeared to be slightly more potent in this regard (Fig. 6A,B). Induction of IFN-γ production was antigen-specific, since co-cultured OT-I TEFF cells of an irrelevant specificity did not respond to either of the APC subsets (Fig. S7A,B). Furthermore, stimulation of IFN-γ production by CD45.2− epidermal APCs was specific for CD8+ TEFF cells since these APCs failed to activate gDT-II TEFF cells (Figs. 6B and S7C). In line with this, the vast majority of CD45.2− epidermal cells from infected skin lacked expression of MHC-II molecules (Fig. S7D). To better define the nature of epidermal APCs capable of stimulating CD8+ TEFF cells, we sorted these cells into keratinocytes (CD45.2−EpCAM+), DCs (CD45.2+CD11chi), DETCs (CD45.2+Vγ3+), as well as residual CD45.2+CD11c− cells. As expected, keratinocytes induced moderate levels of IFN-γ production by gBT-I TEFF cells, and so did epidermal DCs (Fig. 6C,D), in contrast to their counterparts isolated from total skin preparations and LNs (Fig. 5C–E). While residual CD45.2+ epithelial cells had only a weak stimulatory capacity, remarkably, DETCs were by far the most potent APCs triggering IFN-γ production by gBT-I TEFF cells (Fig. 6C,D).

Only infected APCs activate CD8+ TEFF cells
Given that the epidermis was the predominant site of viral replication in vivo, we hypothesized that the stimulatory capacity of epidermal APCs resulted from their direct infection. In agreement, intravital two-photon microscopy of skin infected with a cyan fluorescent protein (CFP)-expressing HSV-1 strain revealed that slow-moving gBT-I TEFF cells were swarming around virally infected epidermal cells (Movie S1). By contrast, gBT-I TEFF cells more distal to infection foci displayed significantly higher mean velocities (Fig. 7A,B). Importantly, using IFM, we observed that IFN-γ+ gBT-I cells co-localized with HSV-infected cells in the epidermis and hair follicle epithelium (Fig. 7C). To further identify infected cells, we inoculated mice with a recombinant strain of HSV-1 expressing green fluorescent protein (HSV.GFP) and analyzed epidermal cells 5 days post-infection using flow cytometry. We observed small numbers of GFP-expressing cells amongst various populations of epidermal cells, including keratinocytes (CD45.2−EpCAM+), DETCs (CD45.2+Vγ3+), Langerhans cells (CD45.2+EpCAM+MHC-IIhi), other DCs (CD45.2+EpCAM−MHC-IIhi), as well as undefined CD45.2+MHC-II− cells (Fig. 7D). As expected, GFP+ cells were absent after infection with the wild-type HSV-1 KOS strain. Next, we sorted GFP+ and GFP− DETCs, MHC-IIhi DCs and keratinocytes from epidermal sheets (Fig. 7E) and tested their ability to stimulate TEFF cells ex vivo. Strikingly, all GFP+, presumably infected, APCs were able to trigger IFN-γ production by gBT-I TEFF cells (Fig. 7F,H). In contrast, IFN-γ production was not elicited by their GFP− counterparts. Finally, DCs, but not DETCs, elicited IFN-γ production by gDT-II TEFF cells, irrespective of their GFP-expression status (Fig. 7G,H). Together, these results indicated that various types of epidermal APCs induced cytokine production by CD8+ TEFF cells. Importantly, direct viral infection was a strict requirement for their stimulatory capacity. Overall, our data highlight a previously unappreciated complexity in the regulation of T-cell effector activity that was subset-specific, microanatomically distinct and involved largely non-overlapping subsets of professional and nonprofessional APCs for CD4+ and CD8+ T-cell responses (Fig. S8).

Discussion
Our results highlight a stringent and complex regulation of TEFF-cell responses that targets effector activities strictly to the site of infection and related lymphoid tissues. Thus, IFN-γ production was limited to the time of acute infection and occurred in an antigen-dependent fashion, requiring in situ restimulation via peptide-MHC complexes on bone marrow-derived professional APCs for CD4+ TEFF cells and on directly infected tissue cells for CD8+ TEFF cells. Consistent with a critical involvement of DCs in peripheral CD4+ TEFF-cell responses, we observed a pronounced accumulation of monocyte-derived inflammatory DCs in infected skin. Such DCs are thought to exert multiple functions, including the local production of inflammatory mediators [42], trafficking of antigen to lymph nodes [43] and replenishment of peripheral DC populations following the resolution of infection [35], [44]. In addition, our experiments employing genetic approaches, ex vivo stimulation assays and costimulation blockade provide compelling evidence that DCs in inflamed skin play a key role in stimulating CD4+ T-cell effector activity. These results reinforce the concept that DCs regulate various aspects of peripheral T-cell responses [2], [15], [20], [21], [45], [46]. In fact, the presence of DCs appeared to be essential for CD4+ TEFF-cell responses as non-professional APCs, such as keratinocytes and DETCs, largely lacked MHC-II expression and failed to elicit IFN-γ production in ex vivo assays. It is possible that certain APC subsets may dominate the regulation of peripheral CD4+ TEFF-cell activities, as suggested for CD11chiCD11bhi dermal DCs after skin injection of model antigens [21] or CCR2-depedent monocyte-derived inflammatory DCs during mucosal HSV-2 infection [20]. Nevertheless, our study revealed a considerable degree of redundancy in this regard, with various DC populations from skin and LNs displaying strong stimulatory capacities for CD4+ TEFF cells. These results imply that all DC subsets, relative to their abundance in infected skin, contribute to CD4+ TEFF activation in vivo. Consistent with this, we observed normal IFN-γ+ production in mice deficient in specific APC subsets, such as monocyte-derived DCs, Langerhans cells, CD103+ dermal DCs or B cells. According to our analysis, monocyte-derived inflammatory DCs are by far the most abundant DC subtype in HSV-infected skin [35] and therefore, may be the major drivers of peripheral CD4+ TEFF-cell responses during HSV-1 skin infection. Nevertheless, our results demonstrate that they may not be essential in this regard as other DC subsets may compensate for their absence.
The IFN-γ response by CD4+ T cells occurred in both draining LNs and the epithelial and dermal layers of infected skin, including regions a considerable distance away from infection foci in the epithelium. This remarkably broad distribution echoes the diverse functions of CD4+ TEFF cells in infection control, ranging from the initiation of antibody class-switching in LNs to the regulation of inflammatory cell infiltration and activity as well as direct antimicrobial effects within infected tissues [5]. Interestingly in this respect, IFN-γ can exert long-range effects on target cells located as far as 80 µm from CD4+ TEFF-cell-APC conjugates, as recently shown for skin infection with Leishmania major [47]. Thus, DC-mediated CD4+ TEFF-cell activation in the dermis may be an essential component of the host defense that restricts infection to the skin epithelium and limits its spread after virus reemergence in sensory nerve endings. The importance of the CD4+ TEFF-cell response is further illustrated by the lack of CD8+ TEFF-cell IFN-γ production in the dermis, as shown here. Supporting this notion, CD4+ TEFF-cell responses are thought to dominate the clearance of HSV-1 from the skin [22], [23], most likely via antibody-independent functions such as direct inflammatory and antiviral activities [5], [48], [49].
In striking contrast to the stimulation requirements for CD4+ TEFF cells, we identified nonprofessional APCs, such as keratinocytes and DETCs, as the main drivers of IFN-γ production by CD8+ TEFF cells. According to our histological analysis, keratinocytes are the most abundant cell type in infected epidermis suggesting that they may be largely responsible for activating CD8+ TEFF cells. In addition, various types of inflammatory cells that infiltrate the epithelial layer during infection may contribute to this response. Importantly, both keratinocytes and DETCs are highly susceptible to direct infection by HSV-1 in vivo [30], [50]. Indeed, their stimulatory capacity, and surprisingly also that of epidermal DCs, was strictly dependent on direct infection. DETCs are invariant γδ-T cells that form a dense network in the epidermis of mice and have been implicated in both innate and adaptive immune responses [51]. Interestingly, it has been speculated that human γδ-T cells may act as professional APCs capable of stimulating naïve CD4+ T cells [52], although we did not find evidence supporting a similar role for DETCs, as they failed to activate CD4+ TEFF cells. Nevertheless, their contribution to CD8+ T-cell responses may be particularly relevant at lesion borders where high numbers of infected DETCs [50] could elicit strong IFN-γ responses required to curb the lateral spread of infection. In addition to DETCs, other epidermal-infiltrating T cells may also act as potent APCs for local CD8+ TEFF cells upon infection with virus [53]. A role for infected T cells and DCs in triggering local CD8+ TEFF-cell activity is further supported by our observation that chimeric mice, in which only radiosensitive APCs could activate CD8+ T cells, had a residual IFN-γ response.
Despite the fact that all DC subsets could activate CD4+ TEFF-cells, only HSV-infected epidermal DCs were capable of activating CD8+ TEFF cell to produce IFN-γ. Strikingly, uninfected DCs from the same location failed to stimulate CD8+ TEFF cells, highlighting the importance of direct infection in determining the outcome of CD8+ TEFF-cell-DC interactions. Previous studies have suggested that various types of professional and nonprofessional APCs, including inflammatory DCs and neutrophils, can trigger IFN-γ production by CD8+ TEFF cells during pulmonary infection with influenza virus [15], [18]. Given the ability of influenza virus to infect a broad range of target cells in addition to its primary tropism for lung epithelial cells [17], it is tempting to speculate that in this situation, direct infection may also be required for CD8+ T-cell activation. Supporting this notion, a large number of inflammatory DCs and neutrophils from influenza virus-infected lungs express viral antigens, most likely as a consequence of direct infection [15], [18], and infected neutrophils display a far superior ability to elicit CD8+ TEFF-cell cytokine production than their uninfected counterparts [18].
One of the more surprising findings from our study was that uninfected DCs failed to elicit IFN-γ production by CD8+ TEFF cells, even though they had access to viral antigen and efficiently activated CD4+ TEFF cells. We have previously shown that LN DCs are able to activate naïve CD8+ T cells at various stages during skin infection, unequivocally demonstrating that they present viral antigens in the context of MHC-I molecules [3], [4], [54]. The CD8α+ and CD103+ DC subsets are of particular interest in this regard, because those DCs cross-present viral antigens in draining LNs 5 days after HSV-1 infection [4], corresponding to the time point analyzed here. Despite this, CD103+ DCs from skin and LNs failed to trigger IFN-γ production by CD8+ TEFF cells. This finding parallels observations with CD8+ T-cell responses to migratory DCs after influenza virus infection, where naïve but not memory T cells proliferate in response to antigen presented on migratory DCs [55].
Although not directly addressed in our study, it is tempting to speculate that the inability of uninfected DCs to activate CD8+ TEFF cells may be a means to prevent their elimination by triggering cytotoxic effector functions. While T-cell killing of DCs has been proposed to represent a negative feedback regulation on T-cell priming [56], [57], it should be noted that prolonged antigen presentation is a common feature in a variety of infections [58]–[61]. Therefore, CD8+ T-cell-mediated elimination of DCs in vivo may be inefficient at best, which is also consistent with our preliminary data demonstrating the failure of gBT-I TEFF cells to lyse CD103+ DCs from LN of HSV infected mice during short-term co-culture. The DC-dependent production of IFN-γ by CD4+ TEFF cells observed in our study further supports this assumption. Of note in this respect, minute amounts of surface antigen can trigger T-cell cytotoxicity [62], [63]. Furthermore, cross-presenting nonprofessional APC, such as liver sinusoidal endothelial cells, can drive CD8+ T-cell TNFα production during viral hepatitis [64]. Thus, it appears unlikely that quantitative differences in antigen presentation between directly infected and cross-presenting DCs alone can explain the CD8+ TEFF-cell unresponsiveness described here. In the light of this, and given that infected epidermal DCs were indeed able to trigger CD8+ TEFF-cell IFN-γ production, we speculate that direct infection may alter the functional status of DCs, for instance through interference with putative inhibitory pathways [65], to allow for the activation of CD8+ TEFF cells. Such modulation of DC stimulation thresholds may be particularly relevant when low levels of peptide-MHC-I complexes are presented to CD8+ TEFF cells. Further studies will be required to elucidate the precise molecular mechanisms operating in DCs and/or T cells to prevent the activation of CD8+ TEFF cells by uninfected cross-presenting DCs.
Overall, the stringent temporal, cellular and molecular constraints on TEFF-cell responses identified in our study are likely in place to prevent collateral damage and autoimmune inflammation initiated by TEFF-cell activation in tissues not involved in infection. Our results are compatible with a scenario where CD8+ T-cell responses are strictly focused on infected tissue compartments, whereas CD4+ responses may induce a more regional state of antimicrobial protection in tissues surrounding infection foci. Having identified dramatically distinct requirements for CD4+ and CD8+ T-cell effector activity, our study has provided novel insights into the regulation of cellular immune responses in nonlymphoid tissues. Such knowledge has the potential to guide the development of T-cell subset-specific approaches for therapeutic and prophylactic intervention in antimicrobial immunity and autoimmunity.
Materials and Methods
Ethics statement
All experiments were done according to Australian NHMRC guidelines contained within the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and under approvals ID1112038 and ID1112345 from the University of Melbourne Animal Ethics Committee.
Mice
C57BL/6, B6.SJL-PtprcaPep3b/BoyJ (B6.CD45.1), gBT×B6.CD45.1 (gBT-I.CD45.1), gBT-I.EGFP, gBT-I.DsRed, OT-I×B6.CD45.1 (OT-I.CD45.1), gDT-II×B6.CD45.1 (gDT-II.CD45.1), gDT-II.EGFP, C57BL/6Ji-Kbtm1N12 (H-2Kb−/−), B6.129S2-H2dlAb1-Ea/J (MHC-II−/−), CD11c.DTR, B6129.CCR2(Ccr2−/−), Langerin.DTR.EGFP (Lg.DTR.EGFP) and μMT mice were bred in the Department of Microbiology and Immunology. gBT-I.CD45.1 and gDT-II.CD45.1 are F1 generation offspring expressing both CD45.1 and CD45.2.
Viruses
HSV-1 KOS, KOS pCMV/EGFP Cre (HSV.GFP) and 17 HSVgDUL47ΔYFP (HSV.CFP) were grown and titrated as previously described [46]. HSV.GFP expresses an EGFP/Cre fusion gene under the control of the CMV-IE promoter from the intergenic space between UL3 and UL4 resulting in EGFP expression by infected cells. HSV.CFP was derived from HSV-1 gDUL47 (strain 17 expressing YFP from UL47 and CFP-tagged gD protein) [66], from which the YFP was removed and sequences from UL47 restored. HSV.GFP and HSV.CFP were made by homologous recombination between parent viral genomes with appropriate transfer plasmids in 293A cells after which green/yellow fluorescence were selected for and against, respectively. Final recombinants were verified by PCR and sequencing of relevant parts of the genome after at least three rounds of plaque purification.
Bone marrow chimeric mice
Mice were irradiated with two of doses 550 cGy 3 hours apart followed by reconstitution with 5×106 T-cell-depleted donor bone marrow cells and treatment with purified anti-Thy1 antibody (clone T24/31.7 from hybridoma supernatants) 1 day later. Chimeric mice were allowed to reconstitute for at least 8 weeks before experiments. WT→I-A/E−/− chimeras were not treated with anti-Thy1 antibody and received 5×106 enriched splenic CD4+ T cells from wild-type mice 1 day and 4 weeks after irradiation.
Viral infections and DNFB, diphtheria toxin and antibody treatments
Mice were infected on their flanks with 1×106 plaque-forming units of HSV-1, as previously described [25]. For DNFB treatment, 15 µL of 0.5% (w/v) DNFB was applied to flank skin, as previously described [67]. For depletion of CD11c+ cells, CD11c.DTR mice were injected with 200 ng diphtheria toxin (DT) or PBS intraperitoneally and intradermally 4 d post-infection. Langerin.DTR mice were injected with 500 ng DT or PBS control intraperitoneally. For costimulation blockade, mice were injected with 0.25 mg anti-CD80 (16-10A1) and -CD86 (GL1) blocking antibodies or rat IgG2 (B81-3) and IgG2a (R35-95) control antibodies (BD Pharmingen) intraperitoneally 4 d post-infection.
Adoptive transfer of transgenic T cells
gBT-I and gDT-II cells were isolated from lymphoid tissues and gDT-II cells were further enriched by positive and negative selection using magnetic beads, as described previously [4]. 5×104 gBT-I or 1×104 gDT-II cells were transferred into naïve mice intravenously via the tail vein, respectively. For transfer of in vitro activated T cells, 1.5×106 transgenic cells were injected.
In vitro activation of transgenic T cells
Splenic gBT-I or OT-I cells were activated with peptide-pulsed splenocytes, as previously described [25]. Purified gDT-II cells were activated by co-culture for 5 days with 4×107 irradiated wild-type splenocytes pulsed with 10 µM of gD315–327 in the presence of 2 µg LPS (Sigma). Cells were cultured in 20 ml RPMI 1640 (Department of Microbiology and Immunology) supplemented with 10% FCS (CSL), 5 mM HEPES (Gibco), 2 mM glutamine (Gibco), 5×10−5 M 2-β-mercaptoethanol (Sigma), antibiotics (Gibco, CSL) (RP-10) and 4 µg lipopolysaccharide. On days 2–4, respectively, all cultures were diluted 1∶2 in fresh medium containing 20 U/mL recombinant human IL-2 (PeproTech).
Isolation of skin cells and ex vivo analysis of IFN-γ production
As indicated, skin tissue was chopped and digested with 3 mg/ml collagenase type 3 (Worthington Biochemicals, USA) and 5 µg DNase (Roche, Germany) for 90 min at 37°C. Alternatively, skin was digested in 2.5 mg/mL dispase II (Roche) diluted in PBS for 90 min at 37°C. Then, the epidermis and dermis were separated mechanically and epidermal sheets were incubated in trypsin/EDTA (0.25%/0.1%) (SAFC Biosciences), while the dermis was chopped and incubated in collagenase type 3 and DNase, as previously described [49]. When analyzing ex vivo IFN-γ production, 10 µg/mL Brefeldin A (Sigma) was included during each enzymatic digestion step. For some experiments, mice were additionally injected with 0.25 mg BFA intravenously 6 hours prior to sacrifice.
Enrichment of DCs
Axillary lymph nodes of HSV-1-infected mice were desiccated with a scalpel blade and digested with continual mixing in RPMI 1640 containing 1 mg/mL collagenase type 3 and 2 µg/mL DNase for 20 min, prior to the addition of 600 µL 0.1 M EDTA and continual mixing for 5 minutes further. DCs were subsequently enriched for by magnetic beads, as previously described [4].
In vitro cultures of T cells and APCs
APC subsets were stained with the appropriate monoclonal antibodies, purified by cell sorting using a FACSAria III (BD Pharmingen) and then washed and resuspended in RP-10. Increasing concentrations of the APCs were cultured with 1.25×104 in vitro activated transgenic T cells in round bottom plates for 5 to 18 hours, as indicated, in the presence of 10 µg/mL BFA for the last 5 hours.
Flow cytometry and antibodies
Antibodies were from BD Pharmingen: anti-CD3 (145-2C11), -CD4 (RM4-5), -CD8α (53-6.7), -CD11b (M1/70), -CD19 (ID3), -CD45.1 (A20), -CD80 (16-10A1), -CD86 (GL1), -IFN-γ (XMG1.2), -Ly6C (AL21), -NK1.1 (PK136), -Vα2 (B20.1) and -Vβ8 (MR5-2); from eBioscience: anti-CD45.2 (104) and -CD11c (N418); or from BioLegend: anti-CD326 (g8.8) and -Vα3.2 (RR2-16). For intracellular staining, cells were fixed with a Cytofix/Cytoperm kit (BD Pharmingen). A FACSCanto II (BD Pharmingen) and FlowJo software (TreeStar) were used for analysis. Propidium iodide (Sigma Aldrich) and SPHERO calibration particles (BD Pharmingen) were added for identification of live cells and enumeration.
Immunofluorescence and confocal microscopy
Skin was fixed at room temperature for 30 min in PLP buffer (0.2 M NaH2PO4, 0.2 M Na2HPO4, 0.2 M L-lysine and 0.1 M sodium periodate with 2% paraformaldehyde), washed twice with PBS and incubated for 30 min in 20% sucrose, prior to being embedded, frozen, cut and stained as previously described [48]. IFN-γ staining (AlexaFluor647, BD Pharmingen) was performed overnight at 4°C (1∶75 in PBS containing 2.5% [w/v] donkey serum). Anti-keratin-5 and -14 polyclonal antibodies were from Jomar Biosciences; anti-CD4 (RM4-5) from BioLegend; anti-CD8 (53.67) from BD Pharmingen and polyclonal anti-HSV from Dako North America. Slides were mounted with ProLongGold antifade media (Invitrogen), air-dried and viewed using a Zeiss LSM700 confocal microscope and Imaris 7.1 software (Bitplane).
Intravital two-photon microscopy
Mice were anesthetized and HSV-1-infected flank skin was mounted on an imaging platform and acquired with an upright LSM710 NLO multiphoton microscope as described previously [48]. Imaging data was processed and automatic cell tracking aided by manual corrections was performed with Imaris 7.1 software. For movies, image sequences were composed in Adobe After Effects CS5.
Statistical analysis
Graphs were plotted using Prism 5 (Graphpad) and comparison of data sets was performed by one-way analysis of variance followed by Tukey post-test, or Mann-Whitney or student t tests, as indicated. All graphs depict means ± s.e.m..
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HeathWR, CarboneFR (2009) Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol 10 : 1237–1244.
2. BedouiS, GebhardtT (2011) Interaction between dendritic cells and T cells during peripheral virus infections: a role for antigen presentation beyond lymphoid organs? Curr Opin Immunol 23 : 124–130.
3. AllanRS, SmithCM, BelzGT, van LintAL, WakimLM, et al. (2003) Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301 : 1925–1928.
4. BedouiS, WhitneyPG, WaithmanJ, EidsmoL, WakimL, et al. (2009) Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 10 : 488–495.
5. SwainSL, McKinstryKK, StruttTM (2012) Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol 12 : 136–148.
6. WongP, PamerEG (2003) CD8 T cell responses to infectious pathogens. Annu Rev Immunol 21 : 29–70.
7. KagiD, LedermannB, BurkiK, SeilerP, OdermattB, et al. (1994) Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369 : 31–37.
8. KagiD, SeilerP, PavlovicJ, LedermannB, BurkiK, et al. (1995) The roles of perforin - and Fas-dependent cytotoxicity in protection against cytopathic and noncytopathic viruses. Eur J Immunol 25 : 3256–3262.
9. KagiD, LedermannB, BurkiK, ZinkernagelRM, HengartnerH (1996) Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol 14 : 207–232.
10. SlifkaMK, WhittonJL (2000) Antigen-specific regulation of T cell-mediated cytokine production. Immunity 12 : 451–457.
11. GouldingJ, AbboudG, TahilianiV, DesaiP, HutchinsonTE, et al. (2014) CD8 T Cells Use IFN-gamma To Protect against the Lethal Effects of a Respiratory Poxvirus Infection. J Immunol 192 : 5415–5425.
12. SlifkaMK, RodriguezF, WhittonJL (1999) Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 401 : 76–79.
13. FreemanBE, HammarlundE, RaueHP, SlifkaMK (2012) Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci U S A 109 : 9971–9976.
14. KohlmeierJE, CookenhamT, RobertsAD, MillerSC, WoodlandDL (2010) Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33 : 96–105.
15. HuffordMM, KimTS, SunJ, BracialeTJ (2011) Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med 208 : 167–180.
16. KupzA, GuardaG, GebhardtT, SanderLE, ShortKR, et al. (2012) NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nat Immunol 13 : 162–169.
17. BracialeTJ, SunJ, KimTS (2012) Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 12 : 295–305.
18. HuffordMM, RichardsonG, ZhouH, ManicassamyB, Garcia-SastreA, et al. (2012) Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS One 7: e46581.
19. IijimaN, LinehanMM, ZamoraM, ButkusD, DunnR, et al. (2008) Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J Exp Med 205 : 3041–3052.
20. IijimaN, MatteiLM, IwasakiA (2011) Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci U S A 108 : 284–289.
21. McLachlanJB, CatronDM, MoonJJ, JenkinsMK (2009) Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity 30 : 277–288.
22. ManickanE, RouseBT (1995) Roles of different T-cell subsets in control of herpes simplex virus infection determined by using T-cell-deficient mouse-models. J Virol 69 : 8178–8179.
23. NashAA, JayasuriyaA, PhelanJ, CobboldSP, WaldmannH, et al. (1987) Different roles for L3T4+ and Lyt 2+ T cell subsets in the control of an acute herpes simplex virus infection of the skin and nervous system. J Gen Virol 68 (Pt 3): 825–833.
24. SimmonsA, TscharkeDC (1992) Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: implications for the fate of virally infected neurons. J Exp Med 175 : 1337–1344.
25. van LintA, AyersM, BrooksAG, ColesRM, HeathWR, et al. (2004) Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol 172 : 392–397.
26. DobbsME, StrasserJE, ChuCF, ChalkC, MilliganGN (2005) Clearance of herpes simplex virus type 2 by CD8+ T cells requires gamma interferon and either perforin - or Fas-mediated cytolytic mechanisms. J Virol 79 : 14546–14554.
27. BouleyDM, KanangatS, WireW, RouseBT (1995) Characterization of herpes simplex virus type-1 infection and herpetic stromal keratitis development in IFN-gamma knockout mice. J Immunol 155 : 3964–3971.
28. KhannaKM, BonneauRH, KinchingtonPR, HendricksRL (2003) Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18 : 593–603.
29. LiuT, KhannaKM, CarriereBN, HendricksRL (2001) Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol 75 : 11178–11184.
30. SimmonsA, NashAA (1984) Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol 52 : 816–821.
31. MuellerSN, HeathW, McLainJD, CarboneFR, JonesCM (2002) Characterization of two TCR transgenic mouse lines specific for herpes simplex virus. Immunol Cell Biol 80 : 156–163.
32. LiuF, WhittonJL (2005) Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J Immunol 174 : 5936–5940.
33. MeradM, ManzMG, KarsunkyH, WagersA, PetersW, et al. (2002) Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol 3 : 1135–1141.
34. HonjoM, ElbeA, SteinerG, AssmannI, WolffK, et al. (1990) Thymus-independent generation of Thy-1+ epidermal cells from a pool of Thy-1 - bone marrow precursors. J Invest Dermatol 95 : 562–567.
35. EidsmoL, AllanR, CaminschiI, van RooijenN, HeathWR, et al. (2009) Differential migration of epidermal and dermal dendritic cells during skin infection. J Immunol 182 : 3165–3172.
36. PlantingaM, GuilliamsM, VanheerswynghelsM, DeswarteK, Branco-MadeiraF, et al. (2013) Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38 : 322–335.
37. TamoutounourS, GuilliamsM, Montanana SanchisF, LiuH, TerhorstD, et al. (2013) Origins and Functional Specialization of Macrophages and of Conventional and Monocyte-Derived Dendritic Cells in Mouse Skin. Immunity 39 : 925–38 doi: 10.1016/j.immuni.2013.10.004
38. BoringL, GoslingJ, ChensueSW, KunkelSL, FareseRVJr, et al. (1997) Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 100 : 2552–2561.
39. KissenpfennigA, HenriS, DuboisB, Laplace-BuilheC, PerrinP, et al. (2005) Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22 : 643–654.
40. PoulinLF, HenriS, de BovisB, DevilardE, KissenpfennigA, et al. (2007) The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med 204 : 3119–3131.
41. KitamuraD, RoesJ, KuhnR, RajewskyK (1991) A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350 : 423–426.
42. LeonB, ArdavinC (2008) Monocyte-derived dendritic cells in innate and adaptive immunity. Immunol Cell Biol 86 : 320–324.
43. LeonB, Lopez-BravoM, ArdavinC (2007) Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26 : 519–531.
44. GinhouxF, TackeF, AngeliV, BogunovicM, LoubeauM, et al. (2006) Langerhans cells arise from monocytes in vivo. Nat Immunol 7 : 265–273.
45. McGillJ, Van RooijenN, LeggeKL (2008) Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med 205 : 1635–1646.
46. WakimLM, WaithmanJ, van RooijenN, HeathWR, CarboneFR (2008) Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319 : 198–202.
47. MullerAJ, Filipe-SantosO, EberlG, AebischerT, SpathGF, et al. (2012) CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity 37 : 147–157.
48. GebhardtT, WhitneyPG, ZaidA, MackayLK, BrooksAG, et al. (2011) Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477 : 216–219.
49. GebhardtT, WakimLM, EidsmoL, ReadingPC, HeathWR, et al. (2009) Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10 : 524–530.
50. PutturFK, FernandezMA, WhiteR, RoedigerB, CunninghamAL, et al. (2010) Herpes simplex virus infects skin gamma delta T cells before Langerhans cells and impedes migration of infected Langerhans cells by inducing apoptosis and blocking E-cadherin downregulation. J Immunol 185 : 477–487.
51. MacleodAS, HavranWL (2011) Functions of skin-resident gammadelta T cells. Cell Mol Life Sci 68 : 2399–2408.
52. BrandesM, WillimannK, MoserB (2005) Professional antigen-presentation function by human gammadelta T Cells. Science 309 : 264–268.
53. RafteryMJ, BehrensCK, MullerA, KrammerPH, WalczakH, et al. (1999) Herpes simplex virus type 1 infection of activated cytotoxic T cells: Induction of fratricide as a mechanism of viral immune evasion. J Exp Med 190 : 1103–1114.
54. AllanRS, WaithmanJ, BedouiS, JonesCM, VilladangosJA, et al. (2006) Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25 : 153–162.
55. BelzGT, BedouiS, KupresaninF, CarboneFR, HeathWR (2007) Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol 8 : 1060–1066.
56. WongP, PamerEG (2003) Feedback regulation of pathogen-specific T cell priming. Immunity 18 : 499–511.
57. YangJ, HuckSP, McHughRS, HermansIF, RoncheseF (2006) Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci U S A 103 : 147–152.
58. StockAT, MuellerSN, van LintAL, HeathWR, CarboneFR (2004) Cutting edge: prolonged antigen presentation after herpes simplex virus-1 skin infection. J Immunol 173 : 2241–2244.
59. TurnerDL, CauleyLS, KhannaKM, LefrancoisL (2007) Persistent antigen presentation after acute vesicular stomatitis virus infection. J Virol 81 : 2039–2046.
60. ZammitDJ, TurnerDL, KlonowskiKD, LefrancoisL, CauleyLS (2006) Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity 24 : 439–449.
61. LinLC, FleschIE, TscharkeDC (2013) Immunodomination during peripheral vaccinia virus infection. PLoS Pathog 9: e1003329.
62. ValituttiS, MullerS, DessingM, LanzavecchiaA (1996) Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med 183 : 1917–1921.
63. SykulevY, JooM, VturinaI, TsomidesTJ, EisenHN (1996) Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 4 : 565–571.
64. WohlleberD, KashkarH, GartnerK, FringsMK, OdenthalM, et al. (2012) TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep 2 : 478–487.
65. OdorizziPM, WherryEJ (2012) Inhibitory receptors on lymphocytes: insights from infections. J Immunol 188 : 2957–2965.
66. DonnellyM, ElliottG (2001) Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J Virol 75 : 2575–2583.
67. MackayLK, StockAT, MaJZ, JonesCM, KentSJ, et al. (2012) Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109 : 7037–7042.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Disruption of Fas-Fas Ligand Signaling, Apoptosis, and Innate Immunity by Bacterial Pathogens
- Ly6C Monocyte Recruitment Is Responsible for Th2 Associated Host-Protective Macrophage Accumulation in Liver Inflammation due to Schistosomiasis
- Host Responses to Group A Streptococcus: Cell Death and Inflammation
- Pathogenicity and Epithelial Immunity