
Identification of the Regulatory Logic Controlling Pathoadaptation by the SsrA-SsrB Two-Component System
Sequence data from the past decade has laid bare the significance of horizontal gene transfer in creating genetic diversity in the bacterial world. Regulatory evolution, in which non-coding DNA is mutated to create new regulatory nodes, also contributes to this diversity to allow niche adaptation and the evolution of pathogenesis. To survive in the host environment, Salmonella enterica uses a type III secretion system and effector proteins, which are activated by the SsrA-SsrB two-component system in response to the host environment. To better understand the phenomenon of regulatory evolution in S. enterica, we defined the SsrB regulon and asked how this transcription factor interacts with the cis-regulatory region of target genes. Using ChIP-on-chip, cDNA hybridization, and comparative genomics analyses, we describe the SsrB-dependent regulon of ancestral and horizontally acquired genes. Further, we used a genetic screen and computational analyses integrating experimental data from S. enterica and sequence data from an orthologous regulatory system in the insect endosymbiont, Sodalis glossinidius, to identify the conserved yet flexible palindrome sequence that defines DNA recognition by SsrB. Mutational analysis of a representative promoter validated this palindrome as the minimal architecture needed for regulatory input by SsrB. These data provide a high-resolution map of a regulatory network and the underlying logic enabling pathogen adaptation to a host.
Published in the journal:
. PLoS Genet 6(3): e32767. doi:10.1371/journal.pgen.1000875
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000875
Summary
Sequence data from the past decade has laid bare the significance of horizontal gene transfer in creating genetic diversity in the bacterial world. Regulatory evolution, in which non-coding DNA is mutated to create new regulatory nodes, also contributes to this diversity to allow niche adaptation and the evolution of pathogenesis. To survive in the host environment, Salmonella enterica uses a type III secretion system and effector proteins, which are activated by the SsrA-SsrB two-component system in response to the host environment. To better understand the phenomenon of regulatory evolution in S. enterica, we defined the SsrB regulon and asked how this transcription factor interacts with the cis-regulatory region of target genes. Using ChIP-on-chip, cDNA hybridization, and comparative genomics analyses, we describe the SsrB-dependent regulon of ancestral and horizontally acquired genes. Further, we used a genetic screen and computational analyses integrating experimental data from S. enterica and sequence data from an orthologous regulatory system in the insect endosymbiont, Sodalis glossinidius, to identify the conserved yet flexible palindrome sequence that defines DNA recognition by SsrB. Mutational analysis of a representative promoter validated this palindrome as the minimal architecture needed for regulatory input by SsrB. These data provide a high-resolution map of a regulatory network and the underlying logic enabling pathogen adaptation to a host.
Introduction
Precise gene regulation is crucial to the successful activation and execution of virulence programs for all pathogenic organisms. The acquisition of genes through horizontal gene transfer, a widespread means of bacterial evolution [1], requires a process to integrate new coding sequence into pre-existing regulatory circuitry. Silencing of horizontally-acquired genes by DNA binding proteins like H-NS is one way some incoming genes are initially controlled [2],[3], which can then be subject to regulatory evolution by mutating cis-regulatory operator regions to select for optional gene expression. The eventual promoter architecture selected to deploy virulence genes is often modular and should reflect a design that maximizes organismal fitness while limiting fitness trade-offs and antagonistic pleiotropy [4]. Both simulated [5] and functional experiments [6],[7] show that mutation of cis-regulatory sequences can be rapid, and that plasticity - the degree to which regulatory mutation can perturb the larger gene network - can be well tolerated in bacterial systems.
Promoter architectures that control quantitative traits such as bacterial virulence are, in fact, modular and evolvable [8]. For instance, Salmonella enterica has a multi-faceted pathogenic strategy fine-tuned by several transcriptional regulators. Intracellular survival and persistence of Salmonella requires a type III secretion system (T3SS) encoded in a horizontally-acquired genomic island called Salmonella Pathogenicity Island-2 (SPI-2) [9],[10]. T3SS are complex secretion machines that deliver bacterial effector proteins directly into host cells through an injectisome during infection [11],[12]. Several ancestral regulators control the genes in the SPI-2 genomic island including the two-component systems EnvZ-OmpR and PhoQ-PhoP, and the regulatory protein SlyA [13]. SsrA-SsrB is another two-component regulatory system co-inherited by genetic linkage with the SPI-2 locus that is essential for gene expression in SPI-2 [14]–[16]. SsrA is a sensor kinase activated in the host environment that phosphorylates the SsrB response regulator to create an active transcription factor needed for spatiotemporal control of virulence genes [13],[17]. In the Salmonella genus, the SPI-2 genomic island is found only in pathogenic serotypes of Salmonella enterica that infect warm-blooded animals and is absent from Salmonella bongori, which colonizes cold-blooded animals [18]. It is generally accepted that SPI-2 was acquired by Salmonella enterica after divergence from S. bongori, providing a useful pedigree to study regulatory evolution influenced by SsrB.
We recently demonstrated the evolutionary significance of cis-regulatory mutations for pathoadaptation of Salmonella enterica serovar Typhimurium (S. Typhimurium) to an animal host [19]. Our focus was on SsrB because of its broad conservation among the pathogenic Salmonellae and its essentiality for animal infection, suggesting it coordinates fundamental aspects of Salmonella pathogenesis beyond the SPI-2 genomic island. In this study we investigated how regulatory evolution assimilates horizontally acquired and ancestral genes into the SsrB regulon on a genome-wide scale using an integrated set of experimental methods. Combining our data with previous biochemical work, along with comparative genomic analyses with an orthologous T3SS-encoding genomic island in the tsetse fly endosymbiont, Sodalis glossinidius, we reveal the flexible DNA palindrome that distributes SsrB in the genome to influence transcriptional activation of the SPI-2 T3SS and almost all of its accessory effector proteins. Our data uncovers the principal SsrB circuitry that appears to have been conserved to support multiple bacterial lifestyles, including parasitic and mutualist symbioses.
Results
Transcriptional profiling of an ssrB mutant
To begin to understand regulatory evolution and network expansion of the SsrB response regulator, we profiled the transcriptome of an ssrB mutant and compared it to S. Typhimurium wild-type cells grown in an acidic minimal medium that activates the SsrA-SsrB two-component regulatory system [20]. We identified 133 genes that were significantly down-regulated in the ssrB mutant [z <−1.96] [21] (Table 1). This included almost all genes in the SPI-2 genomic island as well as effector genes encoded throughout the genome (Dataset S1). Next, we performed a Clusters of Orthologous Groups (COG) analysis [22] on the 118 genes that had an ortholog in the annotated genome of S. Typhimurium strain LT2 [23]. Among these, 45 genes lacked a functional COG assignment and the 73 remaining genes were distributed among 86 COGs (Figure 1). The majority of functions in the latter groups are in transport, secretion, and trafficking of cellular components in addition to protein and membrane modification.


The SsrA-SsrB system was acquired by horizontal gene transfer into the S. enterica species after divergence from what is now extant S. bongori. As such, S. bongori has evolved in the absence of SsrA-SsrB and its regulatory architecture has not been influenced by it. Orthologous genes ancestral to both species but regulated by SsrB in S. enterica provide evidence for network expansion and regulatory evolution that we previously showed can be mapped to a single cis-input location by using functional and comparative genomics [19]. To expand on this, we used a reciprocal BLAST-based analysis and identified 47 orthologs in S. bongori among the 133 genes whose transcription was down-regulated in an ssrB mutant (Table 1). In ΔssrB cells, the mean fold-change of the orthologous genes (−6.1-fold) was subtler than for the S. enterica-specific gene set (mean −21.2-fold), which included the T3SS and associated effector genes (Dataset S1). We also determined the distribution of down-regulated genes among genomic islands [24], including prophages, pathogenicity islands (SPI-islands) and additional regions of difference (ROD) between S. enterica and S. bongori (Dataset S2). For this we used a BLAST-based comparison of genome-wide synteny between S. Typhimurium and S. bongori and identified 50 ROD that included 17 previously reported SPI-islands and prophages. Of the 133 down-regulated genes identified, 56 were present within genomic islands (Table 1), with a mean change in gene expression of −25.3-fold in ΔssrB cells.
Genome-wide SsrB interactions
To examine SsrB allocation on the chromosome in vivo, we isolated functional SsrB-DNA interactions using chromatin immunoprecipitation and examined the bound DNA by chip analysis (ChIP-on-chip) using an S. Typhimurium SL1344 array containing 44,021 probes. With this method, we identified 256 significant interaction peaks distributed throughout the genome that were enriched under SsrB-activating conditions and with interaction scores three standard deviations greater than the mean probe score (Figure 2A and Table 1). Of these 256 peaks, 126 (49%) occurred within coding regions of genes (CDS) and 130 (51%) were in intergenic regions (IGR). Given the strong influence of SsrB on horizontally acquired genes (Table 1), we plotted the ChIP-on-chip data against all genomic islands in S. Typhimurium SL1344. From this analysis, 62 of 256 SsrB binding peaks (24.3%) occurred within genomic islands (Figure 2B).

SsrB ChIP peaks were observed upstream of previously identified SsrB regulated genes indicating that our ChIP-on-chip data captured functional interactions (Dataset S3). To generate a consensus set of SsrB regulated genes, we performed an analysis to identify operons in the S. Typhimurium SL1344 genome that encoded at least one gene down-regulated in ΔssrB cells and that possessed an SsrB binding peak in the upstream regulatory region as defined by our ChIP-on-chip analysis. From this, the 133 down-regulated genes mapped to 86 operons, 49 of which had an SsrB interaction upstream or within the first gene of the operon (Table 2). This analysis captured all five reported operons in the SPI-2 genomic island in addition to ten operons outside of this island that encode SPI-2 translocated effectors.

SsrB binds to six promoters within the SPI-2 genomic island
In order to rigorously evaluate our genome-wide functional genomics data, we compared it against traditional biochemical experiments describing SsrB-DNA interactions at the SPI-2 locus. Previous data reported SsrB footprints upstream of 6 genes in SPI-2: ssrA, ssrB, ssaB, sseA, ssaG, and ssaM [25],[26]. Our ChIP-on-chip data showed discrete SsrB binding at all of these promoters except for the promoter reported to be between ssrA and ssrB [25] (Figure 3). We attempted to verify functional activity at this site, but could not using transcriptional fusions (data not shown). Our data also identified three additional SsrB binding peaks upstream of sseA, within the CDS of ssaJ, and in the IGR upstream of ssaR (Figure 3). Functional interactions were confirmed for sseA and ssaR in subsequent reporter experiments described below.

Identification of a functional DNA element for SsrB binding
Previous attempts by others to identify a conserved SsrB DNA recognition motif have been unsuccessful. To overcome this, we employed a bacterial one-hybrid screen originally developed to define binding site preferences for eukaryotic transcription factors [27]. We fused the DNA binding domain of SsrB (SsrBc) to the α-subunit of RNA polymerase and screened a prey library of ∼108 DNA molecules previously counter-selected against self-activation (Figure 4A). We used the PhoP response regulator from E. coli as a control because a DNA recognition sequence for it was known [28]. Bait-prey combinations surviving selection on medium lacking histidine were purified, and preys were sequenced and analyzed using the motif-finding program MEME [29]. From 189 unique sequences isolated for SsrBc, over 80% contained a degenerate consensus motif, mCCyTA (Figure 4B). In control screens with the PhoP-αNTD fusion, the PhoP box sequence (G/T)GTTTA was identified in 11% of sequenced preys (12/109, data not shown) but this sequence was never captured by SsrB-αNTD and vice versa, demonstrating specificity of the bacterial one-hybrid system for prokaryotic regulatory proteins.

Next, we examined our ChIP-on-chip data for the presence of a conserved regulatory motif. We extracted sequence data from the local maxima of the 256 binding peaks and analyzed the sequences with the computational program MDscan [30]. Using the highest-ranking probes to generate an initial prediction followed by lower-ranking probes for refinement, this analysis identified motifs that represented either the forward or the reverse complement of the consensus sequence ACmTTA, which shares consensus with the motif identified in the bacterial one-hybrid screen (Figure 4B). We identified variations of this motif within footprinted regions of SsrB-regulated promoters [25],, however sequence degeneracy made it difficult to precisely map the input functions.
Identification of a functional DNA palindrome conserved in S. Typhimurium SPI-2 and the Sodalis glossinidius SSR-3 region
The analysis of regulatory evolution is particularly challenging because it is difficult to distinguish neutral stochastic change from functional divergence. To solve this problem in the context of mapping the SsrB binding element, we used comparative genomics to search for conserved promoter architecture in another organism with a similar genomic island to Salmonella SPI-2. The tsetse fly endosymbiont Sodalis glossinidius contains the Sodalis Symbiosis Region-3 (SSR-3) that is similar in content and synteny with the S. enterica SPI-2 locus [31]. Gene conservation includes the entire T3SS structural module extending to the regulatory genes ssrA-ssrB and all other genes except the effectors sseF and sseG, which are not present in Sodalis SSR-3. We aligned the sequences of the five mapped promoters in SPI-2 with the orthologous SSR-3 regions to identify local conservation. Highly conserved sites within the promoters were restricted to regions previously footprinted by SsrB [25],[26], whereas adjacent sequence showed substantial drift (Figure 4C). Within the conserved sites we identified a heptameric sequence in 7-4-7 tail-to-tail architecture that created an 18-bp degenerate palindrome. This palindrome was found in all SPI-2 and SSR-3 T3SS promoters with the exception of the sseA promoter that had only one reasonably well-conserved heptamer in the footprinted region (Figure 4C and Figure S1). Interestingly, two copies of the palindrome occur upstream of the ssrA-ssrB operon in S. Typhimurium within the same footprint, and the conservation of either site in Sodalis was weak. Evaluation of the heptamer motif in the palindrome showed high similarity to the motifs identified by the bacterial one-hybrid screen and the ChIP-on-chip experiments (Figure 4B and 4D), giving us confidence that we had identified the major recognition module for transcriptional input by SsrB. In accord with a previous observation [26], there was not a strict requirement in the spacing between the SsrB binding site and the downstream transcriptional start site.
SsrB directs transcriptional input using a flexible palindrome architecture
The presence of a conserved palindrome sequence in SPI-2 promoters and in related sequences from the endosymbiont S. glossinidius suggested that regulatory input by SsrB was through a palindrome sequence architecture. However, other lines of evidence suggested that the recognition site architecture was flexible in nature: (i) our bacterial one-hybrid screen isolated functional single hexamer sequences, (ii) the SsrB footprint at the naturally evolved sseA promoter within SPI-2 [26] contained only one reasonably well-conserved heptamer, and (iii) degenerate or non-ideal palindromes exist in the genome. In order to deconstruct this architecture, we designed a set of experiments to test the palindrome's tolerance to mutation. We chose the ssaG promoter for these experiments because 16 of 18 bases were identical between SPI-2 and SSR-3 from S. glossinidius, differing only in the 4-bp spacer between heptamers (Figure 4C). We mutated the palindrome in a series of transcriptional reporters that were otherwise identical to the evolved ssaG promoter (Figure 5A) and promoter activity was compared to that of a wild-type palindrome sequence. Variants in which the first half-site (7′) or second half-site (7″) was deleted produced similar transcriptional activity to the wild-type palindrome, verifying that a single well-conserved heptamer is sufficient for transcriptional input under these experimental conditions (Figure 5B). Deletion of the 4-bp spacer sequence between the heptamers - the most degenerate element of the palindrome - also generated wild-type promoter activity. However, the orientation of individual heptamers was essential for transcriptional input since rewiring the palindrome in any head-to-head orientation produced negligible promoter activity. However, if the two half sites were swapped front-to-back so that they maintained tail-to-tail orientation (construct labelled “Reverse” in Figure 5), wild-type promoter activity was restored. Precise deletion of the entire 18-bp palindrome lead to ∼10% residual activity in wild-type cells, which was reduced to less than 1% in an ssrB mutant (Figure 5C). To determine whether the remaining 10% transcriptional activity was a result of an SsrB-dependent feed-forward mechanism or transcriptional read-through of our chromosomally integrated reporter, we constructed an ectopic deletion reporter. Assessment of reporter activity for this construct in addition to wild-type constructs in wild-type and ssrB mutant backgrounds showed a similar level of activity to the ssrB mutant (Figure 5D).
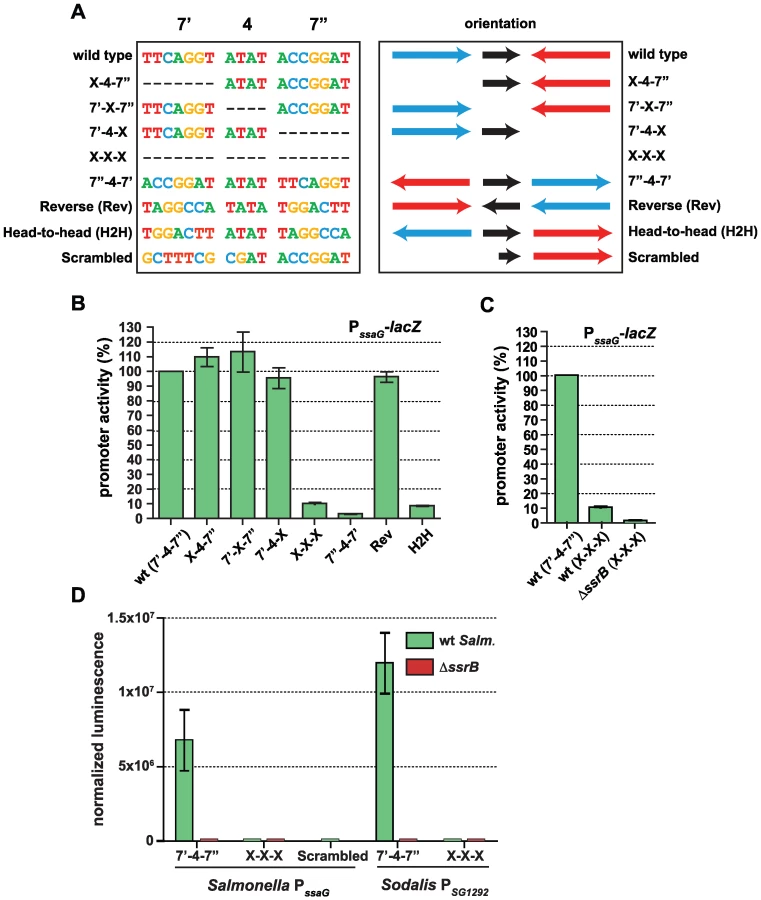
The results for the half-site deletion constructs, which retained activity similar to wild type, were unexpected. Therefore, we compared the sequences generated upon mutation against a consensus palindrome matrix generated from all SPI-2 and other identified putative elements. The 7′-4-X, X-4-7″ and 7′-X-7″ mutations introduced a number of base transitions and transversions never occurring in the matrix, however the modified 7 base pair heptamer retained 4–5 naturally-occurring bases along with the unchanged wild-type sequence in the other heptamer (Figure S2). The possibility existed that this modified heptamer, although now weaker in consensus, could still be sufficient for recruitment of a functional form of SsrB when paired with the other wild-type heptamer. To test this, we created an additional ectopic transcriptional fusion construct in which the left half (7′) of the palindrome was mutated to bases never occurring in the consensus matrix. When tested in promoter activity experiments, this reporter was unable to activate transcription and was identical to the X-X-X mutant construct (Figure 5D).
Salmonella SsrB and the Sodalis ortholog (SG1279) are 69% identical and 81% similar at the amino acid level. All of the critical residues in the dimerization helix and HTH motif required for specific transcriptional activity by SsrB [32] are conserved in the Sodalis ortholog (Figure S3). To demonstrate a functional role for the palindrome identified in Sodalis, we engineered luciferase transcriptional reporters that either contained (7′-4-7″) or lacked (X-X-X) the identified palindrome from the Sodalis SG1292 promoter (ssaG ortholog) and transformed them into wild-type S. Typhimurium and an ssrB mutant. The transcriptional activity from a wild-type Sodalis palindrome sequence was high, but was completely abolished in an ssrB mutant and in experiments where only the palindrome sequence was precisely deleted (Figure 5D). These experiments demonstrated a functional role for the conserved palindrome in Sodalis and the requirement for SsrB for transcriptional activation.
Regulatory evolution of the SPI-2 T3SS effector repertoire
The above results identified the conserved, yet flexible, palindrome sequence defining DNA recognition by SsrB. To examine the extent to which regulatory evolution has been selective for this genetic architecture, we created a position weight matrix (PWM) for the five strongest palindrome sites in SPI-2 and the orthologous sites in Sodalis SSR-3. We then searched for representative candidates of this motif in the S. Typhimurium genome using the simple scoring algorithm MotifLocator [45],[46]. This analysis recovered the motifs upstream of ssaB, ssaG, ssaM, and ssaR that were used to generate the PWM. The palindrome in the ssrA promoter was not used to create the PWM due to its weaker consensus in the left heptamer, however, it was recovered in the computational analysis in a second group of lower-scoring motifs (Figure 6A). We identified 24 palindromes co-occurring with ChIP-on-chip peaks upstream of 24 different SsrB-regulated genes or operons. Applying a stringent threshold to the output allowed us to identify two groups - genes with high-scoring upstream palindromes (ssaB, ssaG, ssaM, ssaR, sopD2, sifA, sifB, sseK2, sseK3, sseL, sseA′, steC, and srcA) and those with medium-scoring palindromes (0.7–0.8 threshold; ssrA, STM1633, sseI, slrP sspH2, pipB, sseJ, pipB2, srfN, sseA and steB) (Figure 6A and Dataset S4) (sseA′ denotes the SsrB palindrome sequence upstream of sseA that falls within the ssaE CDS, while sseA refers to the SsrB-footprinted IGR site with only one conserved heptamer defined in Figure 4C). Remarkably, this accounted for 17 of 22 SL1344 genome-encoded effectors translocated by the SPI-2-encoded T3SS (exceptions are chromosomal steA, gogB, and sseK, and plasmid-encoded spvB and spvC). These genes either lacked an upstream ChIP peak above our 3-standard deviation cut-off (sseK) or had such a peak but did not reach statistical significance in our transcriptional profiling experiments (steA, gogB).

Our ChIP-on-chip data revealed three additional strong SsrB binding peaks within SPI-2: one in the IGR directly upstream of ssaR, a second within the CDS for ssaJ, and a third within the CDS for ssaE that would be predicted to influence transcription of the downstream effector/chaperone operon beginning with sseA. The analysis described above recovered SsrB palindrome sequences at the sseA' and ssaR locations prompting further validation of these sites. No palindrome was identified for the ssaJ interaction peak and so further characterization was not pursued. For the IGR palindrome upstream of ssaR, we tested both a chromosome-integrated transcriptional fusion and an autonomous episomal reporter. In wild-type cells these reporters were as active as other SPI-2 promoters, whereas promoter activity was abrogated in ΔssrB cells, implicating this IGR as a functional promoter for ssaR (Figure 6B and Figure S4). We next tested the function of the intragenic palindrome within ssaE (sseA'). For this, we constructed a single-copy transcriptional reporter that either contained (WT PsseA) or lacked (PsseA del) the single heptamer site in the sseA IGR and integrated this reporter into wild-type cells and mutants lacking either ssrB or the ssaE coding sequence that removed the high-scoring intragenic palindrome sseA'. These experiments showed that the sseA' sequence contributes approximately 75% of transcriptional output at the sseA promoter (Figure 6C), since deleting the single heptamer in the sseA IGR had little effect on transcriptional output in any of the strain backgrounds. These reporter data are in line with the respective binding scores for the ChIP-on-chip interaction peaks (Figure 3) and the sequence similarity for these elements with respect to the consensus palindrome (Figure 6A and Figure 4D). Together, these data provide compelling evidence for the identity of the DNA recognition element that has been selected through evolution to co-regulate an SsrB-dependent gene program involved in adaptation to a host.
Discussion
Horizontal gene transfer is a well-recognized mechanism of bacterial evolution that gives rise to new phenotypes due to the coordinated expression of novel genetic components [1]. A good example of this is acquisition of type III secretion by mutualists and pathogenic bacteria enabling new colonization strategies within a host [33],[34]. Evolved changes to regulatory circuitry can also give rise to phenotypic diversity at the species level [19]. In both cases, regulatory evolution is required to correctly deploy gene products during infection, yet the extent to which regulatory evolution contributes to pathogenic adaptation is only beginning to be realized [8]. The SsrA-SsrB two-component regulatory system in S. enterica has been the focus of our efforts to understand the significance of regulatory evolution for pathogenic adaptation. This regulatory system was co-acquired with a T3SS encoded in the SPI-2 pathogenicity island and likely contributed to immediate and gradual phenotypic diversity as new regulatory nodes were explored and acted upon by natural selection.
Extensive work has been reported on the characterization of SsrB dependent genes, including functional evaluation of genes encoded within SPI-2 in addition to genome-wide transcriptional studies [14],[15]. In this study we identified genes co-expressed under SsrB-inducing conditions and found those with strong levels of expression localized predominantly to mobile genetic elements, recently acquired genomic islands or other annotated islands. We also identified many weakly co-expressed genes, some of which may represent ancestral Salmonella genes recruited into the SsrB regulon like the previously reported srfN [18]. Some of these genes may not be directly regulated by SsrB and will require further experimental investigation.
Direct profiling of SsrB-DNA interactions using ChIP-on-chip was used to identify SsrB binding sites in the genome. This analysis identified many interactions which have not been previously described and interaction sites within coding regions of genes which may represent non-canonical functions for SsrB. Other groups have reported the existence of similar numbers of ChIP-on-chip interactions within intragenic regions for other transcription factors [35],[36] suggesting that this phenomenon is not restricted to SsrB. In light of the disparate number of microarray genes in comparison to ChIP-on-chip peaks we attempted to generate a more comprehensive picture of the SsrB regulon by combining these data sets at the operon level. In doing so we believe that the nineteen operons containing differentially expressed genes determined by microarray and containing a ChIP-on-chip peak three standard deviations above the mean captured by this analysis represent the genes directly activated by SsrB (Table 2). Those operons having a ChIP-on-chip peak directly upstream in the IGR region encompass the majority of known SsrB regulated genes while those possessing a ChIP peak within the CDS of the first gene may represent non-functional interactions that deserve follow-up experimental investigation.
The ChIP-on-chip data not only provided information on the identities of SsrB-regulated genes but also gave insight as to the identity of the SsrB recognition element specified by the interaction site sequences. The regulatory architecture governing SsrB input has been elusive despite several SsrB footprints being defined biochemically [19],[25],[26]. Our ChIP-on-chip data further suggested that SsrB binding within SPI-2 was specific, with binding peaks overlapping precisely with regions of the DNA footprinted by SsrB [25],[26]. By using a genetic screening strategy together with functional and comparative genomics, we were able to define the essential SsrB regulatory element as being an 18-bp palindrome with a conserved 7-4-7 internal organization.
In support of the palindrome as the functional entity we showed the loss of SsrB dependence as a result of deletion of this element for the ssaG promoter. Evaluation of the 7-4-7 palindrome in the ssaG promoter revealed the minimal architecture and sequence orientation required for transcriptional input. Deletion of the entire palindrome resulted in less than 1% activity in wild-type cells, an equivalent level of activity to those lacking ssrB entirely. A search of the S. enterica genome for this palindromic motif revealed candidates upstream of the previously noted SsrB dependent genes, including two additional SPI-2 sites; one IGR site upstream of ssaR that until now had been cryptic, and one intragenic palindrome upstream of sseA in the ssaE CDS. In both cases these input sites were found to be functional.
Although palindrome architecture was conserved upstream of SsrB-regulated genes, degenerate palindromes in which one half-site was more conserved were also functional. As a result of our mutational analyses we conclude that so long as the orientation of a single heptamer of the palindrome is conserved with respect to the downstream gene, SsrB is tolerant of degeneracy in the adjacent spacer and heptamer sequences. While we were able to identify a number of limited palindrome-like sequences from our bacterial one-hybrid screen, this tolerance in addition to the library size required to pull out an 18-bp palindrome in large numbers may explain why we isolated functional single heptamer sequences and why degenerate palindromes naturally exist in the genome. A recent report by Carroll et al, postulated that SsrB first interacts with DNA as a monomer, followed by dimerization [32]. Our findings also suggest that dimerization is likely required for transcriptional activation however strong recognition by one monomer may stabilize interaction of a second monomer with a less than ideal sequence. The finding that a flexible palindromic sequence can be selective for SsrB input raises many interesting questions around the nature of regulatory evolution. The ability to use a short functional half-site adjacent to an uncharacterized threshold level of tolerated bases would reduce the period of neutral evolution required to generate an inverted repeat sequence twice the length [37], and would limit the loss of intermediate variations to drift while a more desirable palindrome is created by regulatory evolution. For bacteria that make use of horizontal gene transfer, this could increase the tempo with which new DNA is integrated into the regulatory circuitry of the cell.
We showed that the SsrB regulatory palindrome is also present in the orthologous SSR-3 island of the endosymbiont Sodalis glossinidius and that the palindrome evolved in Sodalis can act as a cis regulatory input function in Salmonella. Thus, in addition to supporting a pathogenic lifestyle within a host in Salmonella, it seems probable that this common promoter architecture may direct the activation of the SSR-3 T3SS of S. glossinidius in its endosymbiotic relationship with the tsetse fly host, although we acknowledge this requires experimental validation. The SSR-3 region in S. glossinidius is fully conserved in gene synteny and content with that of SPI-2 [31], with the exception of two effector genes missing in SSR-3 (sseF and sseG) that are required to localize vacuolar Salmonella to the perinuclear Golgi in host cells [38],[39]. The SsrB ortholog in S. glossinidius is ∼30% divergent with SsrB at the protein level, initially leading us to think that they might have different binding site preferences. To the contrary, high local conservation in the promoters evolved in Salmonella and Sodalis was the crux in defining the functional SsrB input among stochastic noise. This analysis revealed strong palindrome sequence conservation in five promoters identified in SPI-2 and in the orthologous sequences in Sodalis SSR-3.
Among palindrome-containing promoters, the ssrA promoter is exceptional for two reasons: a lack of conservation between Salmonella and Sodalis, and the evolution of tandem palindromes in Salmonella. One possible interpretation of this divergent regulatory architecture in front of ssrA might relate to bacterial lifestyle. Salmonella may have retained or evolved SsrB input here to create a positive feedback loop on the regulatory system to rapidly adapt to the host environment during infection, similar to transcriptional surge described for the PhoP response regulator [40]. The endosymbiotic relationship of Sodalis with the tsetse fly - where long-term vertical transmission has ostensibly been formative in shaping regulatory circuitry at certain promoters - may obviate the need for rapid transcriptional surge, leading to regulatory drift or selection against positive feedback. With the structure of SsrB available [32] and its recognized sequence now identified, future studies will be able to build a picture of how SsrB interacts with both its target DNA, RNA polymerase and potentially other transcription factors including nucleoid associated proteins in order to direct transcription of its regulon.
In summary, this work highlights the evolutionary significance of cis-regulatory mutation for the adaptation of Salmonella to a host animal. The DNA module that choreographs SsrB-mediated pathogenic behaviour in Salmonella appears to have been conserved for mutualism as well, thereby shedding new light on the significance of cis-regulatory mutations for bacteria evolving in different ecological settings.
Methods
Ethics statement
All experiments with animals were conducted according to guidelines set by the Canadian Council on Animal Care. The Animal Review Ethics Board at McMaster University approved all protocols developed for this work.
Bacterial strains and growth conditions
The Salmonella strain used for microarray and ChIP-on-chip analysis was Salmonella enterica serovar Typhimurium strain SL1344. Bacterial strains and plasmids used in this work are described in Table S1. Primer sequences used to generate constructs are available upon request. Bacteria were grown in LB medium unless otherwise indicated. Low-phosphate, low magnesium (LPM) medium was used as bacterial growth medium for microarray, ChIP-on-chip, and transcriptional reporter experiments [20]. Liquid cultures were routinely grown at 37°C with shaking. Antibiotics were added to media as follows when necessary: ampicillin (Amp, 100 µg/mL), chloramphenicol (CM, 34 µg/mL) kanamycin (Kan, 30 or 50 µg/mL), and streptomycin (SM, 50 µg/mL). NM medium was used in the bacterial one-hybrid experiments as described previously [27].
Transcriptional profiling
Microarray experiments were conducted and analyzed as described previously [16]. cDNA was synthesized from RNA harvested from wild-type cells and an ΔssrB mutant. cDNA from 2 replicate experiments was hybridized to InGen arrays and analyzed using ArrayPipe version 1.309 [21]. Probe signals underwent a foreground-background correction followed by a printTipLoess normalization by sub-grid. Duplicate spots were merged followed by averaging of the two replicates. Local intensity z scores were calculated for determination of significance.
Genome-wide operon and island analysis
For operon analysis, S. Typhimurium SL1344 operons were defined as groups of genes encoded on the same strand with a maximum intergenic distance of 30-bp. Operons selected for further investigation were those possessing at least one significantly down-regulated gene from the cDNA microarray analysis of an ssrB mutant. A cDNA microarray score was assigned based on the average fold-change value of all genes within the operon. For ChIP-on-chip analysis, a top ChIP interaction score was defined as that of the highest scoring probe within the first gene or the intergenic region upstream of the first gene of the operon. For the analysis of regions of difference (ROD) between S. enterica serovar Typhimurium and Salmonella bongori, a reciprocal-best BLAST analysis was performed to identify orthologous genes between S. Typhimurium and S. bongori. Orthologs were defined as reciprocal best BLAST pairs with E-values less than 0.005. Comparison of gene synteny between regions encoding orthologous genes was performed to identify regions of low conservation including gene deletions and insertions. The location and names of genes flanking the comprehensive list of genomic islands is provided in Dataset S2 and were compared to those predicted using IslandViewer [41].
Bacterial one-hybrid screen and analysis
The bacterial one-hybrid (B1H) experiments were conducted as outlined previously using a single-step selection procedure [27]. Full-length phoP from E. coli and the C-terminal domain of ssrB (ssrBc) from S. Typhimurium were cloned into pB1H1 to create a fusion to the αNTD of RNA polymerase. Each bait vector was transformed into E.coli ΔhisB ΔpyrF, purified, and then cells were transformed again with purified prey library that was previously counter-selected for self-activating preys using 5-fluoro-orotic acid. Transformants were recovered for 1 h in SOC medium, washed with NM medium supplemented with 0.1% histidine (NM + his) and allowed to grow for 2 h in this medium. Cells were washed four times with water, once with NM medium lacking histidine (NM –his), then resuspended in NM –his and plated on 150×15 mm dishes containing NM –his media supplemented with either 1 mM (for PhoP screen) or 5 mM (SsrBc screen) 3-aminotriazole. Selection was for ∼48 hours at 37°C. Individual clones were selected from plates containing <600 colonies, the prey plasmids were isolated and sent for sequencing (Macrogen USA). Sequences were parsed to extract the 18-bp prey sequence, then inputted into MEME (version 4.1.1) for motif generation [29]. MEME was run with default parameters and included searching for motifs of length 5–17 bp in either forward or reverse direction and with no limit on the number of occurrences within an input string. Motif logos were generated using Weblogo, version 2.8.2 [42].
Chromatin immunoprecipitation-on-chip (ChIP-on-chip)
Chromatin immunoprecipitation-on-chip (ChIP-on-chip) was conducted as described previously using an SL1344 strain containing an ssrB-3xFLAG allele on the chromosome [19]. The primer sequences used to generate the DNA for recombination were: 5′GAG TTA CTT AAC TGT GCC CGA AGA ATG AGG TTA ATA GAG TAT GAC TAC AAA GAC CAT GAC GG3′ and 5′ATC AAA ATA TGA CCA ATG CTT AAT ACC ATC GGA CGC CCC TGG CAT ATG AAT ATC CTC CTT AG3′. This strain was generated by an allelic replacement method described previously [43] and causes lethal infection of C57BL/6 mice similar to wild-type SL1344 (Figure S5). Immunoprecipitated DNA from three experiments under SsrB-inducing conditions (LPM growth medium) and one experiment under non-inducing conditions (exponential growth in LB medium) was hybridized to a single chip printed with four whole genome arrays designed on S. enterica serovar Typhimurium strain SL1344 (Oxford Gene Technology, Oxford UK). Signals for each probe within an experiment were normalized to the median channel signal for the respective array. Signal ratios were obtained for both inducing and non-inducing conditions by calculating the ratio of the control probe value and experimental probe value. A final interaction score was obtained by taking the log2 value of the ratio between the non-inducing and inducing conditions for each probe to remove SsrB interactions that occur under non-inducing conditions. Parsing and data analyses were performed using the Python scripting language. Genome-wide ChIP-on-chip data was plotted using Circos v.0.51 [44].
Genome-wide motif analysis
ChIP probes were ordered according to their position on the S. Typhimurium SL1344 genome and local maxima for ChIP interaction scores were defined as interaction peaks. Peaks with scores greater than three standard deviations from the mean probe signal were considered significant ChIP interaction peaks and were ranked in order of descending interaction score. The sequence of the top-scoring probe for each peak was exported to a text file and used for analysis by MDscan [30]. The background parameter was run with output generated by the included genomebg program from the S. Typhimurium SL1344 genome sequence. The initial motif was generated from sequences from the top ten SsrB interaction peaks and refined using the top 25 peak sequences.
To identify instances of the palindrome motif in S. Typhimurium, ten 18-bp 7-4-7 palindromic motifs in the promoters of the SPI-2 genes ssrA, ssaB, ssaG, ssaM, ssaR and their orthologous SSR-3 genes were used as input for MDScan to identify a consensus motif and to determine the position specific scoring matrix (PWM). This PWM was used with MotifLocator to identify instances of this motif in the S. Typhimurium SL1344 genome. A background file specific to SL1344 was generated using the associated script called CreateBackgroundModel. A stringent threshold value of 0.8 was used [45],[46].
Transcriptional reporter experiments
Transcriptional fusions to lacZ for the ssaG and sseA promoter palindrome analysis were generated using chemically synthesized double-stranded DNA (Genscript Corp). Promoter DNA was ligated into pIVET5n, then the plasmid was subsequently conjugated into SL1344 to generate single-copy transcriptional fusions integrated on the chromosome as described previously [20]. Luciferase reporter constructs for the Sodalis glossinidius SG1292 and ssaR promoters were generated by PCR amplification of promoter regions from genomic DNA templates. The luciferase reporter construct for the PssaG scrambled substitution was created by PCR product splicing via overlap extension using the existing ssaG promoter cloning primers and two additional internal primers containing the desired mutation sequence (DTM0061R, 5′CGC GAA AGC AAC GAT TAC TCC GGC GCA CG3′ and DTM0061.1F, 5′GAG TAA TCG TTG CTT TCG CGA TAC CGG ATG TTC ATT GCT TTC TA3′). This DNA was ligated into pCS26 and transformed into SL1344 to generate plasmid-based reporters. Overnight cultures of Salmonella were sub-cultured into LPM medium and grown with shaking for 7 h. Samples were removed hourly to measure β-galactosidase activity via a chemiluminescence-based assay as described previously [20] or luminescence directly from cultures (EnVision, Perkin-Elmer). Output was relative light units (RLU) normalized to OD600. Each experiment was performed in triplicate then averaged. Reporter activity from mutant and rewired promoters was normalized to that from wild-type promoters.
Data deposition
All ChIP-on-chip data can be retrieved from the NCBI Gene Expression Omnibus at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE20192. Data files for viewing in Artemis (http://www.sanger.ac.uk/Software/Artemis/) are available upon request.
Supporting Information
Zdroje
1. OchmanH
LawrenceJG
GroismanEA
2000 Lateral gene transfer and the nature of bacterial innovation. Nature 405 299 304
2. NavarreWW
PorwollikS
WangY
McClellandM
RosenH
2006 Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313 236 238
3. LucchiniS
RowleyG
GoldbergMD
HurdD
HarrisonM
2006 H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog 2 e81 doi:10.1371/journal.ppat.0020081
4. Prud'hommeB
GompelN
CarrollSB
2007 Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A 104 Suppl 1 8605 8612
5. StoneJR
WrayGA
2001 Rapid evolution of cis-regulatory sequences via local point mutations. Mol Biol Evol 18 1764 1770
6. MayoAE
SettyY
ShavitS
ZaslaverA
AlonU
2006 Plasticity of the cis-regulatory input function of a gene. PLoS Biol 4 e45 doi:10.1371/journal.pbio.0040045
7. IsalanM
LemerleC
MichalodimitrakisK
HornC
BeltraoP
2008 Evolvability and hierarchy in rewired bacterial gene networks. Nature 452 840 845
8. PerezJC
GroismanEA
2009 Evolution of transcriptional regulatory circuits in bacteria. Cell 138 233 244
9. SheaJE
HenselM
GleesonC
HoldenDW
1996 Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci U S A 93 2593 2597
10. OchmanH
SonciniFC
SolomonF
GroismanEA
1996 Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci USA 93 7800 7804
11. CornelisGR
2006 The type III secretion injectisome. Nat Rev Microbiol 4 811 825
12. GalanJE
Wolf-WatzH
2006 Protein delivery into eukaryotic cells by type III secretion machines. Nature 444 567 573
13. FassE
GroismanEA
2009 Control of Salmonella pathogenicity island-2 gene expression. Curr Opin Microbiol 12 199 204
14. YoonH
McDermottJE
PorwollikS
McClellandM
HeffronF
2009 Coordinated regulation of virulence during systemic infection of Salmonella enterica serovar Typhimurium. PLoS Pathog 5 e1000306 doi:10.1371/journal.ppat.1000306
15. RytkonenA
PohJ
GarmendiaJ
BoyleC
ThompsonA
2007 SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci U S A 104 3502 3507
16. CoombesBK
WickhamME
LowdenMJ
BrownNF
FinlayBB
2005 Negative regulation of Salmonella pathogenicity island 2 is required for contextual control of virulence during typhoid. Proc Natl Acad Sci U S A 102 17460 17465
17. BrownNF
VallanceBA
CoombesBK
ValdezY
CoburnBA
2005 Salmonella Pathogenicity Island 2 is expressed prior to penetrating the intestine. PLoS Pathog 1 e32 doi:10.1371/journal.ppat.0010032
18. OchmanH
GroismanEA
1996 Distribution of pathogenicity islands in Salmonella spp. Infect Immun 64 5410 5412
19. OsborneSE
WalthersD
TomljenovicAM
MulderDT
SilphaduangU
2009 Pathogenic adaptation of intracellular bacteria by rewiring a cis-regulatory input function. Proc Natl Acad Sci U S A 106 3982 3987
20. CoombesBK
BrownNF
ValdezY
BrumellJH
FinlayBB
2004 Expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J Biol Chem 279 49804 49815
21. HokampK
RocheFM
AcabM
RousseauME
KuoB
2004 ArrayPipe: a flexible processing pipeline for microarray data. Nucleic Acids Res 32 W457 459
22. TatusovRL
NataleDA
GarkavtsevIV
TatusovaTA
ShankavaramUT
2001 The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29 22 28
23. McClellandM
SandersonKE
SpiethJ
CliftonSW
LatreilleP
2001 Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413 852 856
24. DobrindtU
HochhutB
HentschelU
HackerJ
2004 Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2 414 424
25. FengX
WalthersD
OropezaR
KenneyLJ
2004 The response regulator SsrB activates transcription and binds to a region overlapping OmpR binding sites at Salmonella pathogenicity island 2. Mol Microbiol 54 823 835
26. WalthersD
CarrollRK
NavarreWW
LibbySJ
FangFC
2007 The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol Microbiol 65 477 493
27. MengX
BrodskyMH
WolfeSA
2005 A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol 23 988 994
28. ZwirI
ShinD
KatoA
NishinoK
LatifiT
2005 Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc Natl Acad Sci U S A 102 2862 2867
29. BaileyTL
WilliamsN
MislehC
LiWW
2006 MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34 W369 373
30. LiuXS
BrutlagDL
LiuJS
2002 An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20 835 839
31. TohH
WeissBL
PerkinSA
YamashitaA
OshimaK
2006 Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res 16 149 156
32. CarrollRK
LiaoX
MorganLK
CicirelliEM
LiY
2009 Structural and functional analysis of the C-terminal DNA binding domain of the Salmonella typhimurium SPI-2 response regulator SsrB. J Biol Chem 284 12008 12019
33. CoombesBK
2009 Type III secretion systems in symbiotic adaptation of pathogenic and non-pathogenic bacteria. Trends Microbiol 17 89 94
34. DaleC
PlagueGR
WangB
OchmanH
MoranNA
2002 Type III secretion systems and the evolution of mutualistic endosymbiosis. Proc Natl Acad Sci U S A 99 12397 12402
35. ShimadaT
IshihamaA
BusbySJ
GraingerDC
2008 The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions. Nucleic Acids Res 36 3950 3955
36. ChoBK
KnightEM
BarrettCL
PalssonBO
2008 Genome-wide analysis of Fis binding in Escherichia coli indicates a causative role for A-/AT-tracts. Genome Res 18 900 910
37. MacArthurS
BrookfieldJF
2004 Expected rates and modes of evolution of enhancer sequences. Mol Biol Evol 21 1064 1073
38. SalcedoSP
HoldenDW
2003 SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J 22 5003 5014
39. DeiwickJ
SalcedoSP
BoucrotE
GillilandSM
HenryT
2006 The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect Immun 74 6965 6972
40. ShinD
LeeEJ
HuangH
GroismanEA
2006 A positive feedback loop promotes transcription surge that jump-starts Salmonella virulence circuit. Science 314 1607 1609
41. LangilleMG
BrinkmanFS
2009 IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25 664 665
42. CrooksGE
HonG
ChandoniaJM
BrennerSE
2004 WebLogo: a sequence logo generator. Genome Res 14 1188 1190
43. UzzauS
Figueroa-BossiN
RubinoS
BossiL
2001 Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98 15264 15269
44. KrzywinskiM
ScheinJ
BirolI
ConnorsJ
GascoyneR
2009 Circos: an information aesthetic for comparative genomics. Genome Res 19 1639 1645
45. ThijsG
LescotM
MarchalK
RombautsS
De MoorB
2001 A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics 17 1113 1122
46. ThijsG
MarchalK
LescotM
RombautsS
De MoorB
2002 A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J Comput Biol 9 447 464
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 3
Nejčtenější v tomto čísle
- Deciphering Normal Blood Gene Expression Variation—The NOWAC Postgenome Study
- Transgenic Rat Model of Neurodegeneration Caused by Mutation in the Gene
- Papillorenal Syndrome-Causing Missense Mutations in / Result in Hypomorphic Alleles in Mouse and Human
- Fatal Cardiac Arrhythmia and Long-QT Syndrome in a New Form of Congenital Generalized Lipodystrophy with Muscle Rippling (CGL4) Due to Mutations