
Nucleoporin 153 Arrests the Nuclear Import of Hepatitis B Virus Capsids in the Nuclear Basket
Virtually all DNA viruses including hepatitis B viruses (HBV) replicate their genome inside the nucleus. In non-dividing cells, the genome has to pass through the nuclear pore complexes (NPCs) by the aid of nuclear transport receptors as e.g. importin β (karyopherin). Most viruses release their genome in the cytoplasm or at the cytosolic face of the NPC, as the diameter of their capsids exceeds the size of the NPC. The DNA genome of HBV is derived from reverse transcription of an RNA pregenome. Genome maturation occurs in cytosolic capsids and progeny capsids can deliver the genome into the nucleus causing nuclear genome amplification. The karyophilic capsids are small enough to pass the NPC, but nuclear entry of capsids with an immature genome is halted in the nuclear basket on the nuclear side of the NPC, and the genome remains encapsidated. In contrast, capsids with a mature genome enter the basket and consequently liberate the genome. Investigating the difference between immature and mature capsids, we found that mature capsids had to disintegrate in order to leave the nuclear basket. The arrest of a karyophilic cargo at the nuclear pore is a rare phenomenon, which has been described for only very few cellular proteins participating in nuclear entry. We analyzed the interactions causing HBV capsid retention. By pull-down assays and partial siRNA depletion, we showed that HBV capsids directly interact with nucleoporin 153 (Nup153), an essential protein of the nuclear basket which participates in nuclear transport via importin β. The binding sites of importin β and capsids were shown to overlap but capsid binding was 150-fold stronger. In cellulo experiments using digitonin-permeabilized cells confirmed the interference between capsid binding and nuclear import by importin β. Collectively, our findings describe a unique nuclear import strategy not only for viruses but for all karyophilic cargos.
Published in the journal:
. PLoS Pathog 6(1): e32767. doi:10.1371/journal.ppat.1000741
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000741
Summary
Virtually all DNA viruses including hepatitis B viruses (HBV) replicate their genome inside the nucleus. In non-dividing cells, the genome has to pass through the nuclear pore complexes (NPCs) by the aid of nuclear transport receptors as e.g. importin β (karyopherin). Most viruses release their genome in the cytoplasm or at the cytosolic face of the NPC, as the diameter of their capsids exceeds the size of the NPC. The DNA genome of HBV is derived from reverse transcription of an RNA pregenome. Genome maturation occurs in cytosolic capsids and progeny capsids can deliver the genome into the nucleus causing nuclear genome amplification. The karyophilic capsids are small enough to pass the NPC, but nuclear entry of capsids with an immature genome is halted in the nuclear basket on the nuclear side of the NPC, and the genome remains encapsidated. In contrast, capsids with a mature genome enter the basket and consequently liberate the genome. Investigating the difference between immature and mature capsids, we found that mature capsids had to disintegrate in order to leave the nuclear basket. The arrest of a karyophilic cargo at the nuclear pore is a rare phenomenon, which has been described for only very few cellular proteins participating in nuclear entry. We analyzed the interactions causing HBV capsid retention. By pull-down assays and partial siRNA depletion, we showed that HBV capsids directly interact with nucleoporin 153 (Nup153), an essential protein of the nuclear basket which participates in nuclear transport via importin β. The binding sites of importin β and capsids were shown to overlap but capsid binding was 150-fold stronger. In cellulo experiments using digitonin-permeabilized cells confirmed the interference between capsid binding and nuclear import by importin β. Collectively, our findings describe a unique nuclear import strategy not only for viruses but for all karyophilic cargos.
Introduction
Most DNA viruses depend on nuclear host factors for their replication. Viruses infecting non-dividing cells have to pass the nuclear envelope through the nuclear pore complexes (NPCs). The NPC is large proteinaceous structure of ∼30 different proteins called nucleoporins (Nups). Due to the eight fold rotational symmetry of the NPC each Nup is present in 8–48 copies, forming a complex of ∼125 MDa. On the cytoplasmic face of the NPC eight fibers extrude from a central ring-like framework, which is embedded in the nuclear envelope. This ring forms openings in the nuclear envelope allowing translocation of cargos with a diameter up to 39 nm [1]. On the karyoplasmic face of the NPC 8 fibers form the cage-like structure of the nuclear basket (reviewed by [2]).
NPCs regulate the traffic of proteins and nucleic acids into and out of the nucleus (reviewed by [3]). While small molecules may diffuse between cytoplasm and nucleus karyophilic macromolecules are transported in a complex with soluble nuclear import receptors. It is estimated that ∼1000 transport complexes pass each NPC per second [4].
The best characterized transport receptors belong to the importin (karyopherin) β superfamily, comprising importin β, transportin 1, 2, transportin SR and exportins. Nuclear import is initiated by binding of the receptors to a signal on the surface of the karyophilic cargo. There is a variety of signals as e.g. M9 domains, interacting with transportins, importin β binding domains and “classical” nuclear localization signals (NLS), which bind to importin β via the adapter molecule importin α.
The driving force of nuclear import and export is a gradient of the small GTPase Ran in its GTP-bound form across the nuclear envelope. RanGTP is enriched in the karyoplasm, where it interacts with the transport receptors of the import complex, leading to its dissociation. While the cargo diffuses deeper into the karyoplasm, the RanGTP-receptor complex is exported into the cytoplasm where it dissociates.
A key component of nuclear import is Nup153 to which the import complex of cargo and importin(s) binds and subsequently dissociates. Nup153 is a 1445 amino acid (aa) protein of the nuclear basket [5], which comprises three domains (reviewed by [6]). The N terminus (aa 1–670) at the nuclear ring of the NPC contains an NPC targeting domain and an RNA binding domain. The N terminus interacts with other proteins of the NPC as Nup107 and Tpr and is required for proper NPC architecture [5],[7],[8]. The zinc finger region (aa 650–880) at the distal ring of the NPC facilitates interactions with Ran. The ∼30 FXFG repeat-containing C terminus is part of the hydrophobic meshwork that forms the “sieve” through which karyophilic cargos have to pass [9]. The participation of Nup153 in vital cellular processes makes it difficult to analyze its functions without interfering with cell viability [10].
Viruses with a nuclear phase in their life cycle make use of this import machinery. Best characterized are adeno-, herpes, influenza and the human immune deficiency virus. The latter two viruses disassemble in the cytoplasm and release the genome in complex with karyophilic viral proteins. These complexes fall below the limit of the NPC diameter and pass the pore like cellular macromolecules (reviewed by [11]) or by transiently interacting with Nups [12] In contrast the genomes of adenoviruses and herpes viruses remain encapsidated within their capsids. Their diameters of 90 and 120 nm exceed the diameter of the nuclear pore. Adenoviruses bind to the cytoplasmic face of the NPC where they disassemble. Genome translocation through the pore involving viral and cellular karyophilic proteins is not well understood [13],[14],[15],[16]. Herpes virus capsids also bind to the exterior of the NPC using importin β [17] but they open at the penton facing the NPC and inject the DNA through the pore. For both viruses the trigger of genome release is unknown.
Similar investigations have been performed in digitonin-permeabilized cells on capsids of the medically important hepatitis B virus (HBV) [18],[19]. Hepatitis B is endemic in large parts of the world. Approximately 350 million people are chronically infected, accounting for 1 million deaths per year. HBV is an enveloped virus comprising an icosaedral capsid, which contains the partially double stranded DNA genome (relaxed circular, rcDNA). The capsid exists in T = 4 (major form) and T = 3 symmetry [20], composed of 240 or 180 copies of the core protein, respectively. The two forms differ in diameter (36 and 32 nm) but functional differences are not known. Entry of the virus into the hepatocyte is not well understood due to low efficiency of the available cell culture systems for HBV infection [21]. Circumstantial evidence however suggests that the capsid enters the cytosol after fusion of the viral envelope with a cellular membrane [22]. In fact lipofection of capsids, which by-passes the rate-limiting natural entry causes productive HBV “infection” of hepatoma cells with in vivo-like efficiency [23]. Like other DNA viruses (with the exception of baculoviruses [24]) HBV capsids are transported towards the NPCs using the microtubule transport system [23].
HBV is a pararetrovirus replicating via an RNA pregenome that is transcribed from the nuclear covalently-closed circular form of the viral genome. Consequently the viral rcDNA has to enter the nucleoplasm upon infection where the rcDNA is converted to the covalently-closed circular form. To allow access of the unknown cellular DNA repair factors the viral genome has to be liberated from the capsid either prior to, during or after transport through the NPC. Transport and genome release are obviously highly efficient and well-coordinated, since ∼80% of virions are infection-competent in vivo [25].
After export to the cytoplasm, the RNA pregenome is translated to core protein and the viral polymerase which binds in situ to an encapsidation signal on the pregenome. This complex is specifically encapsidated within the assembling core protein (reviewed by [26]), which forms an immature capsid (Immat-C). The polymerase converts the RNA into rcDNA, which is found in mature capsids (Mat-C). It is worth noting that genome maturation occurs exclusively inside the capsid and is a prerequisite for envelopment of the capsids by the surface proteins for virion formation [27].
Core proteins assemble spontaneously to HBV capsids, e. g. upon expression in E. coli. The first 140 aa of the core protein are essential for assembly and exhibit an ordered structure. Within a resolution limit of 16 Å E. coli-expressed capsids show the same morphology as virion-derived capsids or capsids from infected cells [28]. The arginine-rich C-terminus (aa 141–185) is flexible [29] and exhibits phosphorylation sites for a cellular protein kinase, which has not yet been unequivocally identified. In E. coli-expressed capsids, which contain unspecific E. coli RNA and which are not phosphorylated, the C terminus is localized within the lumen of the capsid [30].
Some steps of genome maturation depend upon core protein phosphorylation, notably pregenome encapsidation and plus strand DNA synthesis [31],[32]. Both phosphorylation and genome maturation lead to translocation of the core protein C termini, harboring a NLS, to the exterior of the capsids [19]. Consequently, all HBV capsids that have undergone some degree of genome maturation or phosphorylation bind to importin α/β [18] and can be found in the nuclear basket [1],[19]. Early in infection when sufficient amounts of surface proteins have not yet been synthesized, nuclear entry of progeny genomes increases the number of nuclear HBV DNA copies [33], which is generally low but can reach numbers of up to 491 per cell in HBV infected patient [34].
Using Digitonin-permeabilized cells – a frequently used system for analysis of nuclear import [35] – it was however shown that Immat-C and in vitro phosphorylated capsids synthesized in E. coli (P-rC) failed to exit the nuclear basket and thus do not diffuse deeper into the karyoplasm. In contrast, adding Mat-C to the cells led to the presence of intranuclear capsids and genome release [18],[19].
The import strategy of HBV capsids seems thus to follow an entirely different strategy than has been shown for other viruses. In particular the arrest of Immat-C in the basket is unique. To date the only examples for cargos with an aborted nuclear import reaction are some Nups that become incorporated into the NPC after cell division and the protein Ubc9, which partly associates with Nup358 on the cytosolic fibers of the NPC.
Evidently, the viral capsids have to interact with the NPCs upon initial infection but during establishment of the infection as well, at which time the nuclear viral DNA becomes amplified. Due to the incomplete understanding of HBV transport and disassembly, the unique strategy and the medical importance of HBV, we evaluated the molecular background of the capsid arrest in the NPC basket. Using in vitro and in cellulo approaches we identified the cellular interaction partner and identified the underlying mechanism responsible for the selective release of mature viral genomes. Collectively, these findings lead to a model of a multi-step, maturation-regulated nuclear entry of the HBV genome.
Results
Co-immune precipitation of nuclear proteins
Abortion of a nuclear import reaction within the nuclear pore must be based on a direct or indirect interaction with proteins of the NPC. The recently observed arrest of Immat-C and P-rC in the nuclear basket thus most likely involves an association with a Nup on the karyoplasmic face of the NPC. According to the current view on the architecture of the NPC candidates would include Nup50, 54, 58, 62, 93, 96, 98, 107, 133, 153, 160, Rae1, Seh1, Sec13 PBC68, and Tpr (summarized by [36]).
First, we wanted to identify the NPC proteins that are co-precipitated by Immat-C using a nuclear extract of rat liver nuclei. The NPC is composed of tightly interacting mostly hydrophobic proteins requiring denaturating conditions or harsh detergents for separation. As this treatment unfolds proteins we first tested whether refolding restored biological functions. For this purpose, we investigated the importin β binding ability since it is one of the essential functions of Nup153. Affinity of Nup153 to importin β was shown to be stronger than to other nucleoporins (Nup62, Nup214, Nup358 [37]).
To test our experimental system, pull-down was performed by incubating recombinant functional importin β with the extract followed by binding of the nucleoporins to solid-phase bound antibody mAb414. This antibody binds preferentially to the FXFG-repeat containing nucleoporins of vertebrates as e.g. Nup62, Nup153, Nup214 and Nup358 [38],[39] with different efficiency.
Figure 1A depicts that importin β became co-precipitated (lane 1). As the nuclear extract did not contain detectable amounts of importin β (not shown) this result implies that the importin β binding activity of the nucleoporin(s) was maintained or restored during extraction and renaturation. Binding was specific as antibody-coated beads without the extracted proteins and uncoated beads failed to interact with importin β. Binding occurred in the absence of an importin β-bound cargo. This finding is in accordance with the observation that importin β after its dissociation from the cargo does not diffuse deeper into the karyoplasm but remains bound to the NPC before being exported into the cytoplasm [40].

The nuclear extract was then subjected to co-immune precipitations using Immat-C which are arrested within the nuclear basket. These capsids contain DNA replication intermediates of the viral genome; they interact with the NPCs [19] and can thus be responsible for nuclear genome amplification. For visualization of co-precipitated proteins we used Sypro Red, staining all proteins after SDS PAGE. As depicted in Figure 1B no co-precipitation could be observed in the absence of nuclear extract. Faint bands were observed in the absence of capsids. This indicates some unspecific binding of nuclear proteins to the carrier beads, probably due to nonspecific hydrophobic interactions. Immat-C-driven pull-down however showed the precipitation of one dominant protein, strongly enriched in comparison to the control extract and the precipitation controls. This protein showed an apparent molecular weight of a ∼180 kDa typical for Nup153 [41],[42]. To confirm the identity of co-precipitated Nup153 we used the antibody mAb414 in Western blot. Figure 1C confirms the Nup153 co-precipitation by Immat-C and provides evidence that neither Nup214 nor Nup358 were co-precipitated.
To further confirm specific Nup153 capsid-interaction we preincubated the nuclear extract with Immat-C prior to precipitation via mAb414 and detection of co-precipitated Immat-C. Since antibodies against denaturated core proteins require very large amounts of protein to provide adequate signals we labeled Immat-C by 32P using the protein kinase activity inside the capsids. Phosphoimaging showed that a single protein was labeled having the molecular weight of the core protein of 21.5 kDa (Fig. 1D). As depicted in Figure 1E Immat-C could be precipitated by mAb414-bound Nup. No signal was obtained in the absence of nuclear extract or in the absence of mAb414.
Interaction between different capsid species and Nup153 domains
To determine whether the Nup153 binding was selective for Immat-C we included different capsid species in co-immune precipitations. We used Mat-C, which enter the nucleus and comprise an rcDNA genome; P-rC, which contain E. coli-RNA and which is arrested in the basket like Immat-C, and capsids that were formed by C-terminally truncated core proteins (ΔC-rC). ΔC-rC do not contain RNA, cannot be phosphorylated, and cannot enter the basket as these capsids do not contain the NLS. All capsids reacted equally well with the anti-capsid antibody used to precipitate them after preincubation with nuclear extract (not shown). Unexpectedly, all capsids were able to precipitate Nup153 as depicted by immune blot using mAb414 (Fig. 2A).

The Sypro Red stain of co-precipitated proteins was restricted for the molecular weight of the interaction partners. Proteins smaller ∼75 kDa could not be detected as this part of the SDS PAGE was heavily overloaded with high amounts of bead-bound antibodies and BSA used for saturation of unspecific binding sites. We could have thus missed smaller adapter proteins connecting the capsids with Nup153.
To obtain evidence of direct interaction we replaced the nuclear extract by E. coli-expressed Nup153 that was purified under native conditions. The fusion protein was previously shown to be functional on nuclear import and export after integration into the NPCs of reconstituted Xenopus laevis oocyte nuclei [43]. Figure 2B showed that all capsids precipitated GST-Nup153. As GST does not bind to the capsids (not shown) we conclude that Nup153 interacts directly with the surface of the capsid. The signal was not derived from the capsid preparation since no signal was observed in the absence of GST-Nup153. Furthermore, GST-Nup153 precipitation was not observed in the absence of the capsids implying that GST-Nup153 did not bind directly to the antibody coated-bead (Fig. 2C).
We next set out to determine to which Nup153 domain the capsids bind. We expressed large parts of Nup153 in three fragments (N: aa 53–272; Z: aa 272–543; C1: aa 618–999) as GST fusion proteins. The Nup153 fragments were incubated with the different capsids and co-precipitated by biomagnetic bead-bound anti-capsid antibodies. Co-precipitation of the Nup153 fragments was demonstrated by Western blot using anti-GST antibodies. Figure 2D shows that Mat-C, Immat-C and P-rC failed to precipitate the 50 kDa N terminal part of Nup153. A strong band at 54 kDa was visible in all samples to which the biomagnetic beads were added. This band was most likely the product of an unspecific binding of the secondary blot antibody to the heavy chain of the antibodies used in precipitation. Using the zinc finger domain Z (56 kDa) no precipitation could be observed (Fig. 2D). However all three capsid species co-precipitated the 68 kDa C1 fragment. Binding was specific as no precipitates could be observed in control reactions without capsids or with capsids but without anti-capsid antibodies on the beads. This Nup153 fragment comprises an importin β-binding domain [44],[45] thus implying that importin β and capsids compete for Nup153 binding.
A fourth His-tagged fragment (C2: aa 992–1219) containing most of the ∼30 FXFG repeats [46] was analyzed by co-precipitation but we observed an unspecific binding to the beads. To circumvent this technical problem we performed retardation gels. We incubated P-rC with His-Nup153 C2 and separated the complex by native agarose gel electrophoresis. The Nup fragment was visualized by anti-His antibodies, the capsid by anti-capsid antibodies. Control experiments were performed using the N and the C1 fragment. Migration of the fragments was determined by anti-GST antibodies.
Figure 2E confirmed that the N fragment did not interact with the P-rC as no shift of capsid migration could be observed upon the presence of this Nup153 domain. Nup153 C1 caused a retarded migration of the majority of capsids (Fig. 2F) and fragment C2 retarded the migration of all P-rC (Fig. 2F). Although fragment C2 is largely hydrophobic we assume that the capsid binding is not unspecific as the capsids are negatively charged and migrate on native agarose gels as ∼3000 bp linear double stranded DNA fragments.
As both fragments C1 and C2 barely overlap we conclude that there is more than one interaction site on Nup153.
Competition of P-rC, Nup153 and importin β
In vivo, numerous proteins and nuclear factors pass the NPC simultaneously requiring interaction with Nup153. As Immat-C and P-rC are arrested in vivo, one should expect that the affinity of the physiological transport complexes to Nup153 is weaker than that of the capsids. We incubated capsids (P-rC) immobilized on biomagnetic beads with Nup153 and importin β in different ratios. Figure 3A, lane 1, shows that no GST-Nup153 bound to the beads in the absence of capsids thus demonstrating the specificity of Nup153 binding. When incubating Nup153 in parallel to different amounts of importin β with the capsids only molar importin β excesses of more than 150-fold with regard to Nup153 prevented Nup153 binding to the capsids. This result thus confirm that capsid and importin β binding sites on Nup153 overlap and show further that the capsid binding was much stronger than the importin β interaction.

We next asked whether bound capsids can be displaced from Nup153 by importin β. We preincubated biomagnetic bead-bound P-rC with Nup153 and added importin β after removal of unbound Nup153. Figure 3B shows that even 6700-fold molar excesses of importin β did not remove Nup153 from the capsid.
Silencing of Nup153 expression by siRNA
In order to determine whether the capsid arrest in the basket is mediated by Nup153 we suppressed Nup153 expression in HeLa cells by siRNA. We controlled Nup153 expression in parallel to the expression of Fibrillarin, which is a component of a nucleolar small nuclear ribonucleoprotein (SnRNP) involved in ‘house keeping’ for nucleolar integrity [47]. Figure 4A shows that Fibrillarin expression was in fact unaltered but Nup153 reduced by 80%. Suppression was limited because Nup153 is essential for cell viability.

After permeabilization of these cells by digitonin a nuclear import assay using P-rC was performed. We visualized Nup153 and capsids using indirect immune fluorescence. Nup153 staining was weaker in 80–90% of the siRNA-treated cells than in non-silenced cells (Fig. 4B, lower panel). The nuclei of these cells were larger than in mock-transfected cells most probably due to the role of Nup153 in mitotic progression [48]. The nucleoporin Nup153 plays separate roles in both early mitotic progression and the resolution of mitosis.
In the nuclei of the control cells, P-rC accumulated at the nuclear envelope and did not enter the karyoplasm as demonstrated previously [18]. In contrast, a proportion of capsids were found in the karyoplasm of Nup153 siRNA-transfected cells, (still image: Fig. 4B upper panel; for videos see Videos S1, S2, S3 and S4). Quantification on 20 cells revealed that 18–19% of capsids entered the nucleus while ca. 80% were still arrested at the NE.
Effect of cross-linking on nuclear import of Mat-C
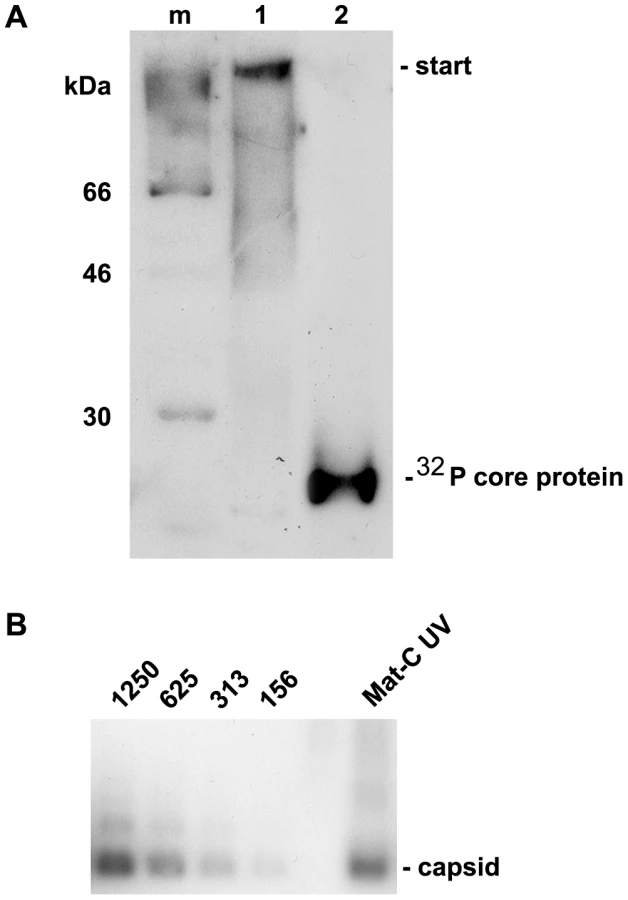
We next asked how Mat-C proteins could enter the nucleus despite the strong binding to Nup153. We followed the hypothesis that capsids should remain intact while genome maturation continues but should liberate the genome from the basket once rcDNA is formed. To test this hypothesis we cross-linked the Mat-C subunits, which were 32P-labelled by UV irradiation (Mat-C UV) and analyzed their integrity. Figure 5A shows that the cross-linking caused a strong reduced migration of the core protein subunits in SDS PAGE. Most of the protein was retained at the entry site of the SDS PAGE; only traces resulted in a smear >45 kDa. To test whether cross-linking occurred between capsids, which would have caused unsuitable particle aggregates we separated Mat-C UV on native agarose gel and detected them by anti-capsid antibody used before. We observed no difference in migration compared to the P-rC standard (Fig. 5B), demonstrating that UV irradiation only induced bonds between subunits of individual capsids. The result further shows that the UV treatment had not changed the surface charge.
We next investigated the transport competence of Mat-C UV in comparison to untreated Mat-C. We injected 1×107 capsids into the cytosolic periphery of 6 Xenopus laevis oocytes and followed the fate of the capsids by electron microscopy. We used Xenopus laevis oocytes sine more NPCs can be analyzed by EM in this system than by using permeabilized cell lines thus giving more reliable results. Combining these two systems and comparing the results is however justified since, as far as is known, nuclear import is identical in both systems [49],[50].
As control, six Xenopus laevis oocytes were injected with Mat-C. We restricted the incubation time after injection to 1 h, the earliest time point at which significant numbers of capsids arrive at the nuclear pore (not shown). Such a short time was chosen in order to detect possible differences in nuclear entry of the capsids. As shown in Figure 6A both types of capsids entered the nuclear basket as intact particles. We determined the number of capsids that arrived at 68 NPCs (Mat-C) and 74 NPCs (Mat-C UV), and determined their location at the NPCs. Figure 6B showed that a similar number of capsids arrived at the nuclear pore indicating that the cross-link neither affected the intracytoplasmic transport capacity nor the interaction with the NPCs. We next analyzed the distribution of the capsids, showing that both capsid species exhibited the same distribution at the NPCs with a majority on the cytoplasmic face. To our experience this dominantly cytoplasmic localization is related to the short incubation time. However, the same proportion of Mat-C and Mat-C UV entered the pore and were found in the nuclear basket, implying that their transport competence was the same.

We next analyzed whether Mat-C UV diffuses deeper into the nucleus like untreated Mat-C or remained arrested at the nuclear envelope like Immat-C and P-rC. These assays were performed with digitonin-permeabilized cells since the relatively low amounts of capsids would have been undetectable in Xenopus laevis oocytes. Capsids were detected by indirect immune fluorescence using the same anti-capsid antibody that showed an unchanged reactivity after cross link. Figure 6C shows that Mat-C entered the nucleus as it was previously reported for different permeabilized cells and in living cells [19],[23]. In contrast Mat-C UV failed to enter the karyoplasm and remained associated at the NPCs and some cytosolic structure. We assume that the cytosolic binding sites are collapsed microtubules as such binding was demonstrated before for digitonin-permeabilized cells [23].
Effect of Nup153 – capsid interaction on nuclear import of other cargos
Figures 2 and 3 showed that the capsids bound to a Nup153 domain that participates in importin β binding and that the interaction competes with importin β in vitro. To obtain in situ evidence on Nup153 that is integrated into the NPCs we analyzed whether binding of capsids to Nup153 interfere with the importin β-dependent nuclear import pathway. These experiments were performed with P-rC because the large numbers of capsids required for Nup153 saturation on the permeabilized cells were only available for this type of capsid. We used the human hepatoma cell line HuH-7 cells which is able to synthesize hepatitis B virions after transfection with HBV DNA.
We incubated digitonin-permeabilized cells with different amounts of P-rC. Nuclear import into the basket was facilitated by means of the nuclear transport receptors in rabbit reticulocyte lysate. After incubation cells were washed. In the last washing buffer no P-rC was detectable.
First we quantified the amount of NPCs and the number of bound capsids. NPC number was calculated from the Nup153 signal obtained by Western blot, which was compared to a dilution series of E. coli-expressed Nup153 (not shown). We determined ∼6000 NPCs per HuH-7 nucleus, which is higher than the ∼2770 NPCs found in HeLa cells [9]. It is however known that the NPC number varies strongly between different cell types (18,451+/−2,336 (Purkinje cells) to 402+/−67 (oligodendrocytes) [51].
We used P-rC preloaded digitonin-permeabilized cells after washing and added new cytosol together with two fluorescent cargos – Alexa594 NLS-BSA and Alexa647 M9-BSA. NLS-BSA is imported by importin β while M9-BSA uses transportin for nuclear entry. Both cargos comprise the same number of nuclear localization signals, as determined previously [18]. After import reaction, capsids and NPCs were stained by indirect immune fluorescence, and nuclear cargo concentrations were determined semi-quantitatively using confocal laser scan microscopy. Quantification was performed in the equatorial region of the nuclei. A positive control was performed on cells that were preincubated in the absence of capsids but to which the cargos were added in a second step and incubated at 37°C. In the negative control no P-rC was added during the first incubation but the following import reaction with the fluorescent cargos was performed on ice, thus inhibiting active nuclear transport [52].
Figure 7 shows no intranuclear fluorescence in the negative control (Fig. 7A) but strong signals for both cargos in the positive control (Fig. 7B). With increasing preload of the nuclei by capsids the import of both cargos became reduced (Fig. 7C–H). At the highest capsid concentration no intranuclear fluorescence could be observed (Fig. 7H). In this sample the average nuclear diameter increased from 200 µm2 in the negative control to 240 µm2. This observation is in accordance with a “plugging” of the NPCs by the capsids, which does not allow exchange of smaller molecules needed for equilibration of the osmotic pressure between nucleus and reaction mixture.

Figures 7B to G show further that the reduction of nuclear import appeared for NLS-BSA at lower preloaded numbers of capsids than for M9-BSA. As no unbound P-rC was present after washing, we can exclude competition between capsid NLS and NLS-BSA for the transport receptors importin α and β.
We next quantified the import in 570 nuclei. The mean intranuclear fluorescence in the positive control was taken as 100%. Figure 8 shows scatter plots obtained for each capsid preload. We observed a significant variation between the nuclei in one sample, for instance between 80% and 120% in the positive control. This can be explained by variable nuclear permeability throughout the cell cycle [53]. Moreover, this observation confirms that the nuclei remained intact during the transport reaction as a disruption would have resulted in equal concentrations. The import via transportin and via importin β was correlated in all cells and followed a Gaussian normal distribution. As indicated by the slopes of the regression lines no significant changes were observed at preloads from 0 to 0.2 capsids per NPC (Fig. 8B–D). With larger numbers of capsids, the importin-mediated import became greatly reduced (Fig. 8E; 0.7 P-rC/NPC) or undetectable (Fig. 8F; 3.3 P-rC/NPC), while the transportin-mediated pathway remained significantly more active (p≤10−9). Concentrations ≥3.3 capsids/NPC blocked both the NLS-and M9-mediated import, most likely the result of steric hindrance by capsids that got stuck in the channels of the NPCs. Collectively the data however indicate that blocking the capsid binding sites on Nup153 interferes with the importin β-mediated nuclear entry but not with the transportin pathway.

Discussion
Karyophilic cargos interact transiently with components of the NPC via transport receptors before they are released to the karyoplasm. HBV capsids are an exception as they remain arrested in the nuclear basket. We showed that HBV capsids bound solubilized and renatured rat Nup153 from a nuclear extract with importin binding activity. This is consistent with the high degree of conservation of Nups among different species and the conserved mechanisms of nuclear translocation [54]. It confirms further the observation that Immat-C is arrested at the NPCs of different cell lines [19].
Neither co-purification of Nup98 or Nup160, which connect Nup153 with the central frame work of the NPC [55] nor of Tpr (268 kDa), which is attached to the NPC via Nup153 [7], was observed. This finding indicates that the NPC components became disassembled during purification and did not reassemble upon renaturation. This interpretation is in accordance with previous findings that NPC reconstitution depends on the presence of RanGTP [56]. RanGTP has a low molecular weight of 25 kDa and is removed from the nuclei prior to disintegration of the NPCs. The absence of a 97 kDa band in the co-precipitation further confirms that the binding was not mediated by importin β, which is in accordance with the direct binding observed in co-precipitation of natively purified GST-Nup153. The latter finding provides further evidence that the binding did not require a protein linker, which may have been below the 75 kDa limit of the Sypro Red stained gel.
HBV capsids undergo complex modifications upon genome maturation which are not well understood. Their changes comprise not only the reverse transcription of the encapsidated RNA to rcDNA but also protein phosphorylation [31],[32] and eventually dephosphorylation. We found that Nup153 was precipitated by Immat-C, containing replication intermediates and possibly empty capsids, and by Mat-C containing rcDNA. This observation suggests that the type of nucleic acid within the capsid has no impact on Nup153 interaction. The observation that E. coli-expressed RNA containing capsids that were phosphorylated in vitro by protein kinase C and empty E. coli-expressed capsids that lacked the RNA-binding and phosphorylated C-terminal domain interacted equally with Nup153 shows that the binding is mediated by the N-terminal assembly domain. It further confirms that importin β, which interacts via importin α with the NLS on the C-terminal domain [18] do not interfer with capsid binding to Nup153.
Searching for the domain on Nup153, which interacts with the capsids we observed that aa 53–543 of Nup153 did not show any interaction. This part of Nup153 comprises a transportin binding site (aa 247–290 [57]). In contrast aa 618–999 and 992–1219 – both comprising FxFG repeats - interacted with the capsids. As other FxFG repeat comprising Nups were not co-precipitated we conclude that the binding is specific for Nup153 and not based on hydrophobic interactions.
To obtain evidence of overlap between importin β and capsid binding sites we performed competition experiments in the presence of excesses of importin β. The strong binding with regard to importin β is consistent with an attachment of the capsids to the FXFG repeats as these sites interact with this import receptor. This strong affinity could explain the mechanism by which the capsids can remain arrested on Nup153 within the cells despite of the 1000 exchange reactions that pass each nuclear pore per second [4].
Partial silencing Nup153 showed that a significant proportion of P-rC entered the nucleus. The incomplete entry is consistent with the high affinity of the capsids to Nup153, assuming that less than 8 copies of Nup153 are sufficient for capsid arrest. Considering that Nup153-linked Tpr was not precipitated the result thus confirms that no other nucleoporin has a significant effect on the abortion of the capsid entry into the nucleus.
Our results showed additionally that the structural part of the capsid caused the interaction, which has a well ordered structure. Within 16 Å resolution no differences could be found between E. coli-expressed capsids, Mat-C and Immat-C [28] although one group reported a hydrophobic pocket on the capsid surface that exists on Mat-C only [58]. We considered structural causes to be an unlikely explanation for the different entry behavior and followed the hypothesis that Immat-C is more stable than Mat-C. Consequently, we assumed that maturation-dependent disintegration of Mat-C leads to capsid subunits which may diffuse from the NPC to the karyoplasm. It was recently shown that capsids may disintegrate to core protein dimers [59]. Irrespective of the T = 3 or T = 4 symmetry of the capsids, the core protein dimers would exceed the number of Nup153 molecules per NPC by a factor of >10. Supernumerous subunits could diffuse into the karyoplasm where they reassemble [59]. Our nuclear import assays with cross linked capsids confirmed this hypothesis showing that disassembly is a prerequisite for intranuclear capsid formation. These data are also consistent with recent observations that nuclear entry of capsid and genome is independent upon RanGTP [19], as a dissociation of the import receptors is not required.
In order to analyze the functional effects of capsid binding to Nup153 in situ we measured nuclear import via the importin β pathway. Our observation that the NLS-BSA import was impaired to a much greater degree than M9-BSA translocation provides evidence for the concept that transport inhibition was specific by blocking importin β binding sites and was not the result of steric interference. The strong inhibition of the importin β pathway is in accordance with observations of Walther et al. [43] who reported that Nup153 is essential for nuclear entry via importin β but not via transportin. In fact our approach of inhibiting Nup153 function with capsids allows for the first time the confirmation of these data without interfering with the NPC architecture, i.e. Nup50 translocation or Tpr depletion [5],[7].
Based on our observations we propose a model of nuclear entry for the HBV genome, which strikingly differs from that for any other virus. According to previous data we postulate that the viral genome is transported within the capsid via microtubules to the nuclear periphery [23]. This assumption does not exclude that the capsids undergo a limited disintegration and re-association of some capsid subunits. Such capsid “breathing” is in accordance with in vitro findings [60],[61] and can explain the observation of polymerase-free hepadnaviral DNA genomes [62], which are nonetheless precipitable by anti capsid antibodies [63]. After binding to importin α/β the capsids are transported into the nuclear basket (Fig. 9). This transport requires the DNA minus strand synthesis which is linked to NLS exposure by some core subunits [19]. The import complex binds to Nup153 via importin β, followed by importin α and importin β dissociation, which is mediated by RanGTP. Due to their affinity to Nup153 capsids interact directly with Nup153. While Mat-C disintegrates, Immat-C remain in the arrested state. In vivo these arrested capsids should not interfere with the viability of the hepatocytes [64] as thousands of capsids would be necessary to significantly block the nuclear pores. In fact, only high level expression of capsids in cell lines causes toxic effects [65]. It is plausible to assume that genome maturation can proceed in Immat-C but experimental evidence is difficult to obtain as genome maturation is slow. At the beginning of infection, when significant amounts of surface proteins have not yet been synthesized, ongoing genome maturation after arrest prevents premature release of the genome into the nucleus. Due to the dependence of the second strand DNA synthesis on the environment within the capsids [66], premature release of minus strand DNA would lead to a loss of viable virus. Thus, the maturation dependent arrest of HBV capsids is probably an essential step in the life cycle of the virus.

Materials and Methods
Preparation of nuclear proteins
Nuclear proteins were prepared from rat liver nuclei using urea [67] followed by protein refolding during dialysis. Eight gram of rat liver were minced in 16 ml of ice-cold 250 mM sucrose in TKM buffer (50 mM Tris-HCl, pH 7.5, 25 mM KCl, 5 mM MgCl2, protease inhibitor mix (complete) (Roche)). All subsequent preparations were performed on ice. The tissue was homogenized using a motor-driven Teflon pestle with 10 strokes at 1700 rpm. The homogenate was filtered through two layers of a filter (59 µm mesh). Three ml of homogenate were mixed with 6 ml 2.3 M sucrose/TKM buffer and loaded on a 1 ml cushion of 2.3 M sucrose/TKM buffer. After centrifugation for 30 minutes at 124,000×g at 4°C, the pellet was resuspended in 200 µl TKM buffer. The resuspended nuclei were treated with 4 ml 8 M urea for 1 h at 65°C and subjected to dialysis at 4°C against 1.5 l 1×PBS/2 mM DTT) for 48 h. During the dialysis the buffer was changed four times. The protein concentration was determined by bicinchonic acid (BCA) assay. The nuclear proteins were frozen in liquid nitrogen and stored in aliquots at −80°C. This lysate did not contain detectable amounts of importin β, presumably excluding interference of importin β with capsid binding (not shown).
Expression and purification of GST-Nup153 and Nup153 fragments
GST-Nup153 was expressed using the vector pGEX 2T-hNup153, which encodes for human wt Nup153 [43]. Expression was performed in E. coli BL21 CodonPlus RIPL (STRATAGENE). Bacteria were grown at 37°C in 2×YT-medium/100 µg/ml ampicillin/30 µg/ml chloramphenicol/ 0.2% glucose to an OD600 of 1.0 and cooled to 18°C prior to induction of the expression by 1 mM IPTG and incubation for 16 h at 18°C. PMSF was added to 2 mM and the bacteria were chilled for 15 min at 4°C followed by sedimentation at 4000 g for 15 min at 4°C. The pellet was resuspended in PBS/1 mM DTT/1×EDTA-free complete protease inhibitors (Roche Applied science). Bacteria were lysed by sonification on ice. The lysate was cleared 30 min 15000×g at 4°C. One milliliter equilibrated GST-Sepharose High Performance (GE-Healthcare) was added to the supernatant and incubated for 1 h at RT followed by o.n. incubation at 4°C. The solution was transferred to a polypropylene column (BIO-RAD). The matrix containing the GST-Nup153 was washed with 30 ml binding-/washing buffer. GST-Nup153 was eluted using 5 ml 20 mM Glutathione/200 mM NaCl/50 mM Tris (pH 7.5)/1 mM DTT/1×EDTA-free complete protease inhibitors. The eluate was dialyzed against 200 mM NaCl/50 mM Tris-HCl pH 7.5/50% Glycerol/250 mM Sucrose/1 mM DTT for 24 h at 4°C. The buffer was changed three times. GST-Nup153 was stored in aliquots at −80°C.
The N terminal human Nup153 fragment was expressed using the vector pGEX-153N (53–272), the zinc finger domain using the vector pGEX-153Z (272–543) and the N terminal half of the C terminus using the vector pGEX-153C1 (618–999). Expression was performed using E. coli BL21 (DE3)Lys using the protocol of [68] Bacteria were grown in NZy (rich) medium and expression was performed for 3 h at 37°C. The sedimented bacteria were lysed as described above. Purification was performed as described above.
The C terminal half of the C terminus (aa 992–1219) was expressed as a His-tagged protein using the vector pET28-153C2 [68]. The His-tagged fragment was expressed by using E. coli BL21 (DE3)Lys in LB/Kanamycin medium. Bacteria were lysed as described above. The precleared lysate was purified by Ni++ agarose (Quiagen) using the protocol of the vendor. After elusion, the His-tagged Nup fragment was renatured by dialysis (1 h against 1 M urea, 0.2 M NaCl, 0.1 M NaH2PO4 pH 7.6, 0.5 mM PMSF, 1 h dialysis against 0.2 M urea, 0.2 M NaCl, 0.1 M NaH2PO4 pH 7.6, 0.5 mM PMSF, 1 h dialysis against 0.2 M NaCl, 0.1 M NaH2PO4 pH 7.6, 0.5 mM PMSF and 1 h against 0.2 M NaCl, 0.1 M NaH2PO4 pH 7.6).
Preparation of capsids
P-rC was generated by in vitro phosphorylation of E. coli-derived capsids using protein kinase C and [γ32P] ATP to determine the success of the reaction [69]. E. coli-derived capsids were expressed and prepared as described previously [20]. Quantification of capsids was done by immune blotting of the capsids after native agarose gel electrophoresis [18]. Preparation of Mat-C from virions from the permanently virion-expressing hepatoma cell line HepG2.2.15 [70] was done according to Rabe et al. [19]. This cell line expresses infectious HBV [70]. Immat-C was purified from the same cell line using the following protocol. Ten 16 cm dishes of HepG2.2.15 cells were grown in DMEM medium, containing 10% FCS until 80% confluence. The medium was replaced by DMEM/1% FCS and cells were grown for an additional 4 days. Cells were washed twice in PBS, harvested by a rubber policeman and sedimented for 5 min at 200×g and 4°C. Cells were resuspended in 0.1% Nonidet P-40/PBS and sonified on ice. Cellular nucleic acids were digested by addition of 20 U/ml DNase I/20 µg/ml RNase A in 15 mM MgCl2 for 15 min at 37°C. This short incubation was chosen as capsid instability may allow entry of nucleases upon longer incubation periods [60],[61]. The lysate was centrifuged for 20 min at 10,000×g. The supernatant was adjusted to 0.75% Nonidet P-40/5 mM CaCl2/20 U/ml S7-Nuklease and incubated for further 15 min at 37°C before EDTA was added to 15 mM. The lysate was centrifuged for 10 min at 4°C and 18,000 g. The capsids in the supernatant were loaded on a 1 ml 25% (w/v) sucrose cushion and sedimented for 2 h at 10°C and 50,000 rpm in an SW60 rotor (Beckman). The sediment was resuspended in 500 µl PBS, centrifuged 5 min at 4°C and 12,500×g. The capsid-containing supernatant was adjusted to 2 mM DTT in order to prevent disulfide bond formation and stored in aliquots at −80°C.
UV-cross linking of mature HBV capsids (Mat-C UV)
Mat-C were labeled by [γ32P] ATP as described elsewhere [69]. Labeled capsids were exposed 6×8 min to 254 nm UV light on ice (Stratagene UV Stratalinker; 1 cm distance to the UV source, ∼4000 µW/cm2). Samples withdrawn before and after cross-linking were analyzed by SDS-PAGE with a subsequent exposure to a phosphoimaging screen. Evaluation of a possible inter-capsid cross-link was done by native agarose gel electrophoresis and subsequent immune detection [18].
Immune precipitation and detection of co-precipitated proteins
For immune precipitation 1.2×107 anti-rabbit antibody-coated biomagnetic beads (Invitrogen) were added to 185 µg anti-HBV capsid antibody (DAKO). The volume was adjusted to 1000 µl by addition of 0.1% BSA/PBS and incubated overnight at 4°C on a rotating wheel. Unbound antibodies were removed by washing the beads three times in 0.1% BSA/PBS. For inverse immune precipitation, 12.5 µg mouse monoclonal antibody 414 (mAb414, HISS) were bound to 1.2×107 anti-mouse antibody-coated biomagnetic beads as described for the anti-capsid antibody.
For co-immune precipitations of the nucleoporins 100 ng of HBV capsids were preincubated with 75 µg nuclear proteins for 2 h at 37°C in transport buffer (20 mM Hepes, pH 7.3, 2 mM MgAc, 110 mM KAc, 5 mM NaAc, 1 mM EGTA, 2 mM DTT, protease inhibitor mix (complete) (Roche)) in a final volume of 250 µl. The precipitation mixture was incubated with constant shaking at 37°C overnight. Afterwards the beads were washed three times in 500 µl 0.1% BSA/PBS, 1×500 µl 0.1% Nonidet P-40/PBS, and transferred into a new cup. After three additional washing steps with 500 µl PBS, the pellet was resuspended in 20 µl 1× loading buffer (Anamed), denatured for 10 min according to the vendor and loaded on the SDS gels. (Invitrogen and Anamed). For detection of total proteins that were co-precipitated Sypro Red staining was performed according the manufacturer.
For immune detection the proteins on the gel were transferred to a PVDF membrane (VWR International) by electroblotting. The membrane was blocked for 1 h at RT in 5% fat-free milk powder in PBS. For detection of co-precipitated nucleoporins the first antibody (mAb414, HISS) was added at a dilution of 1∶3000 in 5% fat-free milk/PBS for 3 h at RT. After washing three times in 0.1% Tween-20/0.5% milk/PBS the membrane was incubated for 1 h with a horse radish peroxidase anti-mouse antibody (0.16 µg/ml; Dianova). Detection was performed by ECL (PerkinElmer).
Co-immune precipitations of Nup153 fragments and gel retardation assay
5 ng P-rC and 75 µg Nup153 fragments were incubated in transport buffer (20 mM HEPES pH 7.3/2 mM Mg acetate/5 mM Na acetate/110 mM K acetate/1 mM EGTA/2 mM DTT/ protease inhibitor [complete, Roche]) for 2 h at 37°C. 1.2×107 anti-GST antibody (MoBiTec)-coated biomagnetic beads (Dynal) were added and incubated over night at 4°C.
The beads were washed 3× with transport buffer including one change of the cups. The beads were incubated for 1 min in PBS/0.1% NP-40, sedimented and washed 4× with PBS. The precipitated proteins were then separated on SDS PAGE and blotted to a PVDF membrane, which was saturated using 5% (w/v) milk powder/1×PBS prior to the addition of the first antibody (anti-GST antibodies, 1∶500, 2 h, RT). The membrane was washed 3×10 min in 1×PBS/0.1% Tween 20/0.5% milk powder and incubated with a peroxidase-labeled anti-rabbit antibody (1∶5000, 1 h, RT). The membrane was washed as described above, followed by an additional 15 min incubation in 1×PBS at RT prior to the visualization by film using ECL.
In gel retardation assays the incubation mixture of capsids and Nup153 fragments were separated on 0.7% agarose/TAE gels and blotted onto PVDF membranes by capillary blotting [71]. Nup-fragments were detected by anti-GST antibodies or and anti-His antibodies (dilution 1∶3000) as described above, HBV capsids were detected by anti-capsid antibodies (DAKO, 1∶10,000).
Competition assays
Two hundred ng P-rC were incubated with 200 ng of GST-Nup153 and different concentrations of importin β for 2 h at 37°C in transport buffer. At the lowest concentration of importin β the ratio was 1 capsid : 28 GST-Nup153 : 33 importin β molecules. The capsids were precipitated by the addition of 1.2×107 anti capsid antibody-saturated biomagnetic beads o.n. at 37°C. The beads were washed 3× with 0.1% BSA/PBS and once with 0.1% Nonidet P-40/PBS. The beads were transferred to a new cup and washed again 3× with PBS before the proteins were separated on a 3–8% SDS PAGE (Tris acetate gel). Co-precipitated GST-Nup153 was detected by Western blot using mAb414 as described above.
Alternatively 200 ng of capsids were bound to 1.2×107 anti capsid antibody-saturated biomagnetic beads o.n. at 37°C followed by an incubation with 200 ng GST-Nup153 in transport buffer for 2 h at 37°C. After washing, varying amounts of importin β were added for 2 h at 37°C. The beads were then washed as described above and subjected to SDS PAGE and immune blot using mAb414.
Microinjection into Xenopus laevis oocytes and electron microscopy
The cytosolic microinjection, preparation of the nuclei and electron microscopy are described elsewhere [72]. Six Xenopus laevis oocytes were used per sample. Per oocyte 1×107 Mat-C or Mat-C UV were microinjected. P-rC, available in much higher concentrations than Mat-C, was microinjected in 2×108 particles per oocyte. For quantification the number of NPC with and without capsids as well as their location at the pore were recorded in 50 nm sections.
Nup153 silencing
HeLa cells were transfected with small interfering RNA (siRNA) (Dharmacon) against Nup153 at a final concentration of 10 nM using lipofectamine RNAiMAX (Invitrogen) according to the manufacture's instructions. The sequence used corresponded to nucleotide 2593–2615 of human Nup153 (AAGGCAGACUCUACCAAAUGUTT), which has been previously shown to accomplish efficient knock down of Nup153 in HeLa cells [10]. Expression of Nup153 was assessed by labeling Nup153 with a monoclonal antibody, SA1 [73]. Cells were analyzed by Western blot and used to assay nuclear import of P-rC two days post transfection. For control, mock transfection of cells without siRNA was performed. Visualization was performed using enhanced chemoluminescence and autoradiography films at different exposure times. The siRNA transfected cells were then used for transport assays after digitonin-permeabilization.
In vitro transport and binding assays
To determine the nuclear import capacity, 25 ng of Mat-C or Mat-C UV were subjected to digitonin-permeabilized HuH-7 cells that were grown on 12 mm collagen-coated cover slips. Growth, permeabilization of the cells and indirect immune stain was described previously [19]. To allow comparison of the results the antibodies were the same as those used for the co-immune precipitations (anti-capsid [DAKO], mAb414). As secondary antibodies an Alexa488-labelled goat-anti-rabbit antibody (Invitrogen) and a Texas Red-labeled goat-anti-mouse antibody were added (Dianova). Microscopy was performed on a Leica DM IRBE confocal laser scan microscope using the filter settings for FITC, TRITC and Cy5 at a pinhole size of 1.
To study the effect of Nup153 capsid-interaction on the nuclear import of other substrates an in vitro transport assay was performed using a modified protocol of Kann et al. [18] In a first step after permeabilization and washing the NPCs were loaded with a geometrical dilution of P-rC (0, 25–12800 ng per 12 mm cover slip) for 30 min at 37°C in the presence of RRL, ATP and an ATP-generating system. Afterwards the cover slips were washed 3 times with 500 µl transport buffer/1×EDTA-free complete protease inhibitors/2 mM DTT to remove unbound capsids. In the last wash less than 3 pg of unbound P-rC were present (<300 pg per 0.5 ml washing solution of the last washing step, ∼5 µl remaining fluid estimated).
The conjugates (50 µg/ml) to be analyzed were added in a second transport reaction with new RRL, ATP and an ATP-generating system for 15 min at 37°C. For analysis of importin-mediated transport we used Alexa594-labelled BSA (Invitrogen) linked to the peptide PKKKRKVED that represents the NLS of the SV40T-Ag [74]. The import by transportin was analyzed using Alexa647-labelled BSA conjugated to the M9-domain of hnRNP A1 protein (YNNQSSNFGPMK) [75]. Both peptides were linked via a CGGG spacer to the BSA. Analysis of the conjugates by MALDI-TOF MS revealed a comparable conjugation of an average of 19 peptides per BSA molecule. Non-imported conjugates were removed by 3 washes in 500 µl transport buffer. Fixation, blocking reaction and immune stains of NPCs and capsids were done as described previously [18] using the mAB414 antibody or the anti-capsid antibody respectively. As second antibodies we used Alexa532-anti-rabbit and Alexa568-anti-mouse antibodies. Confocal laser scan microscopy was done using the TCS SP5 microscope (Leica) equipped with a HCX PLAPO 63×/1.4-0,6 Oil objective (Leica). Images were taken at a zoom of 2 with a size depicting ∼70 µm pinhole size.
Quantification of nuclear import
The intranuclear area, flanked by the rim-like stain of the NPCs or P-rC stain was selected and the fluorescence signals of the imported cargos were determined (software LAS AF version 2.1.0). Calculations were done using Microsoft Excel. Although showing a large linear range absolute fluorophore numbers can be hardly obtained by fluorescence microscopy as the signal depends upon e.g. recording efficiency, filter setting, lens and amplification of the individual dye. To normalize the signals mean values of the positive import reactions were set as 100%. The mean values of nuclei recorded in the negative control at 4°C were set as 0% being only marginally above the background outside the permeabilized cells. Since the evaluated parameters (total nuclear import, concentration and cell size) showed a Gaussian normal distribution within each sample a comparison of the signals was done by Students' T-test.
Quantification of nuclear binding of P-rC
HuH-7 cells (∼1×106 cells) from confluent grown 10 cm dish were treated with trypsin, removed by pipetting in 15 ml D-MEM and sedimented at 4°C for 10 min at 200×g. The cells were resuspended in D-MEM and sedimented as described above. This step was repeated before the cells were resuspended in 5.5 ml D-MEM/40 µg/ml digitonin. After 5 min at 37°C permeabilization was controlled by microscopy of an aliquot. Cells were sedimented in aliquots that reflect the number of cells per cover slip in transport assays and resuspended in 0.5 ml transport buffer/2 mM DTT/PI. This sedimentation and resuspension was repeated twice. The permeabilized cells were resuspended in the transport mixture identical to that in the transport assays using cover slips containing different amounts of P-rC and incubated for 30 min at 37°C. After three washing steps by sedimentation and resuspension in transport buffer/2 mM DTT/1×EDTA-free complete protease inhibitors as above the permeabilized cells in an aliquot were counted. The amount of P-rC was determined as described above by native agarose gel electrophoresis and immune blotting.
Quantification of NPCs
NPCs were quantified by comparing the amount of cellular Nup153 from digitonin-permeabilized cells with a standard dilution series of E. coli-expressed Nup153. Cells were grown and harvested and permeabilized as described for “Quantification of nuclear binding of P-rC”. Aliquots containing the same number of cells were denaturated and loaded on an 8% Tris-Glycin SDS PAGE (Anamed). Proteins were transferred onto a PVDF membrane and Nup153 was detected by mAb414 as described above.
Supporting Information
Zdroje
1. PanteN
KannM
2002 Nuclear Pore Complex Is Able to Transport Macromolecules with Diameters of ∼39 nm. Mol Biol Cell 13 425 434
2. LimRY
AebiU
StofflerD
2006 From the trap to the basket: getting to the bottom of the nuclear pore complex. Chromosoma 115 15 26
3. TerryLJ
ShowsEB
WenteSR
2007 Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 318 1412 1416
4. TranEJ
WenteSR
2006 Dynamic nuclear pore complexes: life on the edge. Cell 125 1041 1053
5. KrullS
ThybergJ
BjorkrothB
RackwitzHR
CordesVC
2004 Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell 15 4261 4277
6. BallJR
UllmanKS
2005 Versatility at the nuclear pore complex: lessons learned from the nucleoporin Nup153. Chromosoma 114 319 330
7. HaseME
CordesVC
2003 Direct interaction with nup153 mediates binding of Tpr to the periphery of the nuclear pore complex. Mol Biol Cell 14 1923 1940
8. MatsuuraY
StewartM
2005 Nup50/Npap60 function in nuclear protein import complex disassembly and importin recycling. Embo J 24 3681 3689
9. RibbeckK
GorlichD
2001 Kinetic analysis of translocation through nuclear pore complexes. Embo J 20 1320 1330
10. HarborthJ
ElbashirSM
BechertK
TuschlT
WeberK
2001 Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114 4557 4565
11. WhittakerGR
KannM
HeleniusA
2000 Viral Entry into the Nucleus. Annu Rev Cell Dev Biol 16 627 657
12. WoodwardCL
PrakobwanakitS
MosessianS
ChowSA
2009 Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J Virol 83 6522 6533
13. WodrichH
CassanyA
D'AngeloMA
GuanT
NemerowG
2006 Adenovirus core protein pVII is translocated into the nucleus by multiple import receptor pathways. J Virol 80 9608 9618
14. HindleyCE
LawrenceFJ
MatthewsDA
2007 A role for transportin in the nuclear import of adenovirus core proteins and DNA. Traffic 8 1313 1322
15. GreberUF
SuomalainenM
StidwillRP
BouckeK
EbersoldMW
1997 The role of the nuclear pore complex in adenovirus DNA entry. EMBO J 16 5998 6007
16. TrotmanLC
MosbergerN
FornerodM
StidwillRP
GreberUF
2001 Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat Cell Biol 3 1092 1100
17. OjalaPM
SodeikB
EbersoldMW
KutayU
HeleniusA
2000 Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20 4922 4931
18. KannM
SodeikB
VlachouA
GerlichWH
HeleniusA
1999 Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol 145 45 55
19. RabeB
VlachouA
PanteN
HeleniusA
KannM
2003 Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A 100 9849 9854
20. CrowtherRA
KiselevNA
BottcherB
BerrimanJA
BorisovaGP
1994 Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 77 943 950
21. GlebeD
UrbanS
2007 Viral and cellular determinants involved in hepadnaviral entry. World J Gastroenterol 13 22 38
22. ChojnackiJ
GrgacicEV
2008 Enveloped viral fusion: insights into the fusion of hepatitis B viruses. Future Virology 3 543 552
23. RabeB
GlebeD
KannM
2006 Lipid-mediated entry of hepatitis B virus capsids in non-susceptible cells allows highly efficient replication and the analysis of the early infection events. J Virol 80 5465 5473
24. van LooND
FortunatiE
EhlertE
RabelinkM
GrosveldF
2001 Baculovirus infection of nondividing mammalian cells: mechanisms of entry and nuclear transport of capsids. J Virol 75 961 970
25. AsabeS
WielandSF
ChattopadhyayPK
RoedererM
EngleRE
2009 The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol 83 9652 9662
26. KannM
GerlichWH
2005 Structure and Molecular Virology.
ZuckermanAJ
ThomasHC
Viral Hepatitis Edinburgh, London, Madrid, Melbourne, New York, Tokyo Churchill Livingston 149 180
27. GerelsaikhanT
TavisJE
BrussV
1996 Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol 70 4269 4274
28. DrydenKA
WielandSF
Whitten-BauerC
GerinJL
ChisariFV
2006 Native hepatitis B virions and capsids visualized by electron cryomicroscopy. Mol Cell 22 843 850
29. WattsNR
ConwayJF
ChengN
StahlSJ
BelnapDM
2002 The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. Embo J 21 876 884
30. ZlotnickA
ChengN
StahlSJ
ConwayJF
StevenAC
1997 Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc Natl Acad Sci U S A 94 9556 9561
31. GazinaEV
FieldingJE
LinB
AndersonDA
2000 Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74 4721 4728
32. MelegariM
WolfSK
SchneiderRJ
2005 Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol 79 9810 9820
33. SummersJ
SmithPM
HorwichAL
1990 Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64 2819 2824
34. BourneEJ
DienstagJL
LopezVA
SanderTJ
LongletJM
2007 Quantitative analysis of HBV cccDNA from clinical specimens: correlation with clinical and virological response during antiviral therapy. J Viral Hepat 14 55 63
35. AdamSA
MarrRS
GeraceL
1990 Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biol 111 807 816
36. KöserJ
MacoB
AebiU
FahrenkrogB
2005 The nuclear pore complex becomes alive: new insights into its dynamics and involvement in different cellular processes. Atlas Genet Cytogenet Oncol Haematol
37. Ben-EfraimI
GeraceL
2001 Gradient of increasing affinity of importin beta for nucleoporins along the pathway of nuclear import. J Cell Biol 152 411 417
38. DavisLI
BlobelG
1987 Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci U S A 84 7552 7556
39. DavisLI
BlobelG
1986 Identification and characterization of a nuclear pore complex protein. Cell 45 699 709
40. GörlichD
VogelF
MillsAD
HartmannE
LaskeyRA
1995 Distinct functions for the two importin subunits in nuclear protein import. Nature 377 246 248
41. SukegawaJ
BlobelG
1993 A nuclear pore complex protein that contains zinc finger motifs, binds DNA, and faces the nucleoplasm. Cell 72 29 38
42. PanteN
BastosR
McMorrowI
BurkeB
AebiU
1994 Interactions and three-dimensional localization of a group of nuclear pore complex proteins. J Cell Biol 126 603 617
43. WaltherTC
FornerodM
PickersgillH
GoldbergM
AllenTD
2001 The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. Embo J 20 5703 5714
44. ShahS
ForbesDJ
1998 Separate nuclear import pathways converge on the nucleoporin Nup153 and can be dissected with dominant-negative inhibitors. Curr Biol 8 1376 1386
45. MoroianuJ
HijikataM
BlobelG
RaduA
1995 Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc Natl Acad Sci U S A 92 6532 6536
46. DenningDP
PatelSS
UverskyV
FinkAL
RexachM
2003 Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc Natl Acad Sci U S A 100 2450 2455
47. LeungAK
Trinkle-MulcahyL
LamYW
AndersenJS
MannM
2006 NOPdb: Nucleolar Proteome Database. Nucleic Acids Res 34 D218 220
48. MackayDR
ElgortSW
UllmanKS
2009 The nucleoporin Nup153 has separable roles in both early mitotic progression and the resolution of mitosis. Mol Biol Cell 20 1652 1660
49. BaptesteE
CharleboisRL
MacLeodD
BrochierC
2005 The two tempos of nuclear pore complex evolution: highly adapting proteins in an ancient frozen structure. Genome Biol 6 R85
50. DeGrasseJA
DuBoisKN
DevosD
SiegelTN
SaliA
2009 Evidence for a shared nuclear pore complex architecture that is conserved from the last common eukaryotic ancestor. Mol Cell Proteomics 8 2119 2130
51. Garcia-SeguraLM
LafargaM
BercianoMT
HernandezP
AndresMA
1989 Distribution of nuclear pores and chromatin organization in neurons and glial cells of the rat cerebellar cortex. J Comp Neurol 290 440 450
52. AdamEJ
AdamSA
1994 Identification of cytosolic factors required for nuclear location sequence-mediated binding to the nuclear envelope. J Cell Biol 125 547 555
53. MillerM
ParkMK
HanoverJA
1991 Nuclear pore complex: structure, function, and regulation. Physiol Rev 71 909 949
54. FeldherrC
AkinD
LittlewoodT
StewartM
2002 The molecular mechanism of translocation through the nuclear pore complex is highly conserved. J Cell Sci 115 2997 3005
55. VasuS
ShahS
OrjaloA
ParkM
FischerWH
2001 Novel vertebrate nucleoporins Nup133 and Nup160 play a role in mRNA export. J Cell Biol 155 339 354
56. D'AngeloMA
AndersonDJ
RichardE
HetzerMW
2006 Nuclear pores form de novo from both sides of the nuclear envelope. Science 312 440 443
57. NakielnyS
ShaikhS
BurkeB
DreyfussG
1999 Nup153 is an M9-containing mobile nucleoporin with a novel Ran-binding domain. Embo J 18 1982 1995
58. RosemanAM
BerrimanJA
WynneSA
ButlerPJ
CrowtherRA
2005 A structural model for maturation of the hepatitis B virus core. Proc Natl Acad Sci U S A 102 15821 15826
59. RabeB
DelaleauM
BischofA
FossM
SominskayaI
2009 Nuclear entry of hepatitis B virus capsids involve disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog 5 e1000563 doi:10.1371/journal.ppat.1000563
60. CeresP
ZlotnickA
2002 Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 41 11525 11531
61. RabeB
DelaleauM
BischofA
FossM
SominskayaI
2009 Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog 5 e1000563 doi:10.1371/journal.ppat.1000563
62. GaoW
HuJ
2007 Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81 6164 6174
63. GuoH
JiangD
ZhouT
CuconatiA
BlockTM
2007 Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81 12472 12484
64. MoriyamaT
GuilhotS
KlopchinK
MossB
PinkertCA
1990 Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science 248 361 364
65. YoakumGH
KorbaBE
LechnerJF
TokiwaT
GazdarAF
1983 High-frequency transfection and cytopathology of the hepatitis B virus core antigen gene in human cells. Science 222 385 389
66. NassalM
1992 The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol 66 4107 4116
67. BlobelG
PotterVR
1966 Nuclei from rat liver: isolation method that combines purity with high yield. Science 154 1662 1665
68. FahrenkrogB
MacoB
FagerAM
KoserJ
SauderU
2002 Domain-specific antibodies reveal multiple-site topology of Nup153 within the nuclear pore complex. J Struct Biol 140 254 267
69. KannM
GerlichWH
1994 Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol 68 7993 8000
70. SellsMA
ZelentAZ
ShvartsmanM
AcsG
1988 Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J Virol 62 2836 2844
71. SouthernEM
1975 Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98 503 517
72. PantéN
2006 Use of intact Xenopus oocytes in nucleocytoplasmic transport studies.
LiuJ
Humana Press Inc 301 314
73. BodoorK
ShaikhS
SalinaD
RaharjoWH
BastosR
1999 Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J Cell Sci 112(Pt 13) 2253 2264
74. LanfordRE
ButelJS
1984 Construction and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell 37 801 813
75. GörlichD
PrehnS
LaskeyRA
HartmannE
1994 Isolation of a protein that is essential for the first step of nuclear protein import. Cell 79 767 778
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils
- CD8+ T Cell Control of HIV—A Known Unknown
- Polyoma Virus-Induced Osteosarcomas in Inbred Strains of Mice: Host Determinants of Metastasis
- The Deadly Chytrid Fungus: A Story of an Emerging Pathogen