
Placental Syncytiotrophoblast Constitutes a Major Barrier to Vertical Transmission of
Listeria monocytogenes is an important cause of maternal-fetal infections and serves as a model organism to study these important but poorly understood events. L. monocytogenes can infect non-phagocytic cells by two means: direct invasion and cell-to-cell spread. The relative contribution of each method to placental infection is controversial, as is the anatomical site of invasion. Here, we report for the first time the use of first trimester placental organ cultures to quantitatively analyze L. monocytogenes infection of the human placenta. Contrary to previous reports, we found that the syncytiotrophoblast, which constitutes most of the placental surface and is bathed in maternal blood, was highly resistant to L. monocytogenes infection by either internalin-mediated invasion or cell-to-cell spread. Instead, extravillous cytotrophoblasts—which anchor the placenta in the decidua (uterine lining) and abundantly express E-cadherin—served as the primary portal of entry for L. monocytogenes from both extracellular and intracellular compartments. Subsequent bacterial dissemination to the villous stroma, where fetal capillaries are found, was hampered by further cellular and histological barriers. Our study suggests the placenta has evolved multiple mechanisms to resist pathogen infection, especially from maternal blood. These findings provide a novel explanation why almost all placental pathogens have intracellular life cycles: they may need maternal cells to reach the decidua and infect the placenta.
Published in the journal:
. PLoS Pathog 6(1): e32767. doi:10.1371/journal.ppat.1000732
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000732
Summary
Listeria monocytogenes is an important cause of maternal-fetal infections and serves as a model organism to study these important but poorly understood events. L. monocytogenes can infect non-phagocytic cells by two means: direct invasion and cell-to-cell spread. The relative contribution of each method to placental infection is controversial, as is the anatomical site of invasion. Here, we report for the first time the use of first trimester placental organ cultures to quantitatively analyze L. monocytogenes infection of the human placenta. Contrary to previous reports, we found that the syncytiotrophoblast, which constitutes most of the placental surface and is bathed in maternal blood, was highly resistant to L. monocytogenes infection by either internalin-mediated invasion or cell-to-cell spread. Instead, extravillous cytotrophoblasts—which anchor the placenta in the decidua (uterine lining) and abundantly express E-cadherin—served as the primary portal of entry for L. monocytogenes from both extracellular and intracellular compartments. Subsequent bacterial dissemination to the villous stroma, where fetal capillaries are found, was hampered by further cellular and histological barriers. Our study suggests the placenta has evolved multiple mechanisms to resist pathogen infection, especially from maternal blood. These findings provide a novel explanation why almost all placental pathogens have intracellular life cycles: they may need maternal cells to reach the decidua and infect the placenta.
Introduction
Infection is a major cause for pregnancy complications including premature labor and resultant maternal and fetal morbidity and mortality (WHO, 2005). Nevertheless, the underlying mechanisms of placental and fetal infection are poorly understood. The placenta and fetus are vulnerable to infection via two different routes: (a) pathogens in the lower genital tract may ascend through the cervix and (b) pathogens in the maternal blood or uterus can colonize the placenta and breach the maternal-fetal barrier. The later group includes many viruses, e.g. cytomegalovirus; protozoan parasites, e.g. Toxoplasma gondii; and bacterial pathogens, e.g. Listeria monocytogenes. It is striking that the majority of pathogens that are able to cross the placenta have either facultative or obligate intracellular life cycles. The reason for the predisposition of placental infection toward intracellular pathogens is unclear. It has been postulated that the unique immunological environment in the placenta—necessary to assure tolerance of the fetal allograft—contributes to this phenomenon [1]–[3], but other aspects of the placenta may also play a role.
L. monocytogenes is a ubiquitous bacterial pathogen that causes food-borne disease in humans and many other mammals [4]–[6]. In pregnant women L. monocytogenes can spread to the placenta and fetus, resulting in spontaneous abortion, stillbirth, or preterm labor, depending on the gestational age [7]. The incidence of L. monocytogenes-induced spontaneous abortion during the first trimester is unknown; such early abortions are often due to chromosomal abnormalities [8] and therefore the aborted tissues are not routinely cultured. During the second trimester, L. monocytogenes has been found to cause ∼3% of spontaneous abortions in humans and cattle [9]–[11]. Clinical infections of the mother at term are rare, but when they occur, they can result in neonatal disease with mortality of up to 50% [12].
Among the intracellular microbes known to cross the maternal-fetal barrier, L. monocytogenes is particularly amenable to experimental analysis. L. monocytogenes has been used for decades as a model system to evaluate intracellular pathogenesis and the host's cell mediated and innate immune response to infection (for recent reviews see [13]–[15]). L. monocytogenes can infect professional phagocytic and non-phagocytic cells in many species. A family of bacterial cell wall surface proteins called internalins (Inl) promote bacterial adherence and internalization into non-phagocytic host cells [16]. Of these, internalin A (InlA) and internalin B (InlB) are the best characterized, binding to E-cadherin and c-Met-tyrosine kinase, respectively [17],[18]. After internalization, the bacterium escapes from the vacuole into the host cell cytoplasm where it multiplies rapidly [19],[20]. The listerial virulence determinant ActA facilitates spread from infected host cells to neighboring cells without bacterial exposure to the extracellular environment [21]–[24]. Thus, L. monocytogenes is able to infect non-phagocytic cells by two different mechanisms: Inl-mediated direct invasion and cell-to-cell spread. In the work described herein, we determine the placental tissue barriers operative against each mechanism and explore how L. monocytogenes might overcome them.
In order to understand the mechanisms leading to placental and fetal infection it is essential to understand the structure and physiology of the placenta. The placenta is made of maternal and fetal tissues. Placentas of different viviparous vertebrates exhibit great variability at the maternal-fetal interface, complicating cross-species comparisons [25]. Humans have a hemomonochorial villous placenta (Figure 1A). Maternal blood from spiral arteries in the decidua (uterine lining during pregnancy) flows into the intervillous space where it surrounds thousands of fetally derived floating villi. Some villi invade the decidua and form anchoring villi. The entire villous surface is covered with a continuous layer of multinucleate syncytiotrophoblast (SYN) (Figure 1B), which is the major fetal surface in contact with maternal blood. The apical side of the syncytium consists of profuse, branched microvilli [26],[27] and provides abundant surface area for gas and nutrient exchange between mother and fetus. The syncytiotrophoblast is undergirded by cytotrophoblasts (CTB) [28], which are separated from fetal capillaries in the villous stroma by a basement membrane. Some cytotrophoblasts leave the basement membrane and differentiate along the invasive pathway to form anchoring villi: columns of unpolarized cytotrophoblasts attach to and then penetrate the uterine wall where they give rise to extravillous cytotrophoblasts [29]. Extravillous cytotrophoblasts commingle with resident decidual, myometrial and immune cells. A subset of extravillous cytotrophoblasts breaches maternal spiral arteries in the decidua and differentiates into endovascular trophoblasts that replace the resident maternal endothelium to direct more blood into the intervillous space [29].

The anatomical site and mechanism by which L. monocytogenes breaches the maternal-fetal barrier are controversial. Of particular interest is whether InlA-mediated binding to E-cadherin is essential for transplacental transmission. Infection of isolated human cytotrophoblasts [30] or the BeWo choriocarcinoma cell line [31] with L. monocytogenes deficient in InlA leads to a 100-fold reduction in invasion. However, in vivo, cytotrophoblasts are covered with syncytiotrophoblast and may not be accessible to the bacteria. Lecuit et al. reported that E-cadherin is expressed at low levels on the apical surface of syncytiotrophoblast in explants from human term placentas [31], and postulated that L. monocytogenes breaches the maternal-fetal barrier by InlA-mediated invasion of the syncytiotrophoblast from the maternal bloodstream [31]. However, other groups have not observed E-cadherin expression on the surface of the syncytium [32]–[36]. Furthermore, InlA or InlB mutants do not affect feto-placental infection in guinea pigs [30] (unpublished observations), and show less than a 5-fold decrease in bacterial numbers in the gerbil placenta and fetus [37]. Wild type InlA does not interact with murine E-cadherin [38], but infection of wild type mice with L. monocytogenes expressing murinized InlA does not influence the course of feto-placental listeriosis [39], and in transgenic mice expressing human E-cadherin, InlA and/or InlB have a <5-fold effect on placental and fetal infection [37]. The minimal or absent in vivo phenotype observed with internalin mutants in these four rodent models is surprising given the strong phenotype in isolated cytotrophoblasts and suggests that the syncytiotrophoblast may not be the initial site of infection.
InlA/E-cadherin is not the only mechanism for infection—L. monocytogenes can spread from cell-to-cell without exposure to the extracellular environment, and there is evidence that L. monocytogenes traffics to the placenta [40] or the brain [41] inside of cells. Furthermore, we and others have found cell-to-cell spread to be important for fetal infection [42],[43].
In this report, we probe the human maternal-fetal barrier using first trimester human placental organ cultures, which allow a detailed examination of the most likely sites of transplacental infection by direct incubation with extracellular L. monocytogenes as well as via co-incubation with infected human cells. We found intact syncytiotrophoblast to be resistant to infection by L. monocytogenes. The portal of entry for L. monocytogenes was instead a small subpopulation of E-cadherin-expressing extravillous cytotrophoblasts in anchoring villi that are not readily accessible from the maternal bloodstream in vivo, and infection of these cells occurred via both InlA-mediated invasion and cell-to-cell spread. Surprisingly, these cells were able to restrict the growth of L. monocytogenes. If infection progressed, the bacteria spread along subsyncytial cytotrophoblasts, mostly sparing the syncytiotrophoblast and villous stroma. Our results clarify the mechanisms of crossing the maternal-fetal barrier and provide a unifying explanation for the conflicting in vitro and in vivo results mentioned above.
Results
Culture of first trimester human placental explants
In order to examine the role of direct invasion and cell-to-cell spread in breaching the human maternal-fetal barrier we turned to first trimester human placental organ cultures, a well-studied model system that allows examination of the trophoblast in a context that retains the cellular architecture of the tissue in vivo [44],[45]. Placental villous trees are dissected and explanted on substrates of extracellular matrix (Matrigel), where they form floating and anchoring villi (Figure 1C-D). All of the tissue is exposed to the media with the exception of the tips of anchoring villi that result from extravillous cytotrophoblast outgrowth and invasion into Matrigel [45],[46]. This mimics the conditions in vivo where extravillous cytotrophoblasts invade the decidua while the rest of the villous tree is bathed in maternal blood [29]. The syncytiotrophoblast covers the villi and remains largely intact for at least 24 h (Susan Fisher, personal communication). First trimester placental explants therefore adequately represent the most probable placental sites that are potentially accessible to L. monocytogenes or infected phagocytes: intact syncytiotrophoblast, subsyncytial cytotrophoblasts underlying damaged syncytiotrophoblast, and extravillous cytotrophoblasts (Figure 1).
L. monocytogenes infection of human placental explants
We infected explants with 2×106 wild type L. monocytogenes (Figure 2). In order to measure intracellular growth we added gentamicin 1 h post-inoculation to eliminate extracellular L. monocytogenes, and subsequently determined numbers of live bacteria per explant over 24 h (Figure 2A). No significant differences in infection were observed between the wild type strains 10403S and EGDe (p = 0.38 by Student's T-test). The average bacterial growth from all placentas was less than 10-fold, which is slow compared to that found in cell lines [20] (Figure 2A), and infection rates were highly variable. Despite the relatively high inoculum, 11% of the explants were not infected and an additional 13% hosted <10 intracellular bacteria at 2 h post-inoculation. The average percentage of intracellular bacteria at 2 h post-inoculation was 0.6%±2% SD of the inoculum (n = 54 explants), similar to the bottleneck in the pregnant guinea pig model of listeriosis [40].

Explants vary in size, shape, age, donor and degree of Matrigel invasion, so variability is expected. However, we were able to distinguish two possible courses of infection by examining three explants from the same placenta at each time-point. Roughly half of the placentas exhibited an increase in bacterial numbers from 2 to 24 h (average = 77-fold, SD = 6.4) while the others showed a decrease (average = 0.25-fold, SD = 0.19).
It has been previously suggested that L. monocytogenes invades the syncytiotrophoblast [31]. If this is true for explants, then larger explants should be more highly infected, since >90% of each explant's bacterially accessible surface area is covered by syncytiotrophoblast. But we found no correlation between colony forming units (CFU) and explant size (r2<0.05) in 30 explants from 11 placentas. Nor did explant age affect CFU (r2<0.02). However, CFU at 2 h post-inoculation did correlate with the number of anchoring villi (r2 = 0.49, Figure 2B), suggesting that extravillous cytotrophoblasts are the preferred sites of L. monocytogenes infection. Examination by immunofluorescence histology revealed only a few foci of infection, usually in extravillous cytotrophoblasts of anchoring villi (Figure 2C).
Syncytiotrophoblast forms a barrier against infection
To better characterize which placental cell types are most vulnerable to L. monocytogenes infection, we increased both the inoculum and the time of incubation without gentamicin. These “permissive infections” increase the probability of infection at vulnerable sites. In addition to wild type bacteria, we also used bacteria deficient in ActA (ΔActA) that are incapable of intercellular spread, thus ensuring that the L. monocytogenes-containing cells we observe are those initially infected by the bacteria. After 8 h, we examined explant sections by immunofluorescence (Figure 3).
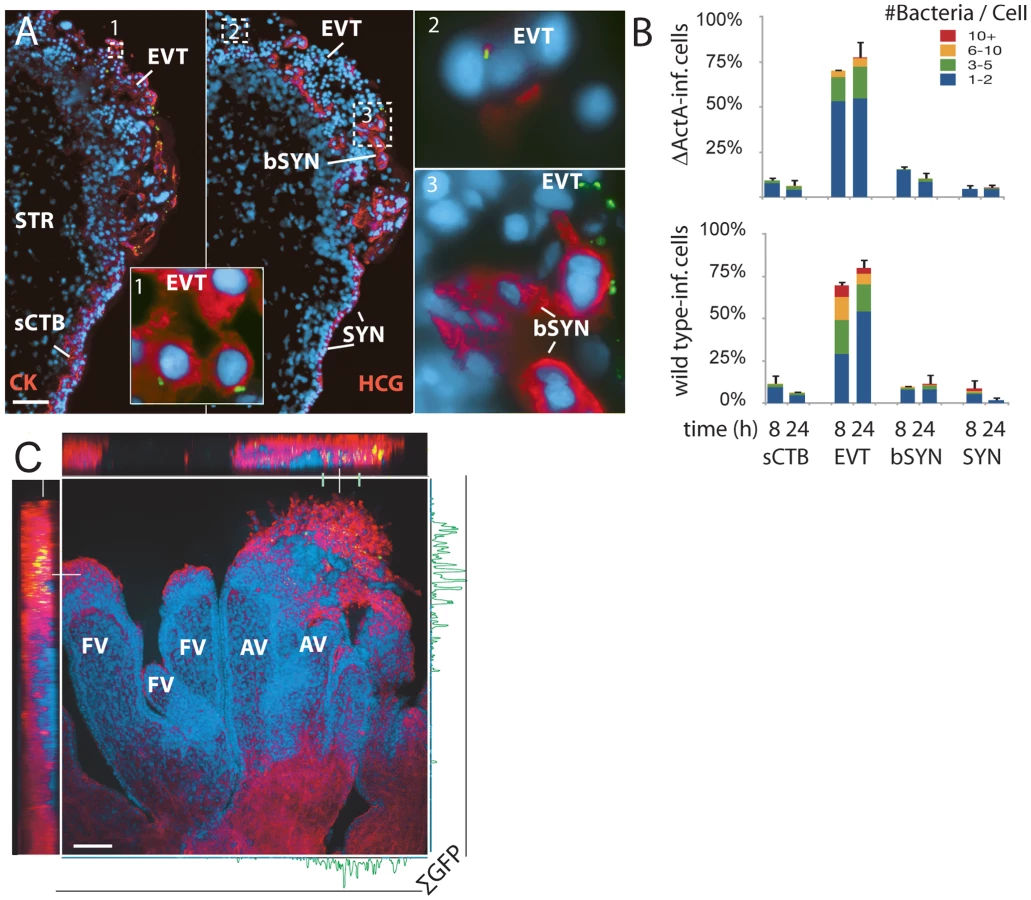
Under these conditions, L. monocytogenes was detectable in three cell types (Figure 3A): subsyncytial cytotrophoblasts, extravillous cytotrophoblasts and syncytiotrophoblast. Infected syncytiotrophoblast could be subdivided into: 1) apparently intact syncytiotrophoblast where only the apical surface is exposed to bacteria; and 2) basolaterally accessible syncytiotrophoblast (bSYN), where the syncytiotrophoblast is naturally terminated by an invading CTB cell column (Figure 3A) or, in rare cases, torn away from the explant, presumably during dissection.
We enumerated the total number of infected cells in explant sections (Figure 3B). For syncytiotrophoblast, a “cell” was defined as a circular region similar in size to a CTB, roughly the area surrounding a single nucleus. Infection of subsyncytial cytotrophoblasts was infrequent, which is unsurprising since unlike syncytiotrophoblast and extravillous cyotrophoblasts they are largely inaccessible to bacteria in the media. However, syncytiotrophoblast infection was also low, even though it covers almost all of the explant surface. Roughly 75% of the infected cells were extravillous cytotrophoblasts, which comprise less than 5% of available surface. Furthermore, these cells were ∼5 times more likely to contain multiple bacteria, possibly indicating multiple infections.
Transverse sections obscure a full view of the syncytiotrophoblast. To ensure that our observations were not a histological artifact, we fixed and mounted whole explants infected with L. monocytogenes expressing GFP. Confocal microscopy of these minimally manipulated explants confirmed that the bacteria are highly localized within the extravillous cytotrophoblasts of anchoring villi (Figure 3C). Together, these results suggest that extravillous cytotrophoblasts may serve as the primary site of infection.
Although in cultured macrophage and epithelial cell lines cell-to-cell spread begins around 4–5 h post-infection [21],[24], we found no significant difference between locations of ΔActA and wild type L. monocytogenes at 8 or even 24 h (Figure 3B, p = 0.99 by chi-squared test), suggesting that the L. monocytogenes life cycle (intracellular growth and/or cell-to-cell spread) is delayed in primary trophoblast cells.
InlA mediates proximal extravillous cytotrophoblast invasion
E-cadherin is an important host cell receptor for L. monocytogenes binding and uptake. Lecuit et al. suggested that L. monocytogenes extracellular invasion of the placenta occurs via L. monocytogenes InlA interactions with host E-cadherin on the apical surface of syncytiotrophoblast [31]. However, other studies of the placenta have failed to find E-cadherin here [32]–[36]. Our results support this: we never observed E-cadherin staining on the apical surface of the syncytiotrophoblast, although it was expressed strongly on the basal surface (Figure 4A). Like others, we found E-cadherin was most abundant on subsyncytial cytotrophoblasts and proximal extravillous cytotrophoblasts, decreasing as cells migrate away from the villus tip.

Since proximal extravillous cytotrophoblasts were the very cells L. monocytogenes infected, we hypothesized that explant infection is InlA-dependent. Indeed, ΔInlA and ΔInlAB mutants were almost completely unable to invade explants (Figure 4B; p<10−20 by Student's T-test). We found no significant difference between invasion of wild type and ΔInlB L. monocytogenes in human explants (Figure 4B; p = 0.68 by Student's T-test) consistent with previous observations in isolated CTB and BeWo cells (human choriocarcinoma cell line) [30].
Damage of syncytiotrophoblast leads to infection of subsyncytial cytotrophoblasts
If the syncytiotrophoblast is relatively resistant to infection, as the preceding data suggest, then removing it should provide L. monocytogenes new sites of invasion. We enzymatically degraded the syncytiotrophoblast by soaking the explants briefly in a collagenase-containing solution before plating [47]. Although the extent of the syncytiotrophoblast removal varied, many subsyncytial cytotrophoblasts were exposed and extravillous cytotrophoblasts increased (Figure 5A). As expected, permissive infections of enzymatically-treated explants allowed for new sites of infection (Figure 5B). The total number of infected cells increased (from an average of 78 in two sections to 228 with enzymatic degradation), and nearly half of the infected cells were now subsyncytial cytotrophoblasts, which express E-cadherin (Figure 5C; p<0.05 by Chi-square test).

Cell-to-cell spread leads to infection of proximal extravillous cytotrophoblasts
Infection of the placenta by extracellular pathogens in the maternal bloodstream must be mediated by the interaction of pathogen virulence determinants, e.g. InlA with host cell receptors like E-cadherin. However, L. monocytogenes also traffics in vivo to the placenta in a gentamicin-resistant manner [40], presumably traveling inside phagocytic leukocytes [41]. We wanted to test whether cell-to-cell spread can mediate placental infection, and, if so, what sites are vulnerable.
We introduced a fluorescent live cell dye to macrophage-like U937 cells (differentiated to adherent cells with PMA) and then infected them with 10403S-sGFP L. monocytogenes. The infected cells were added to explants in the presence of gentamicin to prevent infection of the explant by extracellular bacteria. After 24 h transmission of L. monocytogenes from U937 cells to explants had occurred (Figure 6A-B). As with direct invasion, we found that L. monocytogenes infection by cell-to-cell spread from U937 cells to the placenta was largely confined to extravillous cytotrophoblasts at villous tips (Figure 6C). In fact, the cell populations infected were statistically indistinguishable from InlA-mediated infections after 24 h (p = 0.99 by chi-squared test). Invasive CTB express chemokines that attract cells of the monocyte lineage [48]–[50], and indeed we observed clusters of U937 cells around the extravillous cytotrophoblasts as early as 4–8 h post-inoculation (data not shown). Placental infection was not observed upon co-cultivation with U937 cells carrying ΔActA mutants, which are defective in cell-to-cell spread (data not shown).

Bacterial dissemination occurs along subsyncytial cytotrophoblasts in anchoring villi
Regardless of how L. monocytogenes was introduced, the dominant site of initial infection was the extravillous cytotrophoblast at the tip of anchoring villi. Multiple explants from a single placenta showed strikingly similar progression over the course of infection. In three out of six placentas studied, L. monocytogenes advanced significantly beyond the tips of anchoring villi (Figure 7). By 72 h post-inoculation, subsyncytial cytotrophoblast infection was common in anchoring villi while syncytiotrophoblast remained largely uninfected, suggesting that the syncytium not only resists cell-to-cell spread from macrophage-like cells but also from neighboring cytotrophoblasts (Figure 7A). While infected anchoring villi were always colonized at the distal tips, infection of floating villi always began at proximal junctures shared by anchoring villi. At times floating villi exhibited infection of cytotrophoblasts on both sides of the villus without syncytiotrophoblast infection, indicating that L. monocytogenes trafficked through the subsyncytial cytotrophoblasts (Figure 7B). Spread into the stroma was rare and presumably restricted by the basement membrane underlying subsyncytial cytotrophoblasts. Some stromal cells were infected at later timepoints (Figure 7C). In explants infected with ΔActA L. monocytogenes, bacteria remained in extravillous cytotrophoblasts (Figure 7D).

Overall, ∼75 – 100% of anchoring villi were infected (Figure 7E) and infection of subsyncytial cytotrophoblasts and stroma increased over 72 h (Figure 7F). In contrast, only 22% of explants exhibited any L. monocytogenes in floating villi (Figure 7E). Taken together, these results describe the cell-to-cell path L. monocytogenes follows in disseminating throughout placental explants over three days: from extravillous cytotrophoblasts of anchoring villi along lateral villous subsyncytial cytotrophoblasts and from there into floating villi and/or stroma, all while leaving the syncytiotrophoblast largely uninfected.
Discussion
Pathogens present in the maternal bloodstream may colonize the placenta, causing infection, inflammation, and ultimately spontaneous abortion, preterm labor, and neonatal morbidity and mortality [51]. Many pathogenic microbes are found transiently in maternal blood. For example, the simple daily act of brushing teeth is associated with bacteremia [52],[53], and L. monocytogenes is ingested frequently by healthy adults [54]. Yet neither result in significant maternal-fetal infection the majority of the time. This is surprising considering that twenty percent of maternal blood can be found circulating freely in the placenta's intervillous space, where it bathes fetal villi that are covered by a syncytiotrophoblast whose surface area ranges from 3000 cm2 in the late first trimester to 125,000 cm2 at term [28]. Thus, it seems reasonable to hypothesize that the syncytium forms an extremely effective physical barrier against infection. In this study, we have conclusively shown that the syncytiotrophoblast is resistant to infection by L. monocytogenes and that extravillous cytotrophoblasts are the portal of entry.
For internalin-mediated infections the resistance of the syncytiotrophoblast can be reasonably explained by the tissue's lack of E-cadherin on the apical surface. When syncytiotrophoblast was infected, it was more likely to be basolaterally accessible syncytiotrophoblast, in which an E-cadherin expressing basolateral surface was exposed. Even for infections in uninterrupted syncytiotrophoblast, it remains possible that basolateral access was not apparent in the examined section but was available in an adjacent section. It is interesting to note that InlA and E-cadherin interactions mediating intestinal invasion are confined by anatomical and cellular barriers as well [55].
Even more surprising was the near absence of syncytiotrophoblast infection by cell-to-cell spread, either from U937 cells—which were observed near the syncytiotrophoblast—or from neighboring cytotrophoblasts at later timepoints. Three possible explanations exist: 1) the syncytiotrophoblast under-expresses unknown host molecule(s) required for cell-to-cell spread; 2) attachment of leukocytes to syncytiotrophoblast is insufficiently close in time or space for cell-to-cell spread to occur; or 3) the syncytiotrophoblast membrane is physically inhospitable to L. monocytogenes' actin-mediated protrusions. The last two are especially plausible when considering the profuse covering of branched microvilli on the apical surface [56]–[61]. The basal surface may also be girded against protrusion by the especially dense cytoskeletal network that is presumably required to resist cytosolic surface tension in the laterally vast syncytium [56]–[59]. Interestingly, interaction of the bacterial virulence factor InlC with human actin regulatory proteins has recently been shown to promote cell-to-cell spread by decreasing cortical tension, thereby enhancing the ability of motile bacteria to deform the plasma membrane into protrusions [62].
The syncytiotrophoblast may act as a general barrier. We have observed that T. gondii is not able to efficiently colonize the syncytium either (unpublished observations), and other groups have reported similar results for herpes simplex virus [63] and cytomegalovirus [64],[65].
Instead, we present evidence that extravillous cytotrophoblasts, normally not easily accessible from the intervillous space, are the dominant sites of L. monocytogenes colonization from both extracellular and intracellular compartments. Our results are in accord with Lecuit et al. showing that invasion of placental explants by extracellular L. monocytogenes depends on InlA, but our findings differ on the initial site of invasion (EVT versus SYN, see Figure 8). An important difference is our experimental set-up: we used first trimester placental explants instead of term placentas. Term placental organ cultures do not form anchoring villi after removal from the mother and are maintained in floating culture. Damage of the term explant syncytiotrophoblast has been reported as early as 4 h under selected culture conditions [66] and term explants cannot be used to evaluate extravillous cytotrophoblasts [67]. Thus, first trimester explants better represent the architecture of the maternal-fetal barrier in vivo.

However, placental organ cultures from all gestational ages omit the decidua and the maternal vessels. The later are remodeled by extravillous cytotrophoblasts, which differentiate into endovascular trophoblasts that replace the endothelium of the maternal spiral arteries and therefore are in direct contact with maternal blood. Endovascular trophoblasts do not express E-cadherin [32] but it may be possible that they are targets of cell-to-cell spread in vivo.
It has recently been postulated that the conjugated action of InlA and InlB leads to breaching of the maternal-fetal barrier [37]. This is particularly intriguing since InlA and InlB are in the same operon and expression of these invasion proteins is most likely co-regulated [16]. Disson et al. show a 10-fold reduction in invasion of the human intestinal cell line Caco-2 with L. monocytogenes strain EGDe, deficient in InlA or InlB, and an almost 100-fold reduction with the InlAB double deletion mutant [37]. Other groups using L. monocytogenes strains derived from 10403S have observed a 2–3-fold effect of InlB on intestinal invasion (Amieva, personal communication). We do not observe a difference between wt and ΔInlB in infection of early gestation placental organ cultures. It may be that the variability of the human placental explants is too high to resolve a potentially small effect of InlB on invasion of placental explants.
We did examine the role of cell-to-cell spread from infected U937 cells to the placenta, which seems highly relevant considering the importance of Listeria's intracellular life cycle for virulence [68] and the published evidence that L. monocytogenes traffics between organs inside of cells [40],[41]. It is striking that extravillous cytotrophoblasts remain the primary portal of entry, leading us to hypothesize that access to extravillous cytotrophoblasts represents the first bottleneck for L. monocytogenes infection of the placenta (Figure 8). How can L. monocytogenes overcome this hurdle? Extravillous cytotrophoblasts are present in the decidua and are known to actively recruit macrophages, monocytes and natural killer cells [48]–[50]. Therefore, we postulate that L. monocytogenes reaches the placenta in maternal phagocytes that are recruited to the decidua where they infect extravillous cytotrophoblasts by cell-to-cell spread or internalin-mediated invasion.
Another striking finding was that extraordinarily high doses of L. monocytogenes were required to infect placental explants, and that L. monocytogenes growth rates were relatively slow, on average increasing only ∼10-fold over 24 h. However, we could distinguish two placental populations: in about half of the placentas L. monocytogenes did not grow, while in the other half bacterial numbers increased by ∼77-fold. Lecuit et al. used slightly higher doses and reported an increase of ∼100-fold over 24 h. But they also report having removed gentamicin from the culture medium at 2 h post-inoculation. Therefore, their 24 h CFU and histological assays may have included extracellular bacteria that escaped dying cells. In addition, we found little to no cell-to-cell spread at 24 h, while in most cell lines cell-to-cell spread begins as early as 4 h post-inoculation [21],[24]. In some placentas we observed that infection remained confined to the extravillous cytotrophoblasts for at least 72 h. It is intriguing that the sites of infection correlated with the host cell's proliferative capacity [69],[70], which may provide interesting avenues for future studies. The slow rate of intracellular bacterial growth and cell-to-cell spread suggest the possibility that extravillous cytotrophoblasts restrict the intracellular life cycle of L. monocytogenes, thus representing the second bottleneck in the placenta (Figure 8).
Once the placenta is infected, L. monocytogenes can spread to the fetus. One probable route of spread to the fetus is via the fetal capillaries in the villous stroma. Indeed, we observed low numbers of bacteria that had penetrated the basement membrane and infected the stroma suggesting that this is the third bottleneck L. monocytogenes encounters (Figure 8). It is interesting to note that although some bacteria were observed in syncytiotrophoblast at early timepoints, only anchoring villi acted as an origin of colonization.
Our model is consistent with previous findings in vivo that the guinea pig placenta is colonized by 104 times fewer bacteria than maternal liver and spleen, and subsequently only 1 out of 104 bacteria are able to spread from placenta to fetus [40]. It is also in agreement with the epidemiology of human listeriosis, which is a rare disease during pregnancy despite its ubiquity in the environment, as well as the observation that pregnant animals have to be inoculated with high doses of L. monocytogenes to observe consistent placental and fetal infection [37],[39],[40]. In addition, our results provide an explanation for the absent or minor phenotype the internalin mutants exhibit in multiple different pregnant animal models [37],[39],[40].
Although it may be attractive to describe the route taken by a pathogen as a single mechanism, we do not believe that this accurately reflects what occurs in vivo. There is mounting evidence that pathogens have evolved to exploit multiple strategies to breach the intestinal and blood-brain barriers [71],[72], and it is reasonable to expect the same of the maternal-fetal barrier. The mechanisms by which the placenta excludes most pathogens to generate the maternal-fetal barrier are poorly understood, but our results suggest that the syncytiotrophoblast plays a significant role. Given its extensive contact with the maternal blood, this important tissue may have evolved to exclude pathogens, and our model system offers a powerful way to probe the mechanisms by which this occurs. Pathogens that can breach the syncytiotrophoblast or exploit sites of syncytial damage may colonize the placenta via subsyncytial cytotrophoblasts. Our study of L. monocytogenes, a model pathogen that colonizes the placenta, strongly suggests that the placenta's most vulnerable site is the extravillous cytotrophoblast, where cells anchor the placenta in the maternal decidua but have little to no contact with maternal blood. This finding suggests a reason for the observation that almost all pathogens capable of crossing the maternal-fetal barrier are either facultative or obligate intracellular: dissemination in the blood is not enough.
Methods
Ethics statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Institutional Review Board at the University of California, San Francisco, where all experiments were performed (H497-00836-28). All patients provided written informed consent for the collection of samples and subsequent analysis.
Human tissue collection and culture
Placentas from elective terminations of pregnancy (gestational age 4 to 8 weeks) were collected and prepared as previously described [67]. Briefly, fragments from the surface of the placenta were dissected into 1–3 mm tree-like villi, placed on Matrigel (BD Biosciences, San Jose, CA) coated Transwell filters (Millipore, Bedirica, MA, 30-mm diameter, 0.4 µm pore size) and cultured in Dulbecco's modified Eagle's medium-F12 medium (DMEM-F12; 1∶1, vol/vol) supplemented with 20% fetal bovine serum (FBS, Fisher Scientific), 1% L-glutamine and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA). For surface area and perimeter measurements, cultured explants were photographed pre-infection on a Leica MZ16F stereomicroscope (Leica Microsystems, Wetzlar, Germany) using an Axiocam MR monochrome camera (Carl Zeiss, Munich, Germany). Measurements were made using ImageJ software (NIH, Bethesda, MD).
Removal of syncytiotrophoblast
Syncytiotrophoblast was removed from villous trees as previously described [47]. Briefly, placental explants were soaked for 5–15 min in a solution containing Type IA collagenase (100,000 U), hyaluronidase (150,000 U), DNAse (120,000 U) and 0.1% BSA in PBS without divalent cations (UCSF Cell Culture Facility, San Francisco, CA). Explants were observed continuously via a dissecting microscope and when syncytiotrophoblast degradation was apparent they were transferred to Matrigel.
Pathogen strains and growth conditions
The wild type strain of L. monocytogenes used in this study is 10403S [73]. Mutant strains included ΔInlA (DPL4405), ΔInlB (DPL4406), ΔInlAB (DPL4455) [30], ΔActA (DPL3078) [74], and sGFP-expressing 10403S L. monocytogenes (DH-L1039) [75]. EGDe L. monocytogenes (M. Loessner) was used for some experiments. Bacteria were cultured using brain heart infusion (BHI) broth or agar (Becton Dickenson Company, Sparks, MD).
L. monocytogenes infections of placental explants
Intracellular growth assays of L. monocytogenes were performed as previously described [76] with following modifications: placental explants were incubated in antibiotic free media for 1 h prior to infection, 1×106 bacteria/ml were added for 30 min and gentamicin (50 µg/ml) was added at 60 min post-inoculation. Gentamicin was subsequently maintained in the media, which was refreshed every 24 h. At specified times after infection, explants were removed from Matrigel and homogenized in 1 ml dH2O using a T25 digital Ultra-Turrax (IKA, Staufen, Germany). Aliquots were plated on BHI agar and grown at 37°C. For permissive infections explants were incubated with 2×107 bacteria/ml for 5 h before adding gentamicin.
Infection of explants by cell-to-cell spread
Human macrophage-like U937 cells (ATCC 1593.2 [77]) were grown in RPMI-1640 (UCSF Cell Culture Facility) containing 4500 mg/L glucose, 10% FBS and 1% penicillin/streptomycin (Invitrogen). Forty-eight hours prior to infection, cells were differentiated by addition of phorbol 12-myristate 13-acetate (PMA; concentration 18 nM; Sigma) to the medium. On the day of infection, cells were labeled with CellTracker Green CMFDA (Invitrogen) and infected with L. monocytogenes for 1 h at an MOI of 1∶1. Cells were washed once with PBS and lifted from culture plates by incubation in ice cold PBS without divalent cations for 5 min. U937 cells were resuspended in explant medium containing 50 µg/ml gentamicin, and 1×106 cells per transwell were added to the explants. Every 24 h, fresh media containing gentamicin was added.
Immunofluorescence and histology
Explants were removed at the times indicated and placed into vinyl cryomolds (Ted Pella, Redding, CA), then covered with optimal cutting temperature (OCT) media (Ted Pella) and flash-frozen. Histological slicing was performed using a Hacker-Slee cryostat. Glass slides with sections were incubated ∼5 min in acetone at 4°C. All antibody staining was conducted at room temperature. When dry, slides were soaked 60 min in blocking solution (1% bovine serum albumin (BSA, Sigma) in PBS), then rinsed and exposed to primary antibodies in 0.5% BSA/PBS. Slides were rinsed three times for 5 min each in 0.5% BSA/PBS, then secondary antibodies were added at the indicated concentrations and incubated for 60 min. After three rinses, coverslips were affixed over Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Uninfected explants did not stain with anti-Listeria antibodies. Primary antibodies: polyclonal rabbit Listeria O antiserum (1∶1000 Becton, Dickenson), monoclonal mouse anti-human cytokeratin 7 (1X, Clone OV-TL, Dako, Carpinteria, CA), monoclonal mouse anti-human E-cadherin (1∶200, Clone NCH-38, Dako), monoclonal mouse anti-human βHCG (1∶500, clone SPM105, Neomarkers, Fremont, CA) and monoclonal mouse anti-human EGFR (1∶250, Clone cocktail R19/48, Biosource, Camarillo, CA). Secondary antibodies: Alexa Fluor 594 goat anti-mouse IgG (1∶500) and Alexa Fluor 488 goat anti-rabbit IgG (1∶1000, both Invitrogen). All immunofluorescence conditions were compared to no-primary controls to ensure that non-specific binding did not occur.
Slides were viewed using an inverted TE2000-E microscope (Nikon, Tokyo, Japan) equipped with a 12-bit cooled CCD camera (Q Imaging, Surrey, Canada). Images were collected using Simple PCI software (Hamamatsu, Sewickley, PA). Counts of L. monocytogenes localization were made by tallying every infected cell in each section at 100X magnification.
Confocal microscopy
Whole mount explants were prepared by rinsing explants with PBS and then soaking in 3% paraformaldehyde in PBS (Ted Pella) for 12 h at 4°C. Explants were then rinsed three times with PBS and suspended in 1∶100 Alexa Fluor 594 phalloidin and 1∶100 DAPI (both Invitrogen) for 24 h at 4°C. Explants were mounted onto glass slides in Vectashield and sealed under coverslips. Imaging was performed at the Nikon Imaging Center at UCSF using an upright Nikon C1 spectral confocal microscope equipped with 405, 488 and 561 nm lasers.
Image processing for figures
Images were prepared using Photoshop and Illustrator (Adobe, San Jose, CA). RGB color hues were linearly adjusted for better CMYK printing but no non-linear alterations were performed.
Zdroje
1. MoffettA
LokeC
2006 Immunology of placentation in eutherian mammals. Nat Rev Immunol 6 584 594
2. TrowsdaleJ
BetzAG
2006 Mother's little helpers: mechanisms of maternal-fetal tolerance. Nat Immunol 7 241 246
3. MedawarPB
1953 320 338 Some immunological and endocrinological problems raised by the evolution of viviparity in vertebrates
4. DrevetsDA
BronzeMS
2008 Listeria monocytogenes: epidemiology, human disease, and mechanisms of brain invasion. FEMS Immunol Med Microbiol
5. SwaminathanB
Gerner-SmidtP
2007 The epidemiology of human listeriosis. Microbes Infect 9 1236 1243
6. LecuitM
2007 Human listeriosis and animal models. Microbes Infect 9 1216 1225
7. MylonakisE
PaliouM
HohmannEL
CalderwoodSB
WingEJ
2002 Listeriosis during pregnancy: a case series and review of 222 cases. Medicine (Baltimore) 81 260 269
8. KajiiT
FerrierA
NiikawaN
TakaharaH
OhamaK
1980 Anatomic and chromosomal anomalies in 639 spontaneous abortuses. Hum Genet 55 87 98
9. KirkbrideCA
1993 Bacterial agents detected in a 10-year study of bovine abortions and stillbirths. J Vet Diagn Invest 5 64 68
10. KaurS
MalikSV
VaidyaVM
BarbuddheSB
2007 Listeria monocytogenes in spontaneous abortions in humans and its detection by multiplex PCR. J Appl Microbiol 103 1889 1896
11. LallemandAV
GaillardDA
ParadisPH
ChippauxCG
1992 Fetal listeriosis during the second trimester of gestation. Pediatr Pathol 12 665 671
12. TebergAJ
YonekuraML
SalminenC
PavlovaZ
1987 Clinical manifestations of epidemic neonatal listeriosis. Pediatr Infect Dis J 6 817 820
13. PamerEG
2004 Immune responses to Listeria monocytogenes. Nat Rev Immunol 4 812 823
14. PortnoyDA
2005 Manipulation of innate immunity by bacterial pathogens. Curr Opin Immunol 17 25 28
15. CossartP
Toledo-AranaA
2008 Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect 10 1041 1050
16. BierneH
SabetC
PersonnicN
CossartP
2007 Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect 9 1156 1166
17. MengaudJ
OhayonH
GounonP
MegeRM
CossartP
1996 E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84 923 932
18. ShenY
NaujokasM
ParkM
IretonK
2000 InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103 501 510
19. O'RiordanM
PortnoyDA
2002 The host cytosol: front-line or home front? Trends Microbiol 10 361 364
20. PortnoyDA
JacksPS
HinrichsDJ
1988 Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167 1459 1471
21. RobbinsJR
BarthAI
MarquisH
de HostosEL
NelsonWJ
1999 Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J Cell Biol 146 1333 1350
22. KocksC
GouinE
TabouretM
BercheP
OhayonH
1992 L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68 521 531
23. TheriotJA
MitchisonTJ
TilneyLG
PortnoyDA
1992 The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357 257 260
24. TilneyLG
PortnoyDA
1989 Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109 1597 1608
25. CarterAM
2007 Animal models of human placentation--a review. Placenta 28 Suppl A S41 47
26. CantleSJ
KaufmannP
LuckhardtM
SchweikhartG
1987 Interpretation of syncytial sprouts and bridges in the human placenta. Placenta 8 221 234
27. SchieblerTH
KaufmannP
1981
BeckerV
SchieblerTH
KubliF
Reife Placenta Stuttgart Thieme
28. BenirschkeK
KaufmannP
BaergenRN
2006 Pathology of the human placenta. 30 41 p
29. Red-HorseK
ZhouY
GenbacevO
PrakobpholA
FoulkR
2004 Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest 114 744 754
30. BakardjievAI
StacyBA
FisherSJ
PortnoyDA
2004 Listeriosis in the pregnant guinea pig: a model of vertical transmission. Infect Immun 72 489 497
31. LecuitM
NelsonDM
SmithSD
KhunH
HuerreM
2004 Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E-cadherin. Proc Natl Acad Sci U S A 101 6152 6157
32. ZhouY
FisherSJ
JanatpourM
GenbacevO
DejanaE
1997 Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest 99 2139 2151
33. CoutifarisC
KaoLC
SehdevHM
ChinU
BabalolaGO
1991 E-cadherin expression during the differentiation of human trophoblasts. Development 113 767 777
34. BrownLM
LaceyHA
BakerPN
CrockerIP
2005 E-cadherin in the assessment of aberrant placental cytotrophoblast turnover in pregnancies complicated by pre-eclampsia. Histochem Cell Biol 124 499 506
35. VicovacL
AplinJD
1996 Epithelial-mesenchymal transition during trophoblast differentiation. Acta Anat (Basel) 156 202 216
36. FloridonC
NielsenO
HolundB
SundeL
WestergaardJG
2000 Localization of E-cadherin in villous, extravillous and vascular trophoblasts during intrauterine, ectopic and molar pregnancy. Mol Hum Reprod 6 943 950
37. DissonO
GrayoS
HuilletE
NikitasG
Langa-VivesF
2008 Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455 1114 1118
38. LecuitM
DramsiS
GottardiC
Fedor-ChaikenM
GumbinerB
1999 A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. Embo J 18 3956 3963
39. WollertT
PascheB
RochonM
DeppenmeierS
van den HeuvelJ
2007 Extending the host range of Listeria monocytogenes by rational protein design. Cell 129 891 902
40. BakardjievAI
TheriotJA
PortnoyDA
2006 Listeria monocytogenes traffics from maternal organs to the placenta and back. PLoS Pathog 2 e66 doi:10.1371/journal.ppat.0020066
41. DrevetsDA
JelinekTA
FreitagNE
2001 Listeria monocytogenes-infected phagocytes can initiate central nervous system infection in mice. Infect Immun 69 1344 1350
42. BakardjievAI
StacyBA
PortnoyDA
2005 Growth of Listeria monocytogenes in the guinea pig placenta and role of cell-to-cell spread in fetal infection. J Infect Dis 191 1889 1897
43. Le MonnierA
AutretN
Join-LambertOF
JaubertF
CharbitA
2007 ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect Immun 75 950 957
44. FisherSJ
LeitchMS
KantorMS
BasbaumCB
KramerRH
1985 Degradation of extracellular matrix by the trophoblastic cells of first-trimester human placentas. J Cell Biochem 27 31 41
45. MillerRK
GenbacevO
TurnerMA
AplinJD
CaniggiaI
2005 Human placental explants in culture: approaches and assessments. Placenta 26 439 448
46. HunkapillerNM
FisherSJ
2008 Chapter 12. Placental remodeling of the uterine vasculature. Methods Enzymol 445 281 302
47. FisherSJ
CuiTY
ZhangL
HartmanL
GrahlK
1989 Adhesive and degradative properties of human placental cytotrophoblast cells in vitro. J Cell Biol 109 891 902
48. DrakePM
GunnMD
CharoIF
TsouCL
ZhouY
2001 Human placental cytotrophoblasts attract monocytes and CD56(bright) natural killer cells via the actions of monocyte inflammatory protein 1alpha. J Exp Med 193 1199 1212
49. HuangY
ZhuXY
DuMR
LiDJ
2008 Human trophoblasts recruited T lymphocytes and monocytes into decidua by secretion of chemokine CXCL16 and interaction with CXCR6 in the first-trimester pregnancy. J Immunol 180 2367 2375
50. Red-HorseK
DrakePM
FisherSJ
2004 Human pregnancy: the role of chemokine networks at the fetal-maternal interface. Expert Rev Mol Med 6 1 14
51. GoldenbergRL
HauthJC
AndrewsWW
2000 Intrauterine infection and preterm delivery. N Engl J Med 342 1500 1507
52. FornerL
LarsenT
KilianM
HolmstrupP
2006 Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol 33 401 407
53. CobeHM
1954 Transitory bacteremia. Oral Surg Oral Med Oral Pathol 7 609 615
54. NotermansS
DufrenneJ
TeunisP
ChackrabortyT
1998 Studies on the risk assessment of Listeria monocytogenes. J Food Prot 61 244 248
55. PentecostM
OttoG
TheriotJA
AmievaMR
2006 Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2 e3 doi:10.1371/journal.ppat.0020003
56. OcklefordCD
WakelyJ
BadleyRA
1981 Morphogenesis of human placental chorionic villi: cytoskeletal, syncytioskeletal and extracellular matrix proteins. Proc R Soc Lond B Biol Sci 212 305 316
57. BerrymanM
GaryR
BretscherA
1995 Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J Cell Biol 131 1231 1242
58. BehamA
DenkH
DesoyeG
1988 The distribution of intermediate filament proteins, actin and desmoplakins in human placental tissue as revealed by polyclonal and monoclonal antibodies. Placenta 9 479 492
59. KingBF
1983 The organization of actin filaments in human placental villi. J Ultrastruct Res 85 320 328
60. VacekZ
1969 Electron microscopic observations on the filaments in the trophoblast of the human placenta. Folia Morphol (Praha) 17 382 388
61. ScheunerG
RuckhaberleKE
FlemmingG
ReissigD
1980 [Submicroscopic detection of oriented protein filaments in the plasmoditrophoblast of the human placenta (author's transl)]. Anat Anz 147 145 151
62. RajabianT
GavicherlaB
HeisigM
Muller-AltrockS
GoebelW
2009 The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol 11 1212 1218
63. KoiH
ZhangJ
MakrigiannakisA
GetsiosS
MacCalmanCD
2002 Syncytiotrophoblast is a barrier to maternal-fetal transmission of herpes simplex virus. Biol Reprod 67 1572 1579
64. MaidjiE
McDonaghS
GenbacevO
TabataT
PereiraL
2006 Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol 168 1210 1226
65. FisherS
GenbacevO
MaidjiE
PereiraL
2000 Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol 74 6808 6820
66. SoorannaSR
Oteng-NtimE
MeahR
RyderTA
BajoriaR
1999 Characterization of human placental explants: morphological, biochemical and physiological studies using first and third trimester placenta. Hum Reprod 14 536 541
67. GenbacevO
SchubachSA
MillerRK
1992 Villous culture of first trimester human placenta–model to study extravillous trophoblast (EVT) differentiation. Placenta 13 439 461
68. GlomskiIJ
DecaturAL
PortnoyDA
2003 Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71 6754 6765
69. GenbacevO
McMasterMT
FisherSJ
2000 A repertoire of cell cycle regulators whose expression is coordinated with human cytotrophoblast differentiation. Am J Pathol 157 1337 1351
70. GenbacevO
ZhouY
LudlowJW
FisherSJ
1997 Regulation of human placental development by oxygen tension. Science 277 1669 1672
71. BarnesPD
BergmanMA
MecsasJ
IsbergRR
2006 Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med 203 1591 1601
72. KimKS
2008 Mechanisms of microbial traversal of the blood-brain barrier. Nat Rev Microbiol 6 625 634
73. BishopDK
HinrichsDJ
1987 Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol 139 2005 2009
74. SkobleJ
PortnoyDA
WelchMD
2000 Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J Cell Biol 150 527 538
75. ShenA
HigginsDE
2005 The 5′ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol 57 1460 1473
76. JonesS
PortnoyDA
1994 Intracellular growth of bacteria. Methods Enzymol 236 463 467
77. SundstromC
NilssonK
1976 Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int J Cancer 17 565 577
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils
- CD8+ T Cell Control of HIV—A Known Unknown
- Polyoma Virus-Induced Osteosarcomas in Inbred Strains of Mice: Host Determinants of Metastasis
- The Deadly Chytrid Fungus: A Story of an Emerging Pathogen