
Integration of UPR and Oxidative Stress Signaling in the Control of Intestinal Stem Cell Proliferation
Loss of proper protein homeostasis (proteostasis) as well as increased production of reactive oxygen species (ROS) is a hallmark of aging. In complex metazoans, these processes can result in proliferative diseases and cancers. The protein folding capacity of the endoplasmic reticulum (ER) is monitored and maintained by the unfolded protein response of the ER (UPRER). In this study, we identify a coordinated role of UPRER and oxidative stress signaling in regulating the proliferation of intestinal stem cells (ISCs). We find that the ER-stress responsive transcription factor Xbp1 and the ER-associated degradation pathway component Hrd1 are sufficient and required cell autonomously in ISCs to limit their proliferative activity. This function is dependent on the activities of the stress sensor JNK and the redox-responsive transcription factor CncC, which we have previously identified as regulators of ISC proliferation. We further show here that promoting ER homeostasis in aging ISCs is sufficient to limit age-associated epithelial dysplasia. Our results establish the integration of UPRER and oxidative stress signaling as a central mechanism promoting regenerative homeostasis in the intestinal epithelium.
Published in the journal:
. PLoS Genet 10(8): e32767. doi:10.1371/journal.pgen.1004568
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004568
Summary
Loss of proper protein homeostasis (proteostasis) as well as increased production of reactive oxygen species (ROS) is a hallmark of aging. In complex metazoans, these processes can result in proliferative diseases and cancers. The protein folding capacity of the endoplasmic reticulum (ER) is monitored and maintained by the unfolded protein response of the ER (UPRER). In this study, we identify a coordinated role of UPRER and oxidative stress signaling in regulating the proliferation of intestinal stem cells (ISCs). We find that the ER-stress responsive transcription factor Xbp1 and the ER-associated degradation pathway component Hrd1 are sufficient and required cell autonomously in ISCs to limit their proliferative activity. This function is dependent on the activities of the stress sensor JNK and the redox-responsive transcription factor CncC, which we have previously identified as regulators of ISC proliferation. We further show here that promoting ER homeostasis in aging ISCs is sufficient to limit age-associated epithelial dysplasia. Our results establish the integration of UPRER and oxidative stress signaling as a central mechanism promoting regenerative homeostasis in the intestinal epithelium.
Introduction
Long-term homeostasis of high-turnover tissues relies on the precise regulation of stem cell (SC) activity that allows tailoring regenerative responses to the needs of the tissue. Regenerative processes in barrier epithelia, such as the intestinal epithelium, are particularly vulnerable to exogenous insults. Understanding how cellular stress responses of intestinal epithelial cells (IECs) and intestinal stem cells (ISCs) coordinate and maintain regenerative processes in the gut will provide insight into the etiology of pathologies ranging from inflammatory bowel diseases (IBDs) to colorectal cancers.
The unfolded protein response of the ER (UPRER) plays a central role in the control of homeostasis of the intestinal epithelium. Loss of protein folding capacity in the ER of IECs results in complex cell-autonomous and non-autonomous activation of stress signaling pathways, triggering an inflammatory condition that severely perturbs proliferative homeostasis, innate immune function and cell survival in the epithelium, and has been implicated in IBDs [1]–[7].
The UPRER is triggered by the accumulation of misfolded proteins in the ER [8], which activate three highly conserved UPRER sensors: the PKR-like ER kinase PERK, the transcription factor ATF6, and the endoribonuclease IRE1 (Figure 1B). These sensors make up the three branches of UPRER signaling, which consists of IRE1-mediated splicing of the mRNA encoding the bZip transcription factor X-Box binding protein 1 (Xbp1), phosphorylation of the translation initiation factor 2 alpha (eIF2α) by PERK, and cleavage and activation of ATF6, resulting in its nuclear translocation and activation of stress response genes, including Xbp1 [1]–[7], [9]. Xbp1 regulates transcription of ER components, and the resulting transcriptional induction of ER chaperones and of genes encoding ER components enhances ER folding capacity, and the reduction in protein synthesis (by eIF2α) alleviates the protein load in the ER. Furthermore, factors required to degrade un/misfolded proteins through ER-associated degradation (ERAD) are induced [8], [10]–[12]. The accumulation of un/misfolded proteins in the ER is further associated with increased production of reactive oxygen species (ROS), most likely due to the production of hydrogen peroxide as a byproduct of protein disulfide bond formation by protein disulfide isomerase (PDI) and ER oxidoreductin 1 (Ero1) [13]–[15].

Recent studies suggest that the UPRER may influence regenerative processes in the gut directly, as it is engaged in cells transitioning from a stem-like state into the transit amplifying state in the small intestine of mice [16]. Regeneration is also influenced by the intracellular redox state of stem cells, and changes in intracellular ROS production play an important role in the regulation of SC pluripotency, proliferative activity, and differentiation [17]–[20]. Coordinated control of cellular protein and redox homeostasis by the UPRER and other stress signaling pathways is therefore critical to maintain SC function. Exogenous ER stress likely disrupts this coordination, perturbing regeneration and proliferative homeostasis. Consistent with this model, excessive UPRER activity has been implicated in tumorigenesis [2], [21].
To understand the long-term maintenance of epithelial homeostasis in the intestine, detailed insight into the regulation and function of the UPRER and its coordination with the redox response in the intestinal epithelium, in a cell-type specific and temporally resolved manner, is required. Here, we have initiated such an analysis, using the Drosophila intestinal epithelium as a model system. The Drosophila ISC lineage exhibits a high degree of functional and morphological similarities with the ISC lineage in the mammalian small intestine [22]–[24]. When a regenerative response is induced in the intestinal epithelium, ISCs self-renew and give rise to transient, non-dividing progenitor cells called EnteroBlasts (EBs), which differentiate into either absorptive EnteroCytes (ECs) or secretory EnteroEndocrine (EEs) cells, triggered by differential Notch signaling. ISCs are the only dividing cells in the posterior midgut of Drosophila and their entry into a highly proliferative state is regulated by multiple stress and mitogenic signaling pathways, including Jun-N-terminal Kinase (JNK), Jak/Stat, Insulin, Wnt, and EGFR signaling [24], [25]. The transcription factor CncC (orthologue of mammalian Nrf2 and worm SKN-1), a master regulator of intracellular redox homeostasis, controls proliferation of ISCs by limiting ROS accumulation [19]. Interestingly, mammalian Nrf2 has been suggested to buffer ROS production during ER stress, while worm SKN-1 has recently been found to coordinate antioxidant gene expression with Xbp1 [26], [27].
During aging, flies develop epithelial dysplasia in the intestine, caused by excessive ISC proliferation and deficient differentiation of EBs [28], [29]. This phenotype is a consequence of an inflammatory condition initiated by dysbiosis of the commensal bacteria, and causes metabolic decline, loss of epithelial barrier function, and increased mortality [30]–[32]. The ISC-intrinsic mechanisms causing the decline of proliferative homeostasis in the aging intestinal epithelium remain unclear.
Here, we have dissected the role of the UPRER and redox signaling in the control of ISC function and epithelial homeostasis at cellular resolution. We find that ER homeostasis is lost in the aging intestinal epithelium, and that this loss correlates with intestinal dysplasia. Activation of the UPRER within ISCs is required and sufficient for ISC proliferation, and excessive ER stress contributes to the age-associated dysplasia observed in the Drosophila gut. These effects are mediated by changes in the intracellular redox state, which perturb Nrf2/CncC and JNK activities. Accordingly, we find that JNK and Nrf2/CncC act epistatically in the control of ISC proliferation by ER stress. Our findings provide an integrated model for the regulation of ISC activity by redox and proteostatic signaling, and highlight the effects of this integration on epithelial homeostasis.
Results
UPRER activation in aging Drosophila intestines
In a recent transcriptome analysis of age-related changes in the Drosophila intestine [32], we noticed that expression of the ER stress-responsive genes Bip/Hsc70-3/Hsc3, the ER chaperone Gp93, and Xbp1 are significantly induced in aging guts (Figure 1A). To confirm these findings, we used an antibody against Hsc3 and a reporter for Xbp1 expression, Xbp1P::dsRed [33], and assessed their expression in young and old guts (Figure 1C, 1D, S1H). Consistent with the RNAseq results, Hsc3 immunoreactivity and Xbp1 expression increased throughout the posterior midgut of aging flies (Figure 1C, 1D, S1H), suggesting that the UPRER is activated in the aging intestinal epithelium. Since ER stress has been implicated in deregulation of mammalian ISC function [2], [16], and since Drosophila ISCs over-proliferate in aging guts, causing epithelial dysplasia [28], [29], [32], we assessed whether the UPRER is activated in aging ISCs. ISCs and EBs can be identified in the posterior midgut of flies by expression of GFP driven by the esg::Gal4 driver, and ISCs can further be identified by expression of the Notch ligand Delta (DI). In young animals, Hsc3 was expressed at lower levels in progenitor cells than in differentiated cells. In old guts however, Hsc3 expression was strongly increased in progenitor cells, suggesting a specific activation of the UPRER in these cells (Figure 1E, Figure S1H). We confirmed this using an Xbp1 splicing reporter [11], [34], which assesses activation of Ire1. When this reporter was expressed in ISCs and EBs using esg::Gal4, no activity was detected in young flies, but GFP fluorescence was readily detectable in old guts (Figure 1F).
Control of ISC proliferation by the UPRER
To test whether the loss of ER homeostasis is a cause or a consequence of the age-associated over-proliferation of ISCs, we examined the proliferative activity of ISCs in which Xbp1 had been knocked down by RNAi (the effectiveness of the RNAi and over-expression constructs used here and below were validated by RT-PCR; Figure S2D). We used the esg::Gal4 driver in combination with tub::Gal80ts, which allows temperature-inducible expression of UAS-controlled dsRNAs in ISCs and EBs (this combination is labeled esgts throughout). Perturbing Xbp1 was sufficient to strongly induce ISC proliferation, as measured by the frequency of cells positive for the mitotic marker phospho-Histone H3 (pH3; Figure 2A), and by the expansion of Dl+/esg+ cells within the epithelium (Figure 2B).

We confirmed the induction of ISC proliferative activity in these conditions by assessing the rate of tissue turnover in flies in which Xbp1RNAi and GFP were heritably expressed in ISCs and their daughter cells in response to an ISC-specific recombination event (using esg::Gal4, tubGal80ts, UAS::GFP, UASFlp, act>STOP>Gal4, also termed esgtsF/O, [35]). Knocking down Xbp1 greatly accelerated epithelial renewal compared to wild type conditions, further highlighting a role for ER stress in promoting ISC proliferation (Figure S1A). The over-proliferation induced by knocking down Xbp1 in ISCs also increased phosphorylation of eIF2α, which is phosphorylated in response to ER stress by PERK, indicating that this induction of ISC proliferation is associated with an activation of the UPRER (Figure 2C).
We asked whether the proliferative response of ISCs in Xbp1 loss of function conditions was induced by changes in ER homeostasis in ISCs specifically, or whether induction of ER stress in EBs or daughter cells was driving ISC proliferation non-autonomously. To address this question, we first restricted expression of Xbp1RNAi to ISCs, by combining esg::Gal4 with a transgenic construct that inhibits Gal4 activity in EBs (Su(H)-Gbe::Gal80; Su(H)-Gbe promoter elements are activated specifically in EBs in response to Dl/N signaling in EBs [36]; Figure S2E). Using 3 different dsRNA constructs against Xbp1, we confirmed that Xbp1 knockdown specifically in ISCs is sufficient to induce ISC proliferation (Figure 2D, Figure S1G). Xbp1 has a complex role in ER homeostasis, serving both as a sensor for ER stress and as a promoter of ER growth and proteostasis [11], [34], [37], and may also act independently of the ER stress response to regulate ISC proliferation. We therefore tested if independently activating the UPRER, by impairing the removal of unfolded proteins in the ER directly, is sufficient to induce ISC proliferation. To this end, we perturbed the ER-associated degradation (ERAD) pathway by knocking down the ERAD-associated E3 ubiquitin ligase Hrd1, which is required for ubiquitination and degradation of unfolded proteins in the ER [38]. Knockdown of Hrd1 also induced eIF2α phosphorylation in ISCs (Figure 2C) and increased ISC proliferation, both when driven by esgts and when driven by esgts in combination with Su (H)::Gal80 (Figure 2E, Figure S1G). We also examined whether knocking down Xbp1 or Hrd1 in other cell types of the gut epithelium is sufficient to promote ISC proliferation. Knocking down Xbp1, but not Hrd1 in EBs (using Su(H)Gbe::Gal4 [39] in combination with tub::Gal80ts) or in ECs (using NP1::Gal4, tub::Gal80ts) increased ISC proliferation (Figure 2F), suggesting the existence of an Xbp1-specific non-autonomous effect on ISC proliferation in this tissue. Taken together, our results indicate that loss of ER homeostasis within ISCs induces ISC proliferation. The non-autonomous feedback of Xbp1 perturbation in ISC daughter cells on ISC proliferative activity is interesting and will be explored mechanistically in a separate study (Wang et al., in preparation).
If activation of the UPRER is required for the regenerative response of ISCs, perturbation of UPRER components should influence the growth of ISC-derived cell clones. To test this idea, and to determine the requirement for UPRER components in the regulation of ISC activity in homeostatic conditions, we performed linage tracing of mutant stem cells via the MARCM system [40]. Clones generated by ISCs homozygous for the Xbp1 loss-of-function allele Xbp1k13803, the Hrd1 loss of function allele hrd1Delta (a deletion that deletes Hrd1 and CG2126, see methods), or clones expressing Xbp1RNAi or Hrd1RNAi grew significantly faster than wild-type controls (Figure 2G, Figure 2H, Figure S1B–E). Accordingly, clones derived from ISCs over-expressing endogenous Xbp1 (using Xbp1d08698, a line in which Xbp1 transcription is induced downstream of a transgenic UAS [37], [41]), a transgene encoding a constitutively active, spliced version of Xbp1 [11], or transgenic Hrd1 [42], grew significantly slower than clones derived from wild-type ISCs (Figure 2G, Figure 2H, Figure S1F). While maintaining ER homeostasis through the UPRER is thus essential to limit ISC proliferation and prevent dysplasia, a functional UPRER is also required for normal homeostatic regeneration.
UPRER as a rheostat for stem cell proliferation
To further confirm that promoting ER homeostasis within ISCs selectively limits their proliferation, we assessed if increasing the expression of UPRER components in ISCs or their daughter cells was sufficient to allay tunicamycin-induced ISC proliferation. Tunicamycin, potently induces ER stress by inhibiting N-linked protein glycosylation and thus impairing protein folding [43]. Feeding tunicamycin very robustly induced ISC proliferation, supporting a role for activation of the UPRER in promoting ISC proliferation (Figure 3A, Figure 3B, Figure 3D, and Figure 3E). Increasing ER stress tolerance by over-expressing endogenous Xbp1, spliced Xbp1, Hrd1, or Hsc3/Bip in ISCs and EBs (using esg::Gal4 and esgts; spliced Xbp1 was expressed only in adults using esgts) is sufficient to significantly reduce tunicamycin-induced ISC proliferation (Figure 3A, Figure 3B, Figure 3D, note that expressing spliced Xbp1, endogenous Xbp1 (using Xbp1d08698 or Xbp1EP2112 [37], [41]), as well as Hsc3/Bip also inhibited proliferation induced by oxidative stress inducer paraquat, Figure 3C, Figure 3D,). This inhibition was also observed when spliced Xbp1 was over-expressed selectively only in ISCs (using esgts; Su(H)Gbe::Gal80; Figure 3E), but not when spliced Xbp1 or Hrd1 were expressed in ECs or EBs only (using the EC-specific NP1::Gal4 or the EB-specific Su(H)Gbe::Gal4, both rendered heat-inducible by combination with tub::Gal80ts; Figure 3F, Figure 3G). Altogether, our data indicate that maintaining ER homeostasis in ISCs is critical for long-term ISC quiescence, while an active UPRER within ISCs is required and sufficient for ISC proliferation under homeostatic conditions, as well as in response to ER or oxidative stress. To assess whether engaging the UPRER is universally required for ISC proliferation, we assessed if reducing ER stress by over-expressing spliced Xbp1 was sufficient to limit ISC proliferation in a range of mitogenic conditions. ISC proliferation can be triggered through the JNK or the insulin/IGF signaling (IIS) pathways by over-expressing the JNK Kinase Hemipterous (Hep) [28] or the Insulin Receptor (InR) [44]–[46]. Over-expression of spliced Xbp1 was sufficient to inhibit ISC proliferation in both conditions (Figure S2A, Figure S2B; this inhibition is not due to apoptosis of ISCs, as ISCs were readily observed even at 14 days after inducing expression of Hep and/or Xbp1spliced).

Modulating Xbp1 activity further influenced the growth of ISC/EE tumors that accumulate due to defective EB differentiation in Notch loss of function conditions: While spliced Xbp1 prevented tumor formation, loss of Xbp1 exacerbated the growth of these tumors (Figure S2C). By regulating ER homeostasis, Xbp1 thus serves as a rheostat broadly controlling ISC proliferative activity.
ROS-mediated control of ISC proliferation by the UPRER
The control of ISC proliferation by the UPRER resembles ISC control by ROS, which can trigger dysplastic over-proliferation of ISCs, but are required for proliferation during homeostatic regeneration [19], [24]. Oxidative stress and ER stress are tightly linked: perturbation of redox homeostasis results in the accumulation of misfolded proteins and activation of the UPRER, and ER stress results in cytosolic oxidative stress [47]–[49]. To explore the relationship between ER stress, oxidative stress, and the UPRER in the ISC lineage, we assessed changes in intracellular redox homeostasis in ISCs deficient in Xbp1 or Hrd1 (Figure 4A). Both conditions resulted in significantly increased fluorescence of dihydro-ethidium (DHE, a redox-sensitive dye that can be used to detect ROS accumulation in live intestines [17], [19]) compared to wild-type progenitor cells (Figure 4A). Over-expression of spliced Xbp1, in turn, resulted in decreased DHE fluorescence in ISCs even under stress conditions (Figure 4B).
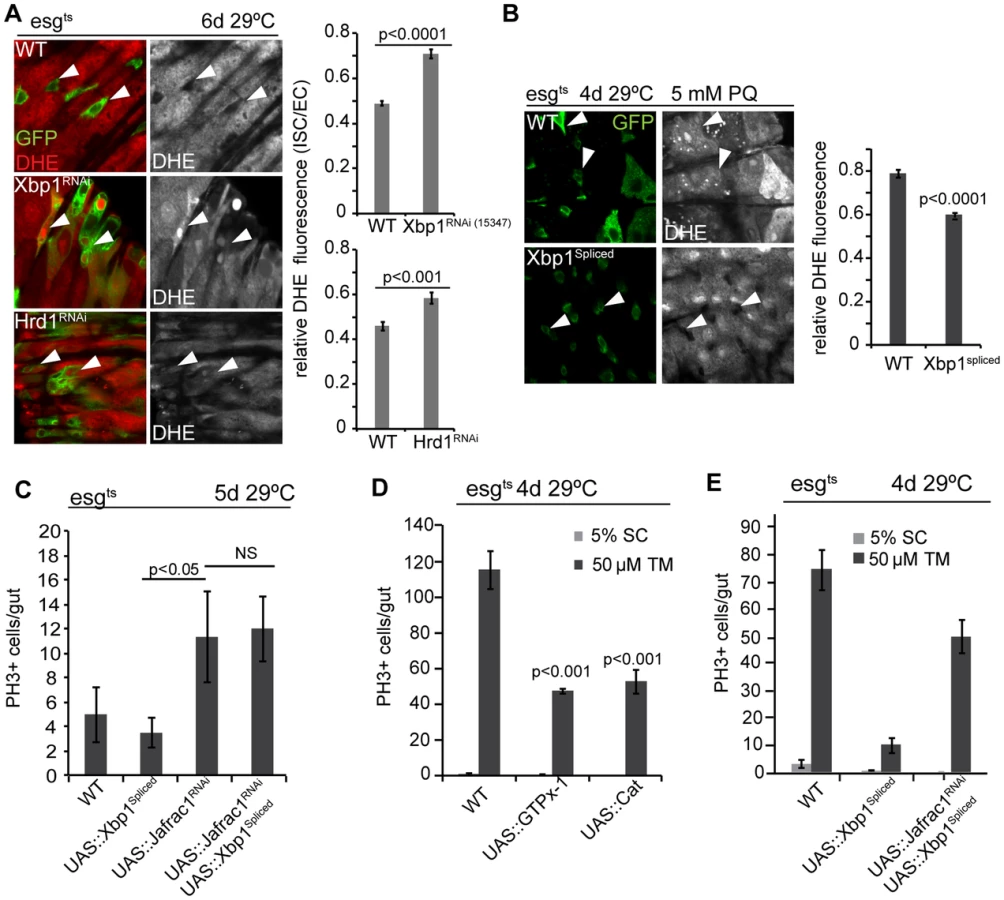
To further dissect the relationship between ER stress and oxidative stress, we perturbed the peroxiredoxin Jafrac1, which strongly influences intracellular redox homeostasis and regulates ISC proliferation [19], [50]. Knockdown of Jafrac1 was sufficient to increase ISC proliferation, and this increase was insensitive to the expression of spliced Xbp1 (Figure 4C). Over-expression of the anti-oxidant enzymes glutathione peroxidase I (GTPx-1) or Catalase (Cat), on the other hand, inhibited tunicamycin-induced ISC proliferation (Figure 4D), while knocking down Jafrac1 in ISCs prevented the inhibition of tunicamycin-induced ISC proliferation by spliced Xbp1 (Figure 4E). Increased ROS production thus acts downstream of Xbp1 in the regulation of ISC proliferation.
Increased ISC proliferation in Xbp1 or Hrd1 loss of function conditions, or in response to tunicamycin treatment, was associated with increased phosphorylation of JNK in Dl+ ISCs (Figure 5A, Figure S3B), and activation of the JNK target gene puckered in all cells of the intestinal epithelium, including ISCs and neighboring ECs (Figure 5B–D, Figure S3A, Figure S3B). This activation can be repressed by over-expressing spliced Xbp1, GTPx-1, or Cat in ISCs, suggesting that JNK is activated in response to ER stress-mediated ROS production (Figure 5D). Since JNK activation in ISCs promotes their proliferative activity [28], we tested whether JNK activity was required for ISC proliferation in Xbp1 loss of function conditions. Indeed, expression of BskRNAi, or of a dominant-negative version of Bsk (BskDN), reduced proliferation of ISCs in which Xbp1 was knocked down, and in animals exposed to tunicamycin (Figure 5E). These results suggest that activation of JNK in response to ER-stress-induced ROS production is required in ISCs to induce proliferation.

Coordinated control of ISC proliferation by the UPRER and the Keap1/CncC pathway
How do Xbp1 and the UPRER regulate ISC proliferation? Since promoting ER homeostasis by increasing Xbp1 activity or by stimulating the ERAD pathway was sufficient to limit ISC proliferation in all tested stress and mitogenic conditions, and since the Nrf2 homologue CncC exerts a similar effect on ISC proliferation [19], we asked whether CncC activity was influenced by the ER stress response.
To test whether the UPRER influences CncC activity in ISCs, we used a gstD1::lacZ construct that responds to CncC activity in ISCs [19], [51]. Strikingly, loss of Xbp1 or Hrd1 was sufficient to inhibit gstD1::lacZ expression in ISCs, while ISCs over-expressing spliced Xbp1 maintained high gstD1::lacZ expression (Figure 6A). We confirmed the modulation of Xbp1 activity in these cells by detecting expression of the Xbp1 target hsc3 (Figure 6A). When ER stress was induced by treating animals with tunicamycin, gstD1::lacZ expression was reduced in ISCs, and this inhibition could be alleviated by over-expressing CncC, Xbp1, or Hrd1 (Figure 6B). The same results were obtained when spliced Xbp1 was expressed (Figure S4A).

Loss of ER homeostasis thus reduces CncC activity in ISCs, suggesting that CncC inhibition is a required component of the ER stress response in the regulation of ISC proliferation. To test this idea, we assessed if ISC proliferation is influenced by the interaction between Xbp1 and CncC. ISCs mutant for the CncC-specific E3 ubiquitin ligase Keap1 do not divide, due to impaired inhibition of CncC activity [19]. ISCs deficient in both Xbp1 and Keap1 did not divide either, suggesting that CncC acts downstream of Xbp1 in the regulation of ISC proliferation (Figure 6C, Figure 6D). Accordingly, knocking down Keap1 or over-expressing CncC was sufficient to rescue ISC over-proliferation caused by loss of Xbp1 (Figure 6E, Figure S4B). Over-expressing CncC was also sufficient to inhibit proliferation induced by tunicamycin treatment (Figure 6F). At the same time, loss of CncC was not sufficient to rescue the proliferation defect of ISCs over-expressing spliced Xbp1, suggesting that Xbp1 inhibits ISC proliferation not only by preventing CncC inhibition, but by additional mechanisms, most likely by inhibiting PERK activation through ER stress (Figure 6G).
Improved ER quality control in ISCs alleviates age-related intestinal dysplasia
The age-associated activation of the UPRER in ISCs, and the control of ISC proliferation by the UPRER, suggested that ER stress in ISCs also plays an important role in promoting age-related dysplasia. To address this question, we asked whether promoting ER homeostasis in progenitor cells is sufficient to limit dysplasia. Xbp1 or Hrd1 over-expression was sufficient to maintain expression of gstD1::lacZ, indicating that CncC activity, which declines with age in ISCs [19] was maintained (Figure 7A). Accordingly, Hrd1 expression prevented the age-related increase in hsc3 expression in ISCs (Figure S5A; CncC or Keap1RNAi expression also preserve gstD1::lacZ expression, confirming the responsiveness of the reporter to CncC activity; Figure S5B). As expected, the same genetic conditions also limit ISC proliferation in aging flies, preventing dysplasia (Figure 7B) [19].

Discussion
Our results establish a critical role for the coordination of oxidative and ER stress responses in the control of stem cell function, proliferative homeostasis and regenerative capacity in the Drosophila intestine. As previously observed for ROS signaling [19], [24], we find that ER stress not only promotes ISC proliferation, but that the UPRER is also required for ISC proliferation under basal, homeostatic conditions. The UPRER thus emerges as a rheostat regulating ISC proliferation under both stress and homeostatic conditions. Our results suggest that the tissue-wide increase in ER stress in the aging intestinal epithelium perturbs this regulation, resulting in intestinal dysplasia.
The consequences of perturbing ER homeostasis in the intestinal epithelium are reminiscent of similar effects in Xbp1-deficient mice, where loss of Xbp1 promotes ISC proliferation and intestinal tumorigenesis [2]. At the same time, a recent study suggests that UPRER components are primarily expressed in transit amplifying cells of the intestinal epithelium, and that activation of the UPRER (specifically the PERK branch) promotes differentiation of intestinal epithelial stem cells [16]. The Drosophila midgut epithelium does not contain a transit amplifying cell population, yet our data suggest that a role for the UPRER in the control of ISC activity is conserved.
The requirement for CncC inhibition in ER stress-mediated activation of ISC proliferation highlights the integrated control of ISC activity by oxidative and ER stress signals. We propose that Xbp1, by promoting ER homeostasis, limits ROS accumulation in ISCs and thus maintains ISC quiescence (Figure 7D, Figure 7E). Excessive ROS results in JNK activation, which in turn activates Fos and inhibits CncC in ISCs, triggering proliferation ([19], [52] and Li, Hochmuth and Jasper, unpublished results).
This coordination of ER and oxidative stress responses by CncC and the UPRER is likely to be complex. In C. elegans the UPRER coordinates transcriptional regulation of anti-oxidant genes with the CncC homologue SKN-1 [27]. Interestingly, SKN-1 can also directly control the expression of UPRER components (including Xbp1, ATF-6 and Bip) by binding to their promoter regions, independent of oxidative stress [27]. Studies in worms have further established the UPRER as a critical determinant of longevity, and Xbp1 extends lifespan by improving ER stress resistance [53], [54]. Strikingly, local activation of the UPRER can trigger UPRER responses in distant tissues, indicating that endocrine processes exist that coordinate such stress responses across cells and tissues [53]–[56]. Our results support the notion that improving proteostasis by boosting ER folding capacity improves long-term tissue homeostasis. These effects seem to be largely mediated by cell-autonomous integration of the UPRER and redox response by JNK and CncC, but we also observe non-autonomous effects of ER stress on ISC proliferation when knocking down Xbp1 in EBs or ECs selectively. Furthermore, JNK is activated broadly in the intestinal epithelium when Xbp1 or Hrd1 are knocked down in ISCs and EBs, suggesting that non-autonomous interactions between cells experiencing ER stress also influence the regenerative response of this tissue. The molecular events regulating the coordination between cell-autonomous and non-autonomous events in the ER stress response of ISCs are subject of current investigation (Wang et al., in preparation).
In the small intestine of mice, the UPRER influences regenerative activity not only by influencing ISCs and transit amplifying cells directly, but also by influencing intestinal immune homeostasis. Loss of Xbp1 in intestinal epithelial cells (IECs) leads to apoptosis of secretory Paneth cells and goblet cells, and this pathology is associated with inflammation and higher risk of IBD [1], [3]. Deregulation of innate immune responses by the UPRER is also found in human patients [1], [3], [57], as well as in C. elegans [3], [58]. It can therefore be anticipated that the age-related increase in ER stress in the fly intestine also influences innate immune homeostasis and may contribute to commensal dysbiosis, which we have recently shown to be a driving factor in the age-related loss of proliferative homeostasis of the fly intestine [32]. It will be intriguing to dissect the interaction between the UPRER machinery, innate immune signaling in ECs, commensal homeostasis and stem cell function in detail, and we anticipate that these interactions have a significant effect on overall lifespan of the organism.
Materials and Methods
Fly lines and husbandry
Fly lines w1118, frt82B, frt40A, UAS::nlsGFP, UAS::hsc3, UAS::Xbp1RNAi (TRip:HMS03015) were obtained from the Bloomington Drosophila stock center. The following RNAi lines were obtained from the Vienna Drosophila RNAi Center: UAS::Xbp1RNAi (v109312, v15347), UAS::Hrd1RNAi (v6870), UAS::bskRNAi.
The following fly lines were generously provided as indicated: y1w1; esg::Gal4/+ by Dr. S Hayashi; UAS::xbp1EP2112 by Dr. Kyoung Sang Cho; UAS::xbp1d08698 by Dr. P. Fernandez-Funez; esgtsF/O by Dr. H. Jiang; Su(H)Gbe::Gal4 by Dr. S. Bray; pucE69::lacZ by Dr. E. Martín-Blanco; UAS::xbp1spliced by Dr. P. Domingos, UAS::bskDN by Dr. M. Mlodzik.
The Hrd1 loss of function allele Hrd1Delta was made by FRT-mediated deletion of sequences between the Pbac insertion lines Pbac{PB}sip3 c00467 and Pbac{PB}faf06363. hs-FLP was expressed in flies in which these Pbac insertions were in trans, deleting Hrd1 and its nearby gene CG2126.
All flies were raised on yeast/molasses-based food at 25°C and 65% humidity on a 12 hr light/dark cycle, unless otherwise noted.
For tunicamycin or paraquat exposure, flies were starved in empty vials for 6–8 hrs and fed with a 5% sucrose solution± 50 µM tunicamycin or ±5 mM paraquat for 24 hrs followed by dissection in PBS.
For TARGET experiments, flies were raised at 18°C and shifted to 29°C at certain time points after eclosion. For MARCM clone induction, adult flies were aged for 1–2 days and then heat shocked at 37°C for 45 min.
Generation of Su (H)GBE-Gal80 transgenic flies
The DNA fragment containing an enhancer of Su(H)GBE and a mini promoter of HSP70 was amplified from Su(H)GBE-Gal4 [39] using PCR, with the following primers:
5′-AGTGAATTCAATTAGGCCTACTAGACTTG-3′ (the 20th nucleotide “T” is replaced by “A” to eliminate the endogenous XbaI site).
5′-AGTTCTAGATCATGATGCGGCCGCTCAGGAGGCTTGCTTCAAGCTTG-3′ (a NotI site was introduced in this primer).
The amplified DNA was cut and ligated into EcoRI and XbaI digested pCasper-Tub-Gal80 [1]–[7], [59] to produce the construct pCasper-Su(H)GBE. Then the DNA fragment containing Gal80 and Sv40 polyA was cut from pCasper-Tub-Gal80 at the NotI and XhoI sites, and ligated into NotI - and XhoI-digested pCasper-Su(H)GBE to produce the Su(H)GBE-Gal80 construct.
The construct was sequenced, purified, and microinjected into embryos using the standard method.
Immunostaining and microscopy
Guts were dissected in PBS, fixed for 45 min at room temperature in 100 mM glutamic acid, 25 mM KCl, 20 mM MgSO4, 4 mM sodium phosphate, 1 mM MgCl2, and 4%formaldehyde, washed for 1 hr, and incubated with primary antibodies and second antibodies in washing buffer (PBS, 0.5% BSA, 0.1% Triton X-100).
The following primary antibodies were used:, guinea-pig anti-hsc3 antibody antibody [60] (1∶150), mouse anti-Delta (Developmental Studies Hybridoma Bank, 1∶100), rat anti-Delta (gift from Dr. MD Rand, University of Rochester, 1∶1000); rabbit anti-PH3 (phosphorylated histone H3, Upstate, 1∶1000), mouse anti-β-galactosidase (Developmental Studies Hybridoma Bank, 1∶500), rabbit anti-β-galactosidase (Cappel, 1∶5000), rabbit anti-peIF2α antibody (Cell Signaling: 3597, 1∶150), mouse anti-pJNK antibody (Cell Signaling: 9255,1∶150).
For Delta antibody staining, guts were fixed using a methanol-heptane method as descried [61].
Fluorescent secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. DNA was stained using DAPI. Confocal imaging was performed on a Zeiss LSM700 confocal microscope and processed using ImageJ and Adobe Illustrator.
qRT-PCR analysis of gene expression
Total RNA from young female samples were extracted using Trizol (Invitrogen) and cDNA was synthesized using Superscript III (Invitrogen). Real time RCR was performed on a Bio-Rad CFX96 detection system. Expression Values were normalized to RP49 expression levels. Primers included: total Xbp1 transcripts (Forward:TGGGAGGAGAAAGTGCAAAG, Reverse:TCCGTTCTGTCTGTCAGCTC), Spliced Xbp1 (Forward: ACCAACCTTGGATCTGCCG, Reverse:CGCCAAGCATGTCTTGTAGA), Hrd1 (Forward:GCAGTTGGTCTTTGGCTTTG, Reverse: ATGGGCAGCGCGTATATTT), RP49(Forward:TCCTACCAGCTTCAAGATGAC, Reverse:CACGTTGTGCACCAGGAACT).
ROS measurement via DHE
ROS levels were measured as described before [19]. Briefly, guts were dissected in Schneider's medium, incubated in 30 µM (Invitrogen) for 5 min at room temperature in the dark, washed twice and mounted to be imaged immediately. GFP expressed under the control of esg::Gal4, Su(H)::Gal80 was used to identify ISCs and/or EBs.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KaserA, LeeA-H, FrankeA, GlickmanJN, ZeissigS, et al. (2008) XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 134 : 743–756 doi:10.1016/j.cell.2008.07.021
2. NiederreiterL, FritzTMJ, AdolphTE, KrismerAM, OffnerFA, et al. (2013) ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. Journal of Experimental Medicine 210 : 2041–2056 doi:10.1038/nature07589
3. GlimcherLH (2009) XBP1: the last two decades. Annals of the Rheumatic Diseases 69: i67–i71 doi:10.1136/ard.2009.119388
4. GarrettWS, GordonJI, GlimcherLH (2010) Homeostasis and inflammation in the intestine. Cell 140 : 859–870 doi:10.1016/j.cell.2010.01.023
5. KaserA, ZeissigS, BlumbergRS (2010) Inflammatory bowel disease. Annu Rev Immunol 28 : 573–621 doi:10.1146/annurev-immunol-030409-101225
6. KaserA, FlakMB, TomczakMF, BlumbergRS (2011) The unfolded protein response and its role in intestinal homeostasis and inflammation. Experimental Cell Research 317 : 2772–2779 doi:10.1016/j.yexcr.2011.07.008
7. AdolphTE, TomczakMF, NiederreiterL, KoH-J, BöckJ, et al. (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503 : 272–276 doi:10.1038/nature12599
8. WalterP, RonD (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334 : 1081–1086 doi:10.1126/science.1209038
9. SchröderM, KaufmanRJ (2005) THE MAMMALIAN UNFOLDED PROTEIN RESPONSE. Annu Rev Biochem 74 : 739–789 doi:10.1146/annurev.biochem.73.011303.074134
10. TraversKJ, PatilCK, WodickaL, LockhartDJ, WeissmanJS, et al. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101 : 249–258.
11. RyooHD, StellerH (2007) Unfolded protein response in Drosophila: why another model can make it fly. Cell Cycle 6 : 830–835.
12. SmithMH, PloeghHL, WeissmanJS (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334 : 1086–1090 doi:10.1126/science.1209235
13. FrandAR, KaiserCA (1999) Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell 4 : 469–477.
14. KimS, SiderisDP, SevierCS, KaiserCA (2012) Balanced Ero1 activation and inactivation establishes ER redox homeostasis. The Journal of Cell Biology 196 : 713–725 doi:10.1083/jcb.201110090
15. GrossE, SevierCS, HeldmanN, VituE, BentzurM, et al. (2006) Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA 103 : 299–304 doi:10.1073/pnas.0506448103
16. HeijmansJ, van Lidth de JeudeJF, KooB-K, RosekransSL, WielengaMCB, et al. (2013) ER Stress Causes Rapid Loss of Intestinal Epithelial Stemness through Activation of the Unfolded Protein Response. Cell reports 3 : 1128–1139 doi:10.1016/j.celrep.2013.02.031
17. Owusu-AnsahE, BanerjeeU (2009) Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461 : 537–541 doi:10.1038/nature08313
18. NobleM, SmithJ, PowerJ, Mayer-ProschelM (2003) Redox state as a central modulator of precursor cell function. Annals of the New York Academy of Sciences 991 : 251–271.
19. HochmuthCE, BiteauB, BohmannD, JasperH (2011) Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell 8 : 188–199 doi:10.1016/j.stem.2010.12.006
20. TothovaZ, GillilandDG (2007) FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1 : 140–152.
21. LuoB, LeeAS (2013) The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 32 : 805–818 doi:10.1038/onc.2012.130
22. MicchelliCA, PerrimonN (2006) Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 439 : 475–479 doi:10.1038/nature04371
23. OhlsteinB, SpradlingA (2006) The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 439 : 470–474 doi:10.1038/nature04333
24. BiteauB, HochmuthCE, JasperH (2011) Maintaining tissue homeostasis: dynamic control of somatic stem cell activity. Cell Stem Cell 9 : 402–411 doi:10.1016/j.stem.2011.10.004
25. BuchonN, BroderickNA, LemaitreB (2013) Gut homeostasis in a microbial world: insights from Drosophila melanogaster. Nature reviews Microbiology 11 : 615–626 doi:10.1038/nrmicro3074
26. CullinanSB, DiehlJA (2006) Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. The international journal of biochemistry & cell biology 38 : 317–332.
27. Glover-CutterKM, LinS, BlackwellTK (2013) Integration of the Unfolded Protein and Oxidative Stress Responses through SKN-1/Nrf. PLoS Genet 9: e1003701 doi:10.1371/journal.pgen.1003701
28. BiteauB, HochmuthCE, JasperH (2008) JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell 3 : 442–455 doi:10.1016/j.stem.2008.07.024
29. ChoiNH, KimJG, YangDJ, KimYS, YooMA (2008) Age-related changes in Drosophila midgut are associated with PVF2, a PDGF/VEGF-like growth factor. Aging Cell 7 : 318–334 doi:10.1111/j.1474-9726.2008.00380.x
30. ReraM, ClarkRI, WalkerDW (2012) Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proceedings of the National Academy of Sciences 109 : 21528–21533 doi:10.1073/pnas.1215849110
31. BiteauB, KarpacJ, SupoyoS, DeGennaroM, LehmannR, et al. (2010) Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet 6: e1001159.
32. GuoL, KarpacJ, TranSL, JasperH (2014) PGRP-SC2 Promotes Gut Immune Homeostasis to Limit Commensal Dysbiosis and Extend Lifespan. Cell 156 : 109–122 doi:10.1016/j.cell.2013.12.018
33. RyooHD, LiJ, KangM-J (2013) Drosophila XBP1 expression reporter marks cells under endoplasmic reticulum stress and with high protein secretory load. PLoS ONE 8: e75774 doi:10.1371/journal.pone.0075774
34. SoneM, ZengX, LareseJ, RyooHD (2013) A modified UPR stress sensing system reveals a novel tissue distribution of IRE1/XBP1 activity during normal Drosophila development. Cell Stress Chaperones 18 : 307–319 doi:10.1007/s12192-012-0383-x
35. JiangH, PatelPH, KohlmaierA, GrenleyMO, McEwenDG, et al. (2009) Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137 : 1343–1355 doi:10.1016/j.cell.2009.05.014
36. OhlsteinB, SpradlingA (2007) Multipotent Drosophila intestinal stem cells specify daughter cell fates by differential notch signaling. Science 315 : 988–992 doi:10.1126/science.1136606
37. Casas-TintoS, ZhangY, Sanchez-GarciaJ, Gomez-VelazquezM, Rincon-LimasDE, et al. (2011) The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Human Molecular Genetics 20 : 2144–2160 doi:10.1093/hmg/ddr100
38. BordalloJ, PlemperRK, FingerA, WolfDH (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Molecular biology of the cell 9 : 209–222.
39. ZengX, ChauhanC, HouSX (2010) Characterization of midgut stem cell - and enteroblast-specific Gal4 lines in drosophila. Genesis 48 : 607–611 doi:10.1002/dvg.20661
40. LuoL (2007) Fly MARCM and mouse MADM: genetic methods of labeling and manipulating single neurons. Brain Res Rev 55 : 220–227 doi:10.1016/j.brainresrev.2007.01.012
41. ChoiG, ParkSH, HwangS, HanSY, HongYK, et al. (n.d.) Interference in xbp1 gene expression induces defective cell differentiation and sensory organ development in Drosophila. Genes & Genomics 32 : 233–238 doi:10.1007/s13258-010-0002-0
42. KangM-J, RyooHD (2009) Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proceedings of the National Academy of Sciences 106 : 17043–17048 doi:10.1073/pnas.0905566106
43. DornerAJ, BoleDG, KaufmanRJ (1987) The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. The Journal of cell biology 105 : 2665–2674.
44. BiteauB, KarpacJ, HwangboD, JasperH (2010) Regulation of Drosophila lifespan by JNK signaling. Exp Gerontol 46 : 349–354 doi:10.1016/j.exger.2010.11.003
45. KapuriaS, KarpacJ, BiteauB, HwangboD, JasperH (2012) Notch-mediated suppression of TSC2 expression regulates cell differentiation in the Drosophila intestinal stem cell lineage. PLoS Genet 8: e1003045.
46. AmcheslavskyA, JiangJ, IpYT (2009) Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell 4 : 49–61 doi:10.1016/j.stem.2008.10.016
47. HardingHP, ZhangY, ZengH, NovoaI, LuPD, et al. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11 : 619–633.
48. SevierCS, KaiserCA (2008) Ero1 and redox homeostasis in the endoplasmic reticulum. Biochimica et biophysica acta 1783 : 549–556 doi:10.1016/j.bbamcr.2007.12.011
49. TuBP, WeissmanJS (2004) Oxidative protein folding in eukaryotes: mechanisms and consequences. The Journal of cell biology 164 : 341–346 doi:10.1083/jcb.200311055
50. DeGennaroM, HurdTR, SiekhausDE, BiteauB, JasperH, et al. (2011) Peroxiredoxin stabilization of DE-cadherin promotes primordial germ cell adhesion. Dev Cell 20 : 233–243 doi:10.1016/j.devcel.2010.12.007
51. SykiotisGP, BohmannD (2008) Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell 14 : 76–85 doi:10.1016/j.devcel.2007.12.002
52. BiteauB, JasperH (2011) EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 138 : 1045–1055 doi:10.1242/dev.056671
53. Henis-KorenblitS, ZhangP, HansenM, McCormickM, LeeSJ, et al. (2010) Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proceedings of the National Academy of Sciences 107 : 9730–9735 doi:10.1073/pnas.1002575107
54. TaylorRC, DillinA (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153 : 1435–1447 doi:10.1016/j.cell.2013.05.042
55. KourtisN, TavernarakisN (2011) Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J 30 : 2520–2531 doi:10.1038/emboj.2011.162
56. TaylorRC, DillinA (2011) Aging as an event of proteostasis collapse. Cold Spring Harbor perspectives in biology 3 doi:10.1101/cshperspect.a004440
57. GrootjansJ, HodinCM, de HaanJ-J, DerikxJPM, RouschopKMA, et al. (2011) Level of activation of the unfolded protein response correlates with Paneth cell apoptosis in human small intestine exposed to ischemia/reperfusion. Gastroenterology 140 : 529–539.e3 doi:10.1053/j.gastro.2010.10.040
58. RichardsonCE, KooistraT, KimDH (2010) An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463 : 1092–1095 doi:10.1038/nature08762
59. LeeT, LuoL (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 24 : 251–254.
60. RyooHD, DomingosPM, KangM-J, StellerH (2006) Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J 26 : 242–252 doi:10.1038/sj.emboj.7601477
61. LinG, XuN, XiR (2008) Paracrine Wingless signalling controls self-renewal of Drosophila intestinal stem cells. Nature 455 : 1119–1123 doi:10.1038/nature07329
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 8
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in African Americans Provides Insights into the Genetic Architecture of Type 2 Diabetes
- KDM6 Demethylase Independent Loss of Histone H3 Lysine 27 Trimethylation during Early Embryonic Development
- The RNA Helicases AtMTR4 and HEN2 Target Specific Subsets of Nuclear Transcripts for Degradation by the Nuclear Exosome in
- EF-P Dependent Pauses Integrate Proximal and Distal Signals during Translation