
Platelet-Activating Factor Receptor Plays a Role in Lung Injury and Death Caused by Influenza A in Mice
Influenza A virus causes annual epidemics which affect millions of people worldwide. A recent Influenza pandemic brought new awareness over the health impact of the disease. It is thought that a severe inflammatory response against the virus contributes to disease severity and death. Therefore, modulating the effects of inflammatory mediators may represent a new therapy against Influenza infection. Platelet activating factor (PAF) receptor (PAFR) deficient mice were used to evaluate the role of the gene in a model of experimental infection with Influenza A/WSN/33 H1N1 or a reassortant Influenza A H3N1 subtype. The following parameters were evaluated: lethality, cell recruitment to the airways, lung pathology, viral titers and cytokine levels in lungs. The PAFR antagonist PCA4248 was also used after the onset of flu symptoms. Absence or antagonism of PAFR caused significant protection against flu-associated lethality and lung injury. Protection was correlated with decreased neutrophil recruitment, lung edema, vascular permeability and injury. There was no increase of viral load and greater recruitment of NK1.1+ cells. Antibody responses were similar in WT and PAFR-deficient mice and animals were protected from re-infection. Influenza infection induces the enzyme that synthesizes PAF, lyso-PAF acetyltransferase, an effect linked to activation of TLR7/8. Therefore, it is suggested that PAFR is a disease-associated gene and plays an important role in driving neutrophil influx and lung damage after infection of mice with two subtypes of Influenza A. Further studies should investigate whether targeting PAFR may be useful to reduce lung pathology associated with Influenza A virus infection in humans.
Published in the journal:
. PLoS Pathog 6(11): e32767. doi:10.1371/journal.ppat.1001171
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001171
Summary
Influenza A virus causes annual epidemics which affect millions of people worldwide. A recent Influenza pandemic brought new awareness over the health impact of the disease. It is thought that a severe inflammatory response against the virus contributes to disease severity and death. Therefore, modulating the effects of inflammatory mediators may represent a new therapy against Influenza infection. Platelet activating factor (PAF) receptor (PAFR) deficient mice were used to evaluate the role of the gene in a model of experimental infection with Influenza A/WSN/33 H1N1 or a reassortant Influenza A H3N1 subtype. The following parameters were evaluated: lethality, cell recruitment to the airways, lung pathology, viral titers and cytokine levels in lungs. The PAFR antagonist PCA4248 was also used after the onset of flu symptoms. Absence or antagonism of PAFR caused significant protection against flu-associated lethality and lung injury. Protection was correlated with decreased neutrophil recruitment, lung edema, vascular permeability and injury. There was no increase of viral load and greater recruitment of NK1.1+ cells. Antibody responses were similar in WT and PAFR-deficient mice and animals were protected from re-infection. Influenza infection induces the enzyme that synthesizes PAF, lyso-PAF acetyltransferase, an effect linked to activation of TLR7/8. Therefore, it is suggested that PAFR is a disease-associated gene and plays an important role in driving neutrophil influx and lung damage after infection of mice with two subtypes of Influenza A. Further studies should investigate whether targeting PAFR may be useful to reduce lung pathology associated with Influenza A virus infection in humans.
Introduction
Influenza A viruses belong to the Orthomixoviridae family of RNA single-stranded, negative-sense viruses and cause epidemics, leading to about 250,000 to 500,000 deaths and 3 to 5 million severe cases annually worldwide [1], [2]. The new Influenza A H1N1 pandemic and the warning of a possible avian H5N1 pandemic increased the search for new vaccines and therapies [2], [3]. Antiviral drugs are an attractive possibility [4]. However, the need for the initiation of treatment very early in the course of infection [5] and the possibility of resistance suggest that novel alternatives are necessary [6]. A promising approach to reduce flu morbid is targeting immune molecules and cells related to disease severity (reviewed in [7]).
The immune system is activated shortly after respiratory epithelial cells have been infected by Influenza A. The single stranded RNA of Influenza virus is sensed inside endosomes by Toll like receptor 7 (TLR7) and TLR8 [8] and in cytoplasm by the helicase RIG-I (retinoic acid inducible gene-I) [9] and the inflammasome protein NLRP3 [10], [11]. TLR3 recognizes a intermediate of double strand RNA during Influenza replication [12]. The recognition of infection leads to alveolar macrophage recruitment and release of cytokines and chemokines with antiviral and proinflammatory actions (reviewed by [13]). NK cells are recruited in the first days of infection and are important for the initiation of adaptive immune responses against Influenza virus via IFN-γ production [14]. Neutrophils are another important leukocyte population involved in Influenza control [15]. Influenza A virus is a potent stimulus for neutrophil activation in the lungs and airways [16]. In addition to the antiviral action of neutrophils, excessive lung inflammation may result in lung damage, disruption of alveolar epithelial barrier and fluid leakage that limits respiratory capacity [17], [18].
Platelet Activating Factor (PAF) is a phospholipid mediator involved in many physiological and pathological conditions. The synthesis of PAF under inflammatory conditions is mediated by an acetyl-CoA:lyso-PAF acetyltransferase, named LysoPAFAT/LPCAT2 [19]. PAF acts through a single G protein-coupled receptor (PAFR) expressed in the plasma and nuclear membranes of leukocytes, endothelial cells and platelets [20]. Several inflammatory events have been associated with the administration of PAF, including increase of vascular permeability and lung edema [21], and leukocyte recruitment and activation [20], [22]. Moreover, blockade of the PAFR has been shown to decrease edema formation and/or leukocyte recruitment in several models of inflammation [23], [24], [25]. Phosphatidilcoline oxidation may also lead to the generation of PAF-like lipids that can activate the PAFR [26] and that have been reported to trigger lung injury after Influenza infection [27], [28]. Furthermore, upregulation of PAFR mRNA is seen during Influenza A virus infection [29], which is consistent with its expression on leukocytes and increase of these cells during Influenza A virus infection. Because of the involvement of PAFR during inflammatory responses and its expression during Influenza A virus infection, we hypothesized that PAFR activation may play an important role in driving pulmonary inflammation and injury in the context of Influenza A virus infection. To test our hypothesis, PAFR deficient mice or mice treated with PAFR antagonists were infected with two subtypes of Influenza A virus: a mouse-adapted Influenza virus A/WSN/33 H1N1 or a less virulent Influenza virus, H3N1. Our studies demonstrate that absence or antagonism of PAFR protects against Influenza A related lethality and inflammatory injury.
Results
Influenza A/WSN/33 H1N1-associated progressive weight loss, pulmonary inflammation and death
In order to determine the lethal inoculum of Influenza A/WSN/33 H1N1 virus in C57BL6/J mice, we infected the animals with four different inocula – 103, 104, 105 and 106 PFU – and accompanied them for 21 days after infection. Weight loss occurred over time after infection with 105 and 106 PFU and culminated with 100% death in 9 or 7 days, respectively. There was substantial weight loss in mice infected with 104 PFU until the eighth day of infection; thereafter, mice which survived (45%) recovered weight gradually. There were no weight loss and deaths associated with infection with 103 PFU (data not shown).
Based on the previous findings, we chose 104 as the mild inoculum and 106 PFU as the lethal inoculum to assess pulmonary inflammation associated with the infection. Since weight loss after infection with 104 PFU was slower, we chose to evaluate lung specimens at 3-day intervals from the first to the tenth day of infection. On the other hand, shorter 2-days intervals were chosen in the experiments with 106 PFU, from day one to five of infection.
We found substantial accumulation of neutrophils in BALF and in lung tissue, as assessed by myeloperoxidase (MPO) activity and histology, following infection (Figure 1). After 104 PFU, influx of neutrophils in the lungs (Fig 1A) and airways (Fig 1B) was first observed at day 4 after infection and returned to basal levels at day 10. When infected with the lethal inoculum (106 PFU), neutrophil infiltration in the lungs and BAL fluid was already detectable at day 1 and was more pronounced than with the lower inoculum (Figure 1 D,E). Pulmonary infiltration tended to resolve by day 5 after lethal infection. Consistent with the early peak of neutrophil accumulation at one day after infection with the lethal inoculum, mRNA expression of LPAFAT/LPAFAT2, the enzyme responsible for PAF synthesis in inflammatory conditions [19], was upregulated 2.5 fold at the first day of lethal infection (Fig 1 G).

Quantification of protein in BALF may be used as a marker of plasma leakage and, consequently, of lung injury [30]. There was progressive protein accumulation in the airways after infection with both inocula (Fig 1 C, F). Lung injury could also be observed by histological evaluation in animals infected with Influenza A (Fig 1 H, I). There was peribronchiolar and perivascular infiltration at 10 days after infection with 104 PFU, but, as seen by the normal thickness of alveoli, the infiltration was not present in the majority of alveolar walls (Fig 1H). Inflammation appeared to be more pronounced after infection with 106 PFU with significant peribronchial, perivascular and perialveolar inflammation, with thickening of alveolar walls, edema and mucus production on bronchioles at day 5 (Fig 1I).
There was a good correlation between the levels of CXCL1 and CXCL2 in the lungs (Fig S1A, B and Fig S2A, B) and the recruitment of neutrophils (Fig 1) after infection with 104 and 106 PFU. This is in agreement with studies showing the role of these chemokines and their receptors for neutrophil recruitment during Influenza A virus infection [31], [32]. Levels of the chemokine CCL2 were increased above baseline from day 4 after infection with 104 PFU and from day 1 after 106 PFU (Fig S1C, S2C). Thereafter, levels of CCL2 remained high throughout the observation period after infection with the lethal inoculum. Levels of the pro-inflammatory cytokine TNF-α were only increased at the beginning of the infection, i.e. at days 1 and 4 after 104 PFU and day 1 after 106 PFU (Fig S1D, S2D).
Decreased lethality rate and pulmonary injury in PAFR deficient mice infected with Influenza A/WSN/33 H1N1 virus
Aiming to evaluate a possible role of the inflammatory mediator PAF in Influenza A virus infection, PAFR and WT mice were infected with 104 PFU or 106 PFU of Influenza A/WSN/33 H1N1 virus. Infection of WT mice with 104 PFU resulted in 35% lethality rate. In contrast, only 7% of PAFR KO mice died after infection with the same inoculum (Fig 2A). Inoculation of 106 PFU caused 100% death by day 9 after infection of WT mice. In contrast, 23% PAFR KO infected with 106 were alive at day 21 after infection (Fig 2A).

As the absence of PAFR resulted in partial protection from Influenza A-associated lethality and in an attempt to seek for mechanisms of protection, we evaluated several parameters of inflammation in the lungs of mice after infection with 106 PFU. After infection with 106 PFU, there was decreased recruitment of total leukocytes (Fig 2B), mononuclear cells (Fig 2C) and neutrophils (Fig 2D) in the airways of PAFR KO mice when compared to WT mice. Neutrophil accumulation in lungs of WT and PAFR KO infected mice was similar at the evaluated time point (Fig 2E), as assessed by MPO quantification. The concentration of Evans' blue in BALF, a marker of vascular permeability, was reduced by approximately 35% in PAFR KO when compared to WT mice infected mice (Fig 2F). To examine in greater detail pulmonary changes and inflammation induced by infection with Influenza A, lung sections stained with H&E were analyzed and graded by a pathologist blind to the experimental situations. Leukocyte infiltration into bronchioles and alveoli, hyperemia, exudation, edema and bronchial mucus production were observed in most tissue sections from infected WT mice (Fig 3E, F). Despite of similar myeloperoxidase levels (Fig 2E) and neutrophilic infiltrates (Fig 3I) in lungs of infected groups, in PAFR KO mice, cellular infiltrates were restricted to conducting airways, in peribronchiolar and perivascular areas and did not reach distal airways like alveoli and the lung parenchyma, where gas exchange occurs (Fig 3G, H). Grading scores confirmed the limitation of infiltrates around the airways, the vessels and showed that these infiltrates were present in smaller areas of the parenchyma of PAFR KO infected mice – ranging from 1 to 29% of affected parenchyma in PAFR KO infected mice against 10 to 69% of affected parenchyma in WT infected mice (p<0.05, Fig 3I). Thus, the inflammatory infiltration in the larger airways was similar in WT and PAFR KO mice infected with the lethal inoculum; however, there was more infiltration of respiratory alveoli, with thickening of alveolar walls and pneumonitis in lungs of WT mice. Experiments in animals infected with 104 PFU showed decreased neutrophil influx and less protein leakage, hence less pulmonary injury, in PAFR KO mice when compared to WT infected mice after 8 days of infection (data not shown). Pulmonary inflammation after 5 days of infection with 104 PFU was discrete and restricted to neutrophil infiltration and compromised a percentage of lung parenchyma between 10 and 29%, which is much less severe than lesions developed after lethal infection. Pulmonary inflammation in PAFR KO mice infected with the low inoculum was also discrete and similar to that found in lungs of WT mice (data not shown).

Because there was a significant change in the pathological score, we conducted a more detailed analysis of leukocyte populations in BALF and in lungs of infected mice. In the airways, there was an increase in the number of all leukocyte populations evaluated at day 5 after infection – CD4+ T cells, CD8+ T cells, NKT cells, NK cells, macrophages and neutrophils. CD8+ and CD4+ lymphocytes, NK and NKT cells were equally increased in WT and PAFR KO infected mice (data not shown). Number of granulocytes (Fig 4A) and macrophages (Fig 4B) were increased after infection of WT mice but this increase was lower in PAFR KO mice infected with the same inoculum.

We performed Annexin V binding assay to evaluate whether increased apoptosis could account for the decreased accumulation of neutrophils and macrophages in the airways after flu infection of PAFR KO mice. Influenza A induced a marked increase in the number of apoptotic cells in the airways at day 5 after infection. Overall, the number of apoptotic cells was greater in infected WT than PAFR KO mice (Fig 4C). The number of apoptotic granulocytes was similar in both infected groups (Fig 4D). Therefore, the lower number of neutrophils in the airways of PAFR KO infected mice was due to decreased recruitment of these cells and not due to a higher degree of cellular death. In contrast, there was decreased number of apoptotic macrophages and lymphocytes in infected PAFR KO than in WT mice (Fig 4E, F).
Leukocyte populations were also evaluated in lung homogenates. The percentage of lung CD3+CD8+ T cells did not increase after infection (data not shown). CD3+CD4+ T cells increased after infection in WT but not in PAFR KO mice (Fig 4G). F4-80+ CCR5+ (macrophage) and GR1+ CXCR2+ (neutrophils) cells in lung homogenates increased after infection in both WT and PAFR mice to a similar extent (data not shown). NK populations (CD3+NK1.1+ and CD3 − NK1.1+) enhanced after infection in WT mice but the enhancement was significantly greater in PAFR KO mice (Fig 4H, I, J).
Levels of cytokines and chemokines after Influenza A virus infection in WT and PAFR KO mice
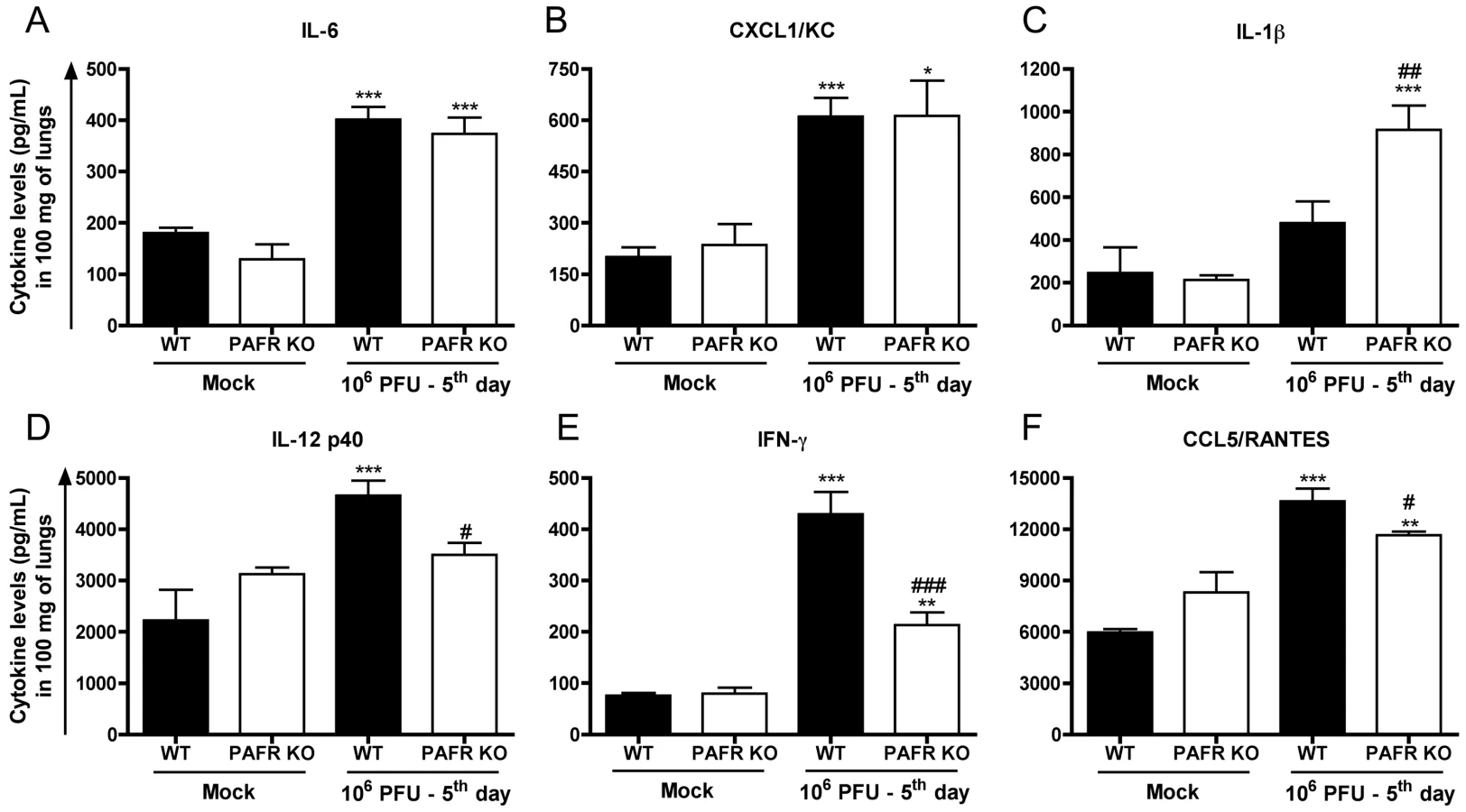
The concentrations of the cytokines IL-1β, IL-6, TNF-α, CXCL1, CXCL2, CCL5, IFN-γ and IL-12p40 were evaluated in lung homogenates of control and Influenza infected mice. As seen in Figure S2, levels of TNF-α and CXCL2 were not increased in the lungs at day 5 after infection with 106 PFU infection in both WT and PAFR KO groups (data not shown). Levels of IL-6 (Fig 5A) and CXCL1 (Fig 5B) were increased after infection but there was no difference between WT and PAFR KO mice. Levels of IL-1β were also enhanced by infection but the increase was more pronounced in PAFR KO than WT infected mice (Fig 5C). Levels of IL-12p40 (Fig 5D), IFN-γ (Fig 5E) and CCL5 (Fig 5F) increased after infection and the increase was more pronounced on WT that PAFR KO mice.
PAFR deficiency did not impair the ability of the immune system to deal with Influenza A/WSN/33 virus
Because pulmonary inflammation and levels of some cytokines known to be important for host resistance to viral infection, including IFN-γ and IL-12p40, were decreased in PAFR KO mice, we investigated whether this reduction was sufficient to impair viral clearance and adaptive responses. As seen in Figure 6A, there was no difference in viral load at day 5 in the lungs of WT and PAFR KO mice infected with 106 PFU (Fig 6A) or 104 PFU (Fig 6B). In animals infected with 104 PFU, viral load was significantly lower in PAFR KO than WT mice at day 8 after infection (Fig 6C).

Next we investigated humoral adaptive immune responses in the presence or the absence of PAFR. In a re-infection protocol, animals were initially infected with a non-lethal (103 PFU) inoculum of Influenza A/WSN/33 H1N1. After 14 days, animals were subjected to another infection with the same virus, at this time using the lethal inoculum, 106 PFU. No weight loss or lethality was observed in WT or PAFR KO mice using this protocol (data not shown). Measurement of anti-Influenza A WSN/33 IgG revealed no differences in antibody titer between the groups at day 21 after re-infection. The titer was 1/250 in both groups and the optical densities at that dilution are shown in Fig 6D.
Normal virus propagation in epithelial cells
Because there was decreased survival and pulmonary inflammation in PAFR-deficient mice and this was associated with decreased viral load after infection with the lower inoculum at day 8 after infection, we assessed whether virus propagation was altered by the absence of PAFR. To this end, we infected A549 cell line, a human alveolar basal epithelial cell, with Influenza A virus expressing red fluorescent protein (RFP). Using this methodology, we found no difference in the ability of influenza virus to infect epithelial cells in vitro in the absence or presence of a PAFR antagonist PCA 4248 (50 µM) (data not shown).
We also measured the height of bronchiolar epithelium after infection with 106 PFU to determine whether infection of epithelial cells, the primary target of influenza infection, was similar in the absence or presence of PAFR. Lethal infection promoted pronounced reduction in epithelial height of bronchiole and absence of PAFR did not alter the susceptibility of epithelial injury (Fig S3). Therefore, virus entry and replication and subsequent cell injury in its primary target, epithelial cells, is not affected when PAFR is absent.
Activation of TLR7/8 but not TLR3 and NLRP3 was dependent of PAFR
TLR3, TLR7/8 and NALP3 are thought to be major intracellular recognition molecules used by the host to detect Influenza A infection [14]. To investigate whether these activation of these pathways could lead to inflammation in a PAFR-dependent manner, we studied the effects of synthetic agonists of TLR7/8 (R848) and TLR3 and NLRP3 [poly(I∶C)] in WT and PAFR KO mice.
Intratracheal instillation of R848, a TLR7/8 agonist, induced LPAFAT/LPAFAT2 mRNA in lungs of WT mice (Fig 7A). This was accompanied by infiltration of neutrophils in lungs (Fig 7B) and airways (Fig 7C), and increased levels of IL-12p40 (Fig 7D), IL-6 (Fig 7E) and CXCL1 (Fig 7F) in lungs of WT mice. In PAFR KO mice, R848 induced similar amount of neutrophil influx in lungs (Fig 7B), but greatly reduced neutrophil recruitment to the airways (Fig 7C). Levels of IL-12p40 (Fig 7D) and CXCL1 (Fig 7F) were reduced in lungs of PAFR-KO mice instilled with R848 when compared to WT.

Poly(I∶C) stimulation did not induce expression of LPAFAT/LPAFAT2 mRNA in WT mice (Fig S4A). Instillation of poly(I∶C) induced neutrophil influx in the airways (Fig S4C), but not lungs (Fig S4B) and CXCL1 production in lungs (Fig S4D) of WT mice, a response that was similar or slightly greater in PAFR KO mice.
Treatment with a PAFR antagonist protected against Influenza A/WSN/33 H1N1 infection
To test the potential use of PAFR as a pharmacological target against Influenza A-associated disease, we used the selective PAFR antagonist PCA 4248 in a therapeutic protocol. The treatment was started 3 days after infection of mice with 104 or 106 PFU and was continued until day 10. Day 3 was chosen because this is the time at which weight loss and neutrophil influx peaked in the higher inoculum (Fig 1B, G). Treatment with PCA 4248 significantly enhanced survival after infection with 106 PFU of Influenza A/WSN/33 H1N1 virus (Fig 8A). Akin to the experiments in PAFR KO mice, higher survival was accompanied by reduction in total leukocyte (Fig 8B), mononuclear cells (Fig 8C) and neutrophil (Fig 8D) accumulation in the airways. Neutrophilic accumulation in lung parenchyma after Influenza infection was not affected by PCA 4248 treatment (Fig 8E) as observed in PAFR KO (Fig. 2E). Plasma leakage in the airways, as assessed by measuring total protein in BAL was significantly lower in PCA 4248-treated than vehicle-treated animals (Fig 8F).

PCA treatment failed to alter significantly (from 55% to 90% survival, p = 0.10) the lethality rate caused by 104 PFU infection (Fig 8A), but there was a significant decrease in weight loss from the ninth to the eleventh day of infection, when compared to the vehicle-treated group (Table 1).

PAFR deficient mice were protected from infection caused by another subtype - Influenza A H3N1
To assess whether the protection observed in PAFR deficient mice and WT mice treated with a PAFR antagonist infected with Influenza A H1N1 was virus-specific, we used another virus strain, a reassortant Influenza A H3N1 subtype to infect WT and PAFR KO mice. Since mouse infection with different Influenza virus subtypes requires adaptation [33], H3N1 virus was subjected to three lung passages in C57BL/6J in order to cause disease in these animals. After three lung passages the virus was able to multiply and increased viral titer (data not shown), a sign of adaptation. As a result, the inoculum of 106 PFU of H3N1 virus was enough to cause 15% of weight loss on average after three days of infection. PAFR KO mice were protected from the disease caused by Influenza A H3N1, since they start to regain weight, from day ten of infection, faster than WT animals (Fig 9).

Discussion
Severe inflammation caused by highly pathogenic Influenza A strains was described to be an important cause of death during 1918 pandemics and highly pathogenic avian Influenza H5N1 infections [34]. Using a mouse-adapted Influenza A/WSN/33 H1N1 strain we show a correlation between viral load, pathogenesis and lethality – i.e. the higher the viral load, there is greater lung injury and death. More importantly, the present work demonstrates that the course of Influenza A virus infection is less severe in the absence of PAFR. Mechanistically, it appears that activation of TLR7/8 by Influenza A explains the induction of LPAFAT/LPAFAT2 mRNA and consequent activation of PAFR. Further, absence or blockade of PAFR during infection is associated with decreased neutrophil and macrophage influx into airspaces, decreased production of certain pro-inflammatory mediators and decreased lung edema, parameters which are commonly increased after pulmonary administration of PAF to rodents or humans [35], [36]. Lack of PAFR did not increase viral load or prevent specific anti-Influenza antibody production. Finally, we demonstrate that administration of PAFR antagonist 3 days after infection also causes similar protection as observed in PAFR-deficient mice.
Neutrophils and neutrophil-active chemokines peaked very early in the course of Influenza A/WSN/33 H1N1 infection and decreased thereafter. On the contrary, lung edema and injury was progressive. Thus, it seems that inflammation is self-resolving but causes lung damage which is more pronounced in the last days of infection, leading to progressive weight loss and death. As inflammation is important to trigger lung injury in Influenza A infected mice and PAF attracts neutrophils into the lung [36], we evaluated whether the receptor for PAF was involved in H1N1-associated lung inflammation and injury, and death. A previous study demonstrated up-regulation of PAFR mRNA in lungs following Influenza A/PR8/34 H1N1 infection [29]. This is consistent with the expression of PAFR on neutrophils and other leukocytes [20] and the influx of these cell types during infection. In our experiments, we found increased expression of the enzyme involved in inflammatory synthesis of PAF – LPAFAT/LPAFAT2 – in the first day of lethal infection. Therefore, the release of PAF is an early event that could be involved in the pathogenesis of Influenza A infection. In fact, PAFR deficient mice or mice treated with PAFR antagonist PCA 4248 were protected from death or weight loss caused by Influenza A virus infection. PAFR signaling was also important for pulmonary inflammation and injury.
Neutrophils are required for the clearance of Influenza virus during the early stages of infection [37]. The antiviral actions of neutrophils are clearly demonstrated through the use of RB6-8C5 antibodies to deplete these cells. In the absence of neutrophils, mice are more susceptible to virus growth and associated lethality [15], [38]. Therefore, direct ablation of neutrophils cannot be used therapeutically. On the other hand, Influenza A virus is known to be a potent activator of neutrophil respiratory burst, apoptosis and subsequent deactivation in front of a second stimulus [16]. The neutrophil activation that occurs during infection, despite its role in controlling viral replication, is thought to be very harmful to the host [37]. PAFR deficiency or antagonism increased survival after Influenza A virus infection and this was associated with lower neutrophil recruitment to the airways. There was, however, no inhibition of neutrophil into pulmonary parenchyma. Therefore, reduction instead of ablation of neutrophil recruitment into the airways appears to be an approach which is sufficient to maintain control of viral burden, but, at the same time, avoid excessive neutrophil activation and lung damage.
Bacterial pneumonia, especially by Streptococcus pneumonia, is an important complication following Influenza A virus infection [39]. Pro-apoptotic effects of Influenza A virus on neutrophils are one of the explanations for the increased susceptibility to bacterial pneumonia after Influenza infection [40]. In our model, absence of PAFR did not influence neutrophil apoptosis. The role of PAFR in altering bacterial pneumonia after Influenza infection is controversial. While the study of van der Sluijs and co-workers described protection in PAFR KO mice after Streptococcus pneumoniae following flu infection [29], McCullers and colleagues showed no correlation between the increased pathology after secondary infection and the antagonism [41] or absence of PAFR [42]. Both groups focused on lethality and harm caused by the bacteria, not the virus, and the overall message was that absence or blockade of PAFR was either without effect or protective, but never harmful. Here, we show that targeting PAFR during Influenza A virus infection is protective against viral-induced pneumonia, regardless of the risk of bacterial pneumonia after viral infection.
In addition to neutrophils, endothelial cells express PAFR and are affected by PAFR signaling [22]. Protein leakage to the airways reflects the increased vascular permeability following inflammatory events associated with Influenza A virus infection [30]. In our system, lower protein amounts or Evans' blue extravasation were found in BALF of PCA treated or PAFR KO animals. As mentioned above, neutrophil accumulation in the parenchyma was not affected by PAFR absence or antagonism. Thus, neutrophil transmigration into the alveolar space seems to play an important role in the pathogenesis of Influenza infection. During the transmigration, neutrophils may release proteinases or oxygen reactive species that lead to increase in vascular permeability that is accompanied by significant protein leakage [43]. Hence, lower protein amounts found in lethally infected mice treated with PCA or PAFR KO mice infected with lower infection inoculum compared with their controls corroborates with this hypothesis.
Pulmonary inflammation and injury are common histopathological findings in humans with mild or severe Influenza A infection. Viruses of low pathogenicity affect basically the proximal airways (bronchi and bronchioles), whereas highly pathogenic viruses or infection of immunocompromised people infected is associated with inflammation and injury in the distal airways (alveoli and parenchyma). When distal airways are affected, lung injury is more severe and lead to loss of respiratory capacity and gas exchange [44]. In our system, the extent of distal airways involvement was decreased in PAFR KO mice, suggesting these results may bear relevance to humans with severe Influenza A infection. Evaluation of gas exchange in our mice was not possible as animals needed to be anesthetized for the procedure (data not shown).
NK cells are recruited to the lungs very early in the course of Influenza infection, are involved in initial viral clearance and provide initial signals to the development of protective adaptive responses [45]. NK cells recognize Influenza A virus through its NKp46 receptor and, stimulated by IL-12 released from dendritic cells, mediate cytotoxicity to infected cells and release of IFN-γ that, in turn, mediates adaptive responses [46]. PAF seems to be important for cytotoxic functions of NK cells [47], [48] and PAFR antagonists reduced cytotoxic activity of NK cells [47]. Jin and colleagues showed that inflammation-released PAF recruits NK cells activated by IL-2, IL-12, IL-15 and IFN-α [48]. Despite these studies showing a potential effect of PAF on NK cell function and recruitment, we actually observed that NK cell recruitment to the lungs following Influenza A virus infection was increased in the absence of PAFR. Our studies do not provide a mechanism to this particular finding but do suggest that enhanced NK cell recruitment could be functionally relevant as seen by lower viral loads in the lung in some of the experiments.
In the absence of PAFR, there were reduced pulmonary levels of IFN-γ, IL-12p40 and CCL5. The reduction in these cytokines which are preferentially produced by or activate Th1 lymphocytes is related to the reduced number of CD4+ T cells in the lungs of PAFR-deficient mice infected with influenza A. These results are consistent with previous experiments in mice infected with a different microorganism, Leishmania amazonensis, where we also demonstrated that absence of PAFR decreased CCL5 expression and Th1-associated function [49]. However, in the case of the protozoan infection, the absence of PAFR was deleterious, because of an inability of the host to control the infection [49]. These results suggest that PAF may be important regulator of CCL5 production and consequent infiltration of effectors T cells with Th1 phenotype in vivo. Despite decreased IFN-γ expression and decreased accumulation of CD4+ T cells in the lungs of infected PAFR-deficient mice, viral load in the lung was decreased or at worst similar to that found in WT mice. Reduction of viral load was not associated with decreased propagation in epithelial cells in vitro. Furthermore, the capacity of Influenza virus to cause epithelial cell injury in vivo was unchanged by absence of PAFR. It is unclear why there was a small decrease in viral load in the lungs on day 8 after infection with the low inoculum. It is possible that this may reflect the better clinical status of animals and the decreased pulmonary inflammation in the absence of PAFR. Absence of PAFR was not associated with a decreased specific antibody release, as shown by re-infection studies and measurement of specific IgG titers. Therefore, changes in CD4+ T cell recruitment and related cytokines were not sufficient to modify negatively the course of infection in this model of experimental Influenza A virus infection.
Macrophages are also recruited to the lungs and airways after Influenza infection where they are thought to play an important role in the production of pro-inflammatory cytokines and in the phagocytosis of Influenza virus-induced apoptotic cells [50]. In vivo abrogation of phagocytosis of apoptotic infected cells by macrophages increases lethality rates in Influenza A infected mice [51]. We found a partial reduction of macrophage recruitment to the airways in PAFR deficient mice infected with Influenza A. There was also, a proportional reduction in number of apoptotic macrophages in PAFR deficient mice, showing that the proportion of live active macrophages was similar in both groups. These macrophages appear to be sufficient for clearance of apoptotic infected cells and helping in the resolution of the inflammatory phase of the infection [52], as seen by the reduced lung damage and survival in infected PAFR-deficient mice.
The suggestion that macrophage function in infected PAFR-deficient mice was sufficient to keep immune responsiveness is strengthened by findings that IL-6 expression was maintained and IL-1β expression was actually increased in these mice. IL-1β is produced mainly by macrophages after infection with Influenza A [13]. The role of IL-1β for viral clearance and pathology during Influenza infection is very complex [53]. Indeed, a previous study has shown that IL-1R1 deficient mice had greater lethality rates, but decreased lung damage caused by Influenza A virus infection [53]. Maines and colleagues showed a clear inverse correlation between viral virulence and IL-1β release in lungs [54]. While highly virulent H5N1 virus induces a small increase in IL-1β secretion, low virulent H5N1 stimulates high levels of this cytokine [54]. Therefore, the cytokine IL-1β does appear to contribute to viral clearance, even at the cost of increasing pulmonary recruitment of leukocytes in some models. In our system, enhanced release of IL-1β in PAFR-deficient mice was associated with maintained or decreased viral load, but no enhancement of leukocyte infiltration or pathology. The inability of IL-1β to enhance inflammation in the system may be explained by the role of PAFR in mediating IL-1β-associated inflammation [55], [56].
The recognition of Influenza virus RNA is the main trigger of antiviral and inflammatory responses [14]. Using the synthetic TLR agonist R848, we found that activation of this ssRNA sensor induces the expression of LPAFAT/LPAFAT2 and inflammation that is directly dependent of PAFR. It has been previously shown that LPAFAT/LPAFAT2 expression and enzyme activity may be induced by the TLR9 ligand ODN1826 and the TLR4 ligand LPS [19]. Poly(I∶C), a classical TLR3 ligand and recently described as a NLRP3 activator [10], did not induce LPAFAT/LPAFAT2 mRNA expression and cause pulmonary inflammation which was PAFR-independent. Therefore, of the known major recognition receptors for Influenza, it appears that TLR7/8 is the one capable of inducing LPAFAT/LPAFAT2 expression and triggers pulmonary inflammation in PAFR-dependent manner. Results with R848 were qualitatively similar (LPAFAT/LPAFAT2 mRNA expression, PAFR-dependency of inflammation) to that observed after Influenza infection, suggesting that activation of TLR7/8 is a main mechanism by which Influenza infection leads PAF release and PAFR activation. One alternative possibility to explain the activation of PAFR in our system derives from recent findings that oxidized phospholipids (Ox-PL) were produced during acute lung injury induced by inactivated H5N1 [27]. Ox-PL are PAF-like molecules known to induce PMN migration and protein leakage in pleural cavity, effects which are PAFR-dependent [57]. Ox-PLs levels were also correlated with the reduced severity of flu in IL-17RA KO mice infected with Influenza A/PR/8/34 (H1N1) [28]. Thus, production of Ox-PL may contribute to the activation of PAFR during influenza infection.
The present model of Influenza A/WSN/33 H1N1 infection mimics infections with highly pathogenic strains during pandemics or in immunocompromised people. We used a low inoculum of H1N1 and a low pathogenic Influenza A strain to model the common features of the seasonal flu in humans. Using a low pathogenic Influenza A H3N1 reassortant virus, we showed that PAFR deficient mice had decreased weight loss in comparison to WT mice. Therefore, PAF may be important to the pathogenesis of Influenza A virus infection, regardless of subtype or strain. This feature could be explained because PAFR targeting modulates the inflammatory response to the virus, without affecting, or also improving viral clearance by the host. Recently, our group published similar results in a murine model of Dengue virus infection. In that work, the absence or pharmacological blockade of PAFR resulted in protection against the main symptoms of dengue virus infection and lethality, regardless of viral titer change [58].
In conclusion, our studies clearly show that PAFR-mediated inflammatory events that follow Influenza A virus infection are important for disease pathogenesis and lethality. Mechanistically, protection in PAFR deficient mice was associated with decreased infiltration of neutrophils and macrophages into the airways and decreased lung damage. Importantly, PAFR deficiency tended to enhance the ability of the murine host to deal with the virus and antibody and adaptive response were maintained. Importantly, treatment with PAFR antagonists starting 3 days after infection also protected against Influenza A morbidity and lethality. These studies show that PAFR is a disease-associated gene after Influenza A virus infection and suggest that PAFR antagonism could be a useful therapeutic target to interfere with inflammatory damage that follows infection. It remains to be determined whether this will be a useful therapeutic strategy in humans and whether association with antiviral will enhance benefits provided by each individual strategy, as suggested elsewhere [59].
Methods
Ethics statement
All the experiments were conducted under prior CETEA/UFMG animal ethics committee approval (203/08), according to Brazilian guidelines on animal work.
Virus strains and culture
The strains used in the study were Influenza A WSN/33 H1N1 and Influenza A (H3 of equine/Cordoba/18/1985 and N1 of Yamagata/32/1989) H3N1. Briefly, Influenza A WSN/33 H1N1 was produced in chicken eggs and passed once more in eggs and then cultured in MDCK (Madin-Darby Canine Kidney) cells. The stocks in a final concentration between 4×107 and 2×108 PFU/mL were diluted in sterile phosphate buffered saline prior to infections.
Influenza A H3N1 strain was adapted to mice through three lung passages. Briefly, 104 PFU of Influenza A H3N1virus was inoculated via intranasal in five animals and after 5 days lungs were collected an tittered. The sample that had the higher titer (105 PFU) was passed through a 0.45 µm filter and used to infect another 5 animals (100 PFU/animal). The process of selection was repeated once and the final titer achieved was 5×107 PFU. The stock was diluted in sterile phosphate buffered saline prior to infections.
Animal infections
Male 8–10 weeks C57BL/6J and PAFR deficient mice (PAFR KO) on a C57BL/6J background, maintained in pathogen free conditions at Laboratorio de Imunofarmacologia (UFMG/Brazil) facilities, were used in the infection experiments. Mice anesthetized with ketamine/xylazine received 25µL of Influenza A/WSN/33 H1N1 virus, Influenza A H3N1virus, or sterile phosphate buffered saline (PBS, Mock group), intranasally. The H3N1 virus was previously adapted to mice, through three lung passages as described above.
Drug treatment
The PAFR antagonist PCA 4248 (Tocris Bioscience), 5mg/kg, diluted in 5% of ethanol in PBS was given twice a day, via subcutaneous injections. The control group (vehicle) received the same volume of the solution used to dilute PCA 4248.
In vitro infections
MDCK and the human lung epithelial cell line A549 (ATCC CCL-185) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 6% of fetal bovine serum were cultured at 37°C with 5% of CO2. Cells were seeded at a density of 4×104 cells/well in a 24-well plate and after 24 hours of growth were incubated with 50 µM of PCA 4248 in DMEM or vehicle for 30 minutes. Cells were infected with a multiplicity of infection of 2 with a red fluorescent protein (RFP) labeled Influenza A virus and incubated for 16 hours. Virus propagation was observed through a fluorescence microscopy.
Experimental design
Influenza A infection
Wild Type (WT) mice were infected with distinct Influenza A/WSN/33 H1N1 inocula. PAFR KO and WT mice, PCA 4248 or vehicle treated were infected with lethal (106 PFU) and lower infection (104 PFU) inocula. Mice were monitored by weight loss and lethality for 21 days or sacrificed after indicated days of infection to have cellular recruitment to the airways and to the lungs evaluated, as well as cytokine and chemokine production, and lung injury and viral load assessment. Animals which lost more than 30% of their initial weight were killed; however, the maximum weight loss observed was lower than 30%. LysoPAFAT/LPCAT2 gene expression was assessed in lungs of WT mice infected with the lethal inoculum. WT and PAFR KO mice were infected with the non-lethal inoculum of 103 PFU of Influenza A/WSN/33 H1N1, monitored for 14 days and reinfected with the lethal inoculum of the same virus. After 21 days, reinfected mice were killed and serum samples were used to measure specific IgG titers. Finally, WT and PAFR KO mice were infected with Influenza A (equine/Cordoba/18/1985 - Yamagata/32/1989) H3N1 and monitored for 21 days.
TLR7/8, TLR3 and NLRP3 activation
Anesthetized WT and PAFR KO mice were instilled intratracheally with 40µL containing a sterile PBS with 50 µg of R848 or poly(I∶C) (Invivogen, San Diego, USA), or saline only (controls) and killed at 4 (R848) or 8 [poly(I∶C)] h after instillation. Lungs were washed and removed for evaluation of cell, MPO levels, and cytokine and chemokine production. The lower lobe of the left lung of WT animals was excised for the evaluation of LysoPAFAT/LPCAT2 gene expression by Real Time PCR.
Bronchoalveolar lavage (BAL)
At indicated time points, mice were euthanized with an overdose of ketamine/xylazine solution. Subsequently, a 1.7mm catheter was inserted into the trachea and two 1mL aliquots of PBS were inserted into the lungs and collected three times to acquire leukocytes recruited to the airway space. After centrifugation, the pellet was used to total and differential leukocytes counts of stained slides.
Assessment of pulmonary vascular leakage by Evans blue
At day 5 after infection with 106 PFU of Influenza A WSN/33 or PBS instillation, WT and PAFR were injected with Evans blue dye (50 mg/kg, i.v.) 2 h before they were killed. Animals were subjected to BAL with 1mL of PBS. BALF was then centrifuged and the optical density was determined at 620 nm. The concentration of extravasated EBD (microgram of EBD per gram lung) in lung homogenates was calculated against a standard curve.
Lung myeloperoxidase measurement
After performing BAL, lungs were perfused with 5 mL of PBS to remove circulating blood and frozen. A hundred µl of tissue was homogenized to perform ELISA and MPO assay, as previously described [60].
Measurement of cytokines and chemokines
Lung tissues were homogenized in a PBS-buffer containing antiproteases, as previously described [60], to assess the concentrations of the cytokines IL-1β, IL-6, IL-12 p40, IFN-γ and TNF-α and the chemokines CXCL1, CXCL2, CCL2, CCL5 and serum IL-6 levels by ELISA DuoSet kits (R&D Systems), in accordance to the manufacturer's instructions.
Real time PCR
Total RNA from diaphragmatic lung lobe tissue conserved at −70°C in RNA later (Applied Biosystems, California, USA) was extracted using Trizol (Invitrogen), as described by the manufacturer. The total RNA obtained was suspended in RNAse-free water and stocked at −70°C. Real-time PCR was performed on an ABI PRISM Step-One sequence-detection system (Applied Biosystems) by using SYBR Green PCR Master Mix (Applied Biosystems) after a reverse transcription reaction of 2 µg of RNA by using M-MLV reverse transcriptase (Promega). The relative expression level of LysoPAFAT/LPCAT2 gene was determined by the 2 (−delta delta Ct) method, normalized by ribosomal subunit 18S and expressed as fold change compared with constitutive gene expression. The following primer pairs were used: LysoPAFAT/LPCAT2 forward 5′ GTCCAGCAGACTACGATCAGTG 3′; LysoPAFAT/LPCAT2 reverse 5′ CTTATTGGATGGGTCAGCTTTTC 3′ as described by [19]; and 18S forward 5′ CTCAACACGGGAAACCTCAC 3′; 18S reverse 5′ CGTTCCACCAACTAAGAACG 3′.
Assessment of lung injury and histological analysis
We analyzed lung injury following Influenza A virus infection assessing protein leakage to the airways, histological changes in lung architecture. A protein quantification assay (Bio-Rad Protein Assay) was performed in bronchoalveolar lavage fluid (BALF) supernatant according to manufacturer's instructions. Formalin-fixed lung left lobes were dehydrated gradually in ethanol, embedded in paraffin, cut into 4-µm sections, stained with H&E and examined under light microscopy and scored by a pathologist blinded to the experiment. The score of 18 points was based on Horvat and colleagues paper [61], which evaluates airway, vascular and parenchymal inflammation, added to a 5 points score evaluating general neutrophilic infiltration (0, absent; 1, minimal; 2, slight; 3, moderate; 4, marked; and 5, severe).
Photographs of areas containing bronchioles in H&E stained slides were taken under 200 fold of magnification and were analyzed with an AxioVision software. Epithelial height of bronchioles in areas of inflammatory infiltrates had their length measured. The number of bronchioles analyzed in each slide varied according to their length, to totalize 1500 µm per slide. The mean epithelial height for each animal was used to construct the graph.
Flow-cytometric analysis of lung and airway leukocyte populations
Leukocytes recovered from BALF and from lungs processed with collagenase IV (Sigma) [62] were stained with fluorescent-labeled monoclonal antibodies CD3, CD4, CD8, NK1.1 and GR1 (BD Pharmigen TM); CXCR2 (R&D Systems); F4/80 and CCR5 (Biolegend). Stained cells were acquired in FACScan cytometer and analyzed in FlowJo (Tree Star) software (Text S1).
Apoptosis assay
We performed Annexin V binding assay on BALF leukocytes using Annexin V/Propidium Iodide (PI) kit (Caltag Laboratories) according to manufacturer's instructions. Flow cytometry was carried out in FACScan and analyzed in FlowJo software.
Plaque assay
In order to determine viral load in lungs, we collected the organs in sterile conditions and performed a plaque assay using MDCK cells. Lungs were weighted and homogenized in PBS, plated in MDCK monolayer and after incubation and staining it was possible to count the plaques. The viral titer was expresses as Plaque Forming Units (PFU) per gram of tissue.
Antibody quantification
WSN/33 H1N1 influenza virus stocks were used as antigens in an indirect ELISA to detect specific antibodies in serum samples of reinfected animals (Text S1).
Statistical analysis
All data are presented as the mean ± SEM. All data were tested for normality and found to have a normal distribution. Normal data were tested for significance using ANOVA followed by use of Newman-Keuls post-test, which corrects for multiple comparisons. Unpaired t test was used to compare two groups and Log-rank test for lethality experiments, where appropriate. Statistical significance was set as P<0.05 and all graphs and analysis were performed using Graph Pad Prism 4 software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BouvierNM
PaleseP
2008 The biology of influenza viruses. Vaccine 26 Suppl 4 D49 53
2. MichaelisM
DoerrHW
CinatlJJr
2009 Novel swine-origin influenza A virus in humans: another pandemic knocking at the door. Med Microbiol Immunol 198 175 183
3. WhiteNJ
WebsterRG
GovorkovaEA
UyekiTM
2009 What is the optimal therapy for patients with H5N1 influenza? PLoS Med 6 e1000091
4. RufBR
SzucsT
2009 Reducing the burden of influenza-associated complications with antiviral therapy. Infection 37 186 196
5. AokiFY
MacleodMD
PaggiaroP
CarewiczO
El SawyA
2003 Early administration of oral oseltamivir increases the benefits of influenza treatment. J Antimicrob Chemother 51 123 129
6. KisoM
MitamuraK
Sakai-TagawaY
ShiraishiK
KawakamiC
2004 Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 364 759 765
7. FedsonDS
2009 Confronting the next influenza pandemic with anti-inflammatory and immunomodulatory agents: why they are needed and how they might work. Influenza Other Respi Viruses 3 129 142
8. WangJP
BowenGN
PaddenC
CernyA
FinbergRW
2008 Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood 112 2028 2034
9. OpitzB
RejaibiA
DauberB
EckhardJ
VinzingM
2007 IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol 9 930 938
10. AllenIC
ScullMA
MooreCB
HollEK
McElvania-TeKippeE
2009 The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30 556 565
11. ThomasPG
DashP
AldridgeJRJr
EllebedyAH
ReynoldsC
2009 The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30 566 575
12. Le GofficR
PothlichetJ
VitourD
FujitaT
MeursE
2007 Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178 3368 3372
13. JulkunenI
MelenK
NyqvistM
PirhonenJ
SarenevaT
2000 Inflammatory responses in influenza A virus infection. Vaccine 19 Suppl 1 S32 37
14. KohlmeierJE
WoodlandDL
2009 Immunity to respiratory viruses. Annu Rev Immunol 27 61 82
15. FujisawaH
2008 Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J Virol 82 2772 2783
16. HartshornKL
LiouLS
WhiteMR
KazhdanMM
TauberJL
1995 Neutrophil deactivation by influenza A virus. Role of hemagglutinin binding to specific sialic acid-bearing cellular proteins. J Immunol 154 3952 3960
17. TuvimMJ
EvansSE
ClementCG
DickeyBF
GilbertBE
2009 Augmented lung inflammation protects against influenza A pneumonia. PLoS One 4 e4176
18. ChenXJ
SethS
YueG
KamatP
CompansRW
2004 Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287 L366 373
19. ShindouH
HishikawaD
NakanishiH
HarayamaT
IshiiS
2007 A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem 282 6532 6539
20. IshiiS
ShimizuT
2000 Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res 39 41 82
21. UhligS
GoggelR
EngelS
2005 Mechanisms of platelet-activating factor (PAF)-mediated responses in the lung. Pharmacol Rep 57 Suppl 206 221
22. MontrucchioG
AlloattiG
CamussiG
2000 Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev 80 1669 1699
23. SouzaDG
PinhoV
SoaresAC
ShimizuT
IshiiS
2003 Role of PAF receptors during intestinal ischemia and reperfusion injury. A comparative study between PAF receptor-deficient mice and PAF receptor antagonist treatment. Br J Pharmacol 139 733 740
24. BedirliA
GokahmetogluS
SakrakO
SoyuerI
InceO
2004 Beneficial effects of recombinant platelet-activating factor acetylhydrolase and BN 52021 on bacterial translocation in cerulein-induced pancreatitis. Eur Surg Res 36 136 141
25. LandgrafRG
NossiDF
SiroisP
JancarS
2007 Prostaglandins, leukotrienes and PAF selectively modulate lymphocyte subset and eosinophil infiltration into the airways in a murine model of asthma. Prostaglandins Leukot Essent Fatty Acids 77 163 172
26. PrescottSM
ZimmermanGA
StafforiniDM
McIntyreTM
2000 Platelet-activating factor and related lipid mediators. Annu Rev Biochem 69 419 445
27. ImaiY
KubaK
NeelyGG
Yaghubian-MalhamiR
PerkmannT
2008 Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133 235 249
28. CroweCR
ChenK
PociaskDA
AlcornJF
KrivichC
2009 Critical role of IL-17RA in immunopathology of influenza infection. J Immunol 183 5301 5310
29. van der SluijsKF
van EldenLJ
NijhuisM
SchuurmanR
FlorquinS
2006 Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol 290 L194 199
30. LinKL
SuzukiY
NakanoH
RamsburgE
GunnMD
2008 CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180 2562 2572
31. SakaiS
KawamataH
MantaniN
KogureT
ShimadaY
2000 Therapeutic effect of anti-macrophage inflammatory protein 2 antibody on influenza virus-induced pneumonia in mice. J Virol 74 2472 2476
32. SekiM
KohnoS
NewsteadMW
ZengX
BhanU
Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J Immunol 184 1410 1418
33. BarnardDL
2009 Animal models for the study of influenza pathogenesis and therapy. Antiviral Res 82 A110 122
34. PerroneLA
PlowdenJK
Garcia-SastreA
KatzJM
TumpeyTM
2008 H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4 e1000115
35. LeeYM
HybertsonBM
ChoHG
RepineJE
2002 Platelet-activating factor induces lung inflammation and leak in rats: hydrogen peroxide production along neutrophil-lung endothelial cell interfaces. J Lab Clin Med 140 312 319
36. GabrijelcicJ
AcunaA
ProfitaM
PaternoA
ChungKF
2003 Neutrophil airway influx by platelet-activating factor in asthma: role of adhesion molecules and LTB4 expression. Eur Respir J 22 290 297
37. WhiteMR
CrouchE
VesonaJ
TackenPJ
BatenburgJJ
2005 Respiratory innate immune proteins differentially modulate the neutrophil respiratory burst response to influenza A virus. Am J Physiol Lung Cell Mol Physiol 289 L606 616
38. TumpeyTM
Garcia-SastreA
TaubenbergerJK
PaleseP
SwayneDE
2005 Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79 14933 14944
39. McNameeLA
HarmsenAG
2006 Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun 74 6707 6721
40. ColamussiML
WhiteMR
CrouchE
HartshornKL
1999 Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 93 2395 2403
41. McCullersJA
RehgJE
2002 Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis 186 341 350
42. McCullersJA
IversonAR
McKeonR
MurrayPJ
2008 The platelet activating factor receptor is not required for exacerbation of bacterial pneumonia following influenza. Scand J Infect Dis 40 11 17
43. ChignardM
BalloyV
2000 Neutrophil recruitment and increased permeability during acute lung injury induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol 279 L1083 1090
44. BruderD
SrikiatkhachornA
EnelowRI
2006 Cellular immunity and lung injury in respiratory virus infection. Viral Immunol 19 147 155
45. CulleyFJ
2009 Natural killer cells in infection and inflammation of the lung. Immunology 128 151 163
46. MandelboimO
LiebermanN
LevM
PaulL
ArnonTI
2001 Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409 1055 1060
47. MandiY
FarkasG
KoltaiM
BeladiI
Mencia-HuertaJM
1989 The effect of the platelet-activating factor antagonist, BN 52021, on human natural killer cell-mediated cytotoxicity. Immunology 67 370 374
48. JinY
DamajBB
MaghazachiAA
2005 Human resting CD16−, CD16+ and IL-2-, IL-12-, IL-15 - or IFN-alpha-activated natural killer cells differentially respond to sphingosylphosphorylcholine, lysophosphatidylcholine and platelet-activating factor. Eur J Immunol 35 2699 2708
49. SantiagoHC
Braga PiresMF
SouzaDG
RoffeE
CortesDF
2006 Platelet activating factor receptor-deficient mice present delayed interferon-gamma upregulation and high susceptibility to Leishmania amazonensis infection. Microbes Infect 8 2569 2577
50. McGillJ
HeuselJW
LeggeKL
2009 Innate immune control and regulation of influenza virus infections. J Leukoc Biol 86 803 812
51. WatanabeY
HashimotoY
ShiratsuchiA
TakizawaT
NakanishiY
2005 Augmentation of fatality of influenza in mice by inhibition of phagocytosis. Biochem Biophys Res Commun 337 881 886
52. HashimotoY
MokiT
TakizawaT
ShiratsuchiA
NakanishiY
2007 Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 178 2448 2457
53. SchmitzN
KurrerM
BachmannMF
KopfM
2005 Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79 6441 6448
54. MainesTR
SzretterKJ
PerroneL
BelserJA
BrightRA
2008 Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol Rev 225 68 84
55. NoursharghS
LarkinSW
DasA
WilliamsTJ
1995 Interleukin-1-induced leukocyte extravasation across rat mesenteric microvessels is mediated by platelet-activating factor. Blood 85 2553 2558
56. YoungRE
ThompsonRD
NoursharghS
2002 Divergent mechanisms of action of the inflammatory cytokines interleukin 1-beta and tumour necrosis factor-alpha in mouse cremasteric venules. Br J Pharmacol 137 1237 1246
57. MaratheGK
DaviesSS
HarrisonKA
SilvaAR
MurphyRC
1999 Inflammatory platelet-activating factor-like phospholipids in oxidized low density lipoproteins are fragmented alkyl phosphatidylcholines. J Biol Chem 274 28395 28404
58. SouzaDG
FagundesCT
SousaLP
AmaralFA
SouzaRS
2009 Essential role of platelet-activating factor receptor in the pathogenesis of Dengue virus infection. Proc Natl Acad Sci U S A 106 14138 14143
59. ZhengBJ
ChanKW
LinYP
ZhaoGY
ChanC
2008 Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc Natl Acad Sci U S A 105 8091 8096
60. RussoRC
GuabirabaR
GarciaCC
BarcelosLS
RoffeE
2009 Role of the chemokine receptor CXCR2 in bleomycin-induced pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol 40 410 421
61. HorvatJC
BeagleyKW
WadeMA
PrestonJA
HansbroNG
2007 Neonatal chlamydial infection induces mixed T-cell responses that drive allergic airway disease. Am J Respir Crit Care Med 176 556 564
62. DolgachevVA
UllenbruchMR
LukacsNW
PhanSH
2009 Role of stem cell factor and bone marrow-derived fibroblasts in airway remodeling. Am J Pathol 174 390 400
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 11
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Zn Inhibits Coronavirus and Arterivirus RNA Polymerase Activity and Zinc Ionophores Block the Replication of These Viruses in Cell Culture
- The Female Lower Genital Tract Is a Privileged Compartment with IL-10 Producing Dendritic Cells and Poor Th1 Immunity following Infection
- Crystal Structure and Size-Dependent Neutralization Properties of HK20, a Human Monoclonal Antibody Binding to the Highly Conserved Heptad Repeat 1 of gp41
- The Arabidopsis Resistance-Like Gene Is Activated by Mutations in and Contributes to Resistance to the Bacterial Effector AvrRps4