
The Strain-Encoded Relationship between PrP Replication, Stability and Processing in Neurons is Predictive of the Incubation Period of Disease
Prion strains are characterized by differences in the outcome of disease, most notably incubation period and neuropathological features. While it is established that the disease specific isoform of the prion protein, PrPSc, is an essential component of the infectious agent, the strain-specific relationship between PrPSc properties and the biological features of the resulting disease is not clear. To investigate this relationship, we examined the amplification efficiency and conformational stability of PrPSc from eight hamster-adapted prion strains and compared it to the resulting incubation period of disease and processing of PrPSc in neurons and glia. We found that short incubation period strains were characterized by more efficient PrPSc amplification and higher PrPSc conformational stabilities compared to long incubation period strains. In the CNS, the short incubation period strains were characterized by the accumulation of N-terminally truncated PrPSc in the soma of neurons, astrocytes and microglia in contrast to long incubation period strains where PrPSc did not accumulate to detectable levels in the soma of neurons but was detected in glia similar to short incubation period strains. These results are inconsistent with the hypothesis that a decrease in conformational stability results in a corresponding increase in replication efficiency and suggest that glia mediated neurodegeneration results in longer survival times compared to direct replication of PrPSc in neurons.
Published in the journal:
. PLoS Pathog 7(3): e32767. doi:10.1371/journal.ppat.1001317
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001317
Summary
Prion strains are characterized by differences in the outcome of disease, most notably incubation period and neuropathological features. While it is established that the disease specific isoform of the prion protein, PrPSc, is an essential component of the infectious agent, the strain-specific relationship between PrPSc properties and the biological features of the resulting disease is not clear. To investigate this relationship, we examined the amplification efficiency and conformational stability of PrPSc from eight hamster-adapted prion strains and compared it to the resulting incubation period of disease and processing of PrPSc in neurons and glia. We found that short incubation period strains were characterized by more efficient PrPSc amplification and higher PrPSc conformational stabilities compared to long incubation period strains. In the CNS, the short incubation period strains were characterized by the accumulation of N-terminally truncated PrPSc in the soma of neurons, astrocytes and microglia in contrast to long incubation period strains where PrPSc did not accumulate to detectable levels in the soma of neurons but was detected in glia similar to short incubation period strains. These results are inconsistent with the hypothesis that a decrease in conformational stability results in a corresponding increase in replication efficiency and suggest that glia mediated neurodegeneration results in longer survival times compared to direct replication of PrPSc in neurons.
Introduction
Prion diseases are a group of transmissible, fatal neurodegenerative diseases, which include Creutzfeldt-Jakob disease in humans, bovine spongiform encephalopathy in cattle, and scrapie in sheep. The prion agent is comprised mainly, if not entirely, of PrPSc which is an abnormal isoform of the host encoded prion protein, PrPC [1], [2], [3], [4], [5], [6]. Prion propagation is thought to occur in a two-step process where PrPSc first binds to PrPC followed by a conformational conversion of PrPC to PrPSc [7], [8], [9]. This conversion results in a change in physical properties of PrPC that include an increase in β-pleated sheet content, decreased solubility in non-denaturing detergents and increased resistance to proteolytic degradation [3], [10], [11].
Prion strains are operationally defined by characteristic incubation periods and neuropathological features that are maintained upon experimental passage [12], [13]. The distribution of PrPSc in organs and neuronal populations can differ between strains, suggesting that PrPSc has a distinct strain-specific cellular tropism [14], [15], [16], [17]. The initial uptake of PrPSc by different cell-lines appears to be independent of the particular strain [18], [19] and suggests that cellular factors are responsible for prion strain tropism [17], [20], however, this has not been confirmed in vivo [21].
Prion strain diversity may be encoded by unique strain-specific conformations of PrPSc [15], [22], [23], [24], [25], [26]. Consistent with this, strain specific differences in the molecular weight of PrPSc following limited PK digestion, the relative resistance of PrPSc to degradation by PK, the relative alpha helical and beta sheet content of PrPSc, the resistance of PrPSc to PK digestion in increasing concentrations of a protein denaturant (i.e. conformational stability), and the aggregation state of PrPSc have been observed [15], [22], [27], [28]. The mechanisms underlying how strain-specific conformations of PrPSc result in the distinct biological properties of disease are poorly understood.
The published reports on the relationship between the conformational stability of PrPSc and the length of the incubation period of disease between prion strains are contradictory. In murine prion strains and during adaptation of synthetic prions, a decrease in the conformational stability of PrPSc correlates with a corresponding decrease in the incubation period [5], [24], [29], [30]. One explanation for this observation is that a decrease of PrPSc stability increases PrPSc fragmentation resulting in an increase in agent replication that produces a correspondingly shorter incubation period [31], [32], [33]. Consistent with this, a decrease in Sup35 fiber stability corresponds to an increased rate of fibril fragmentation in yeast prions [33], [34]. These data contrast with what has been observed in hamster-adapted prion strains. Short incubation period prion strains have PrPSc that is conformationally more stable compared to PrPSc from strains with a relatively longer incubation periods in hamsters [27]. However, a direct comparison between PrPSc replication rate and conformational stability has not been investigated.
Both the prion strain and the cell type infected can influence the processing of PrPSc. Studies of sheep infected with different prion strains, either naturally or experimentally, have identified strain-specific patterns of PrPSc truncation in both neurons and glia [35], [36]. Within a given strain the PrPSc truncation pattern can differ between glia and neurons suggesting that factors in addition to the conformation of PrPSc contribute to PrPSc truncation. While it is thought that replication in neurons is more important to disease development compared to glia, the effect of strain-specific processing of PrPSc in these cell types is less clear [37], [38], [39].
To better understand the strain specific relationship between the agent and the host, we evaluated PrPSc amplification efficiency, conformational stability of PrPSc, and susceptibility of PrPSc to endogenous proteolytic processing in vivo in several cell types, of eight hamster-adapted prion strains. Our data indicate that short incubation period strains have correspondingly more efficient replication, a higher conformational stability, and intrasomal accumulation of PrPSc in neurons compared to long incubation period strains. These data suggest that the relationship between agent replication and clearance influence the progression of disease.
Results
The molecular weight and abundance of PrPSc from multiple hamster-adapted prion strains is similar
Brain tissue from hamsters at terminal disease infected with either the HY TME, 263K, HaCWD, 22AH, 22CH, 139H, DY TME or ME7H agents was digested with proteinase K and 250 µg equivalents were analyzed by Western blot (Figure 1). Western blot analysis of PrPSc indicated that the unglycosylated PrPSc glycoform of each strain migrated at 21 kDa with the exception of DY PrPSc, which migrated at 19 kDa (Figure 1A, Table 1). The abundance of PrPSc was determined for each strain (n = 4) and there was less than a 25% difference in the abundance of PrPSc per µg brain equivalent for each prion strain analyzed (Figure 1B).


Short incubation period strains replicate PrPSc more efficiently compared to PrPSc from long incubation period strains
To determine if differences exist in the rate of PrPSc replication between strains, protein misfolding cyclic amplification (PMCA) was performed on eight hamster-adapted prion strains. Brain homogenates were prepared from animals at the clinical stage of disease or from an uninfected (mock) negative control and serial 10-fold serial dilutions of these homogenates were analyzed by Western blot prior to (Figure 2A and 2C) or after one round of PMCA (Figure 2B and 2D). PMCA reactions that were initially seeded with 500 to 5×10−2 µg eq of HY TME infected brain homogenate resulted in detectable amplification of PrPSc, but amplification was not detected in PMCA reactions seeded with lower concentrations of HY brain homogenate (Figure 2B). One round of PMCA using brain homogenate from DY TME infected animals amplified PrPSc to detectable levels in reactions that were initially seeded with 500 or 50 µg eq, but was not detected in PMCA reactions seeded with lower concentrations (Figure 2D). This was the general trend, as the short incubation period strains HY TME, 263K, and HaCWD resulted in detection of amplified PrPSc in reactions seeded with lower ug eq of brain homogenate compared to the longer incubation period strains 22AH, 22CH, 139H, DY TME, and ME7H (Figure S1, Table 1). These data demonstrate that the efficiency of PrPSc amplification corresponds with the incubation period for the prion strains that were analyzed.

PrPSc from short incubation period strains is conformationally more stable than PrPSc from long incubation period strains
The conformational stability of PrPSc for each of the eight hamster-adapted prion strains was determined using either SDS or Gdn-HCl to denature PrPSc. The [SDS]1/2 half values (% w/v) segregated into two groups corresponding to the incubation period of the strain. The short incubation period strains HY TME, 263K, and HaCWD have a [SDS]1/2 values of 1.14±0.03, 1.04±0.06 and 0.78±0.02 respectively compared to the long incubation period strains 22AH, 22CH, 139H, DY TME, and ME7H, which have [SDS]1/2 values of 0.53±0.04, 0.46±0.02, 0.50±0.01, 0.53±0.05 and 0.44±0.02 respectively (Table 1, Figure 3, Figure S2). Similarly, there was a corresponding decrease in the [Gdn-HCl]1/2 values with an increase in the incubation period. To demonstrate that the reduction in PrPSc was not due to an inhibition of PrPSc binding to the PVDF membrane due to the presence of SDS or Gdn-HCl, the PK digestion step of the conformational stability assay was omitted which resulted in the detection of PrPSc (data not shown). Overall, this data demonstrates that PrPSc from the short incubation period strains is more stable than PrPSc from the long incubation period strains (Table 1, Figure 3, Figure S2).

Absence of PrPSc from the soma of neurons corresponds with increased survival times
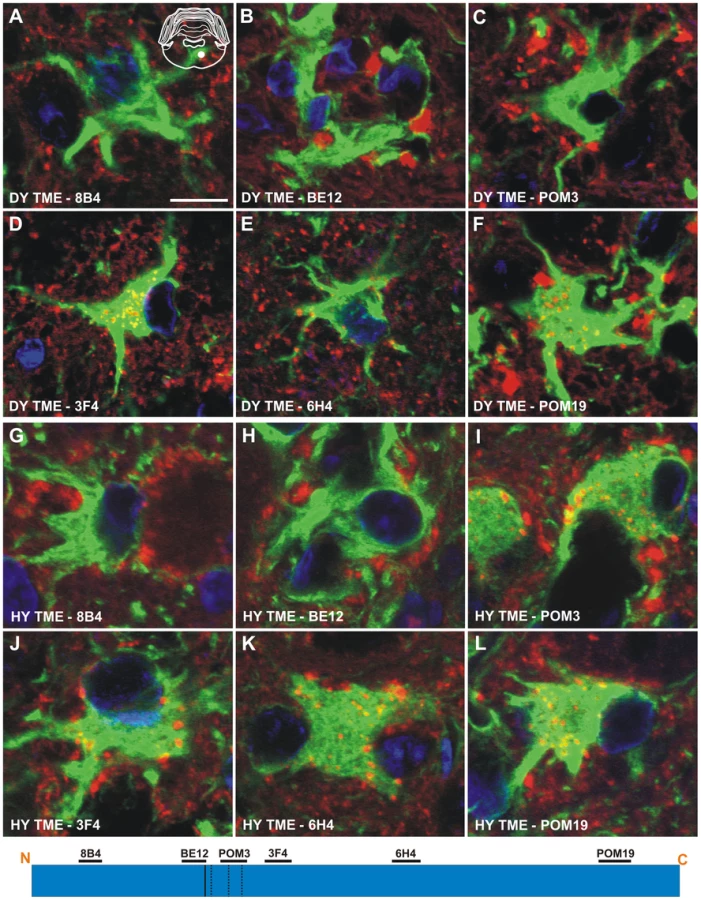
Immunohistochemistry was performed on CNS tissue of hamsters using a panel of six monoclonal anti-PrP antibodies whose epitopes span the length of the hamster PrP protein (Table 2). Immunohistochemistry using this panel of six anti-PrP antibodies on mock-infected tissue sections containing red nucleus neurons failed to detect PrPSc, indicating the specificity of the antibodies for PrPSc (Figure S3). PrPSc deposits were detected perineuronally and within the neuropil of the red nucleus with every anti-PrP antibody tested in animals infected in the sciatic nerve with the DY TME agent at clinical disease suggesting the presence of full length PrPSc (Figure 4A to 4F). Within the soma of neurons, the three antibodies whose epitopes are at the N terminus of PrP (8B4, BE12, and POM3) failed to detect PrPSc deposition (Figure 4A to 4C). The three antibodies whose epitopes are located toward the C-terminal region of PrP (3F4, 6H4, and POM19) occasionally identified PrPSc deposition within the soma of these neurons, however, these DY PrPSc deposits appeared diffuse and faint compared to the PrPSc immunoreactivity in the neuropil (Figure 4D to 4F, Table S1). This same pattern of PrPSc distribution was also observed in VMNs of the lumbar spinal cord, and the neurons in the interposed nucleus, red nucleus and hind limb motor cortex throughout the course of disease and in animals inoculated by the i.c. route at clinical disease (data not shown). The distribution of PrPSc in the red nucleus of hamsters i.c. inoculated with the long incubation period strains 22AH, 22CH, 139H or ME7H was indistinguishable from DY TME agent infected animals (Figure S4, Table S1).


To extend these studies on a short incubation period strains, we performed PrPSc immunohistochemistry on the red nucleus from animals infected in the sciatic nerve with the HY TME agent at clinical disease to ensure a direct comparison could be made with DY TME agent infected animals. The deposition of PrPSc in the neuropil and soma of neurons was similar to what was observed following infection with the DY TME agent and the other the long incubation period strains using the anti-PrP antibodies whose epitopes are located N-terminal to the HY PrPSc PK cleavage site (Figure 4, Panels G–H). However, when using antibodies located C-terminal to the HY PrPSc PK cleavage site, intrasomal and perinuclear PrPSc deposition was detected that was similar in intensity to PrPSc deposition in the neuropil (Figure 4I to 4L, Table S1). This HY TME specific pattern of PrPSc deposition was observed in animals inoculated by either the sciatic nerve or i.c. routes of inoculation at early and late time points post-infection and in the same brain regions that were examined in the DY TME infected animals. This pattern of HY PrPSc truncation was also observed in animals inoculated with the short incubation period strains 263K and HaCWD by the i.c. route at clinical disease (Figure S4, Table S1). These data reveal similarities in the PrPSc deposition patterns in the neuropil and somata of neurons of animals infected with either long or short incubation period strains.
Truncation of HY PrPSc within the soma of neurons
The absence of PrPSc immunoreactivity using antibodies directed against the N-terminal regions of PrPSc suggests that truncated PrPSc is present in these cells. To investigate this possibility, serial sections of red nucleus from clinically-ill HY TME infected hamsters were processed using either the BE12 or POM3 antibodies whose epitopes are N-terminal and C-terminal to the HY PrPSc PK cleavage site respectively. Fiduciary marks, such as blood vessels and white matter tracts, were used to increase the likelihood that the same neurons were analyzed in both sections. The BE12 antibody detected punctate HY PrPSc deposits in the neuropil and perineuronally in the red nucleus but failed to detect intrasomal PrPSc (Figure 5A). However, the POM3 antibody detected coarse, intrasomal PrPSc deposits in these same three neurons (Figure 5B, arrowheads). Additionally, the intrasomal PrPSc formed large perinuclear aggregates (Figure 5B). This demonstrates that the loss of N-terminal epitopes of PrPSc and the aggregation of the C-terminal PrPSc fragments occurs within the same neuron.

Processing of PrPSc in astrocytes and microglia is nearly uniform between strains
The deposition of PrPSc in astrocytes and microglia was investigated using the same panel of anti-PrP antibodies in combination with anti-GFAP and anti-Iba-1, which label astrocytes and microglia, respectively. As negative controls, reactive astrogliosis, microgliosis or PrPSc immunoreactivity was not detected in mock-infected animals (Figure S5A to S5F). Additionally, non-specific binding of the monoclonal antibodies or the fluorescently conjugated secondary antibodies was not detected (Figure S5G and S5H).
The anti-PrP antibodies 8B4, BE12, and POM3 failed to detect PrPSc within astrocytes (Figure 6A to 6C), while the antibodies 3F4, 6H4, and POM19 detected coarse punctate PrPSc deposits in astrocytes of hamsters infected with the DY TME agent at clinical disease (Figure 6D to 6F, Table S1). The anti-PrP antibodies POM3, 3F4, 6H4, and POM19 detected PrPSc within astrocytes, while the antibodies 8B4 and BE12 failed to detect PrPSc within astrocytes of hamsters infected with the HY TME agent at clinical disease (Figure 6G to 6L, Table S1). The same PrPSc truncation pattern detected in astrocytes of HY TME infected animals was also observed in animals infected with the 263K, HaCWD, 22AH, 22CH, 139H or ME7 agents (Figure S6, Table S1). The anti-PrP antibodies 8B4 and BE12 failed to detect PrPSc in microglia, while the anti-PrP antibodies POM3, 3F4, 6H4, and POM19 detected coarse punctate PrPSc deposits within these cells (Figure 7, Figure S7, Table S1).

Discussion
Here we show that short incubation period strains have a more stable PrPSc conformation when compared to long incubation period strains. PrPSc conformational stability assays using either Gdn-HCl or SDS as the denaturant found the same relationship between the conformational stability of PrPSc and incubation period of disease indicating that this relationship is independent of the denaturant used (Table 1). This relationship between PrPSc conformational stability and incubation period is consistent with previous work examining the conformational stability of purified PrPSc from hamster-adapted prion strains [27]. In contrast to what is observed in hamsters, a decrease in the PrPSc conformational stability correlates with a reduction in the incubation period in mice [24], [29], [40]. The results in murine systems suggest that decreasing PrPSc stability increases the fragmentation of PrPSc therefore allowing in the generation of more PrPSc surfaces for PrPC to bind resulting in an increased rate of PrPSc formation and subsequently shortening of the incubation period. Consistent with this hypothesis, studies examining Sup35, PrP, Tau, α-synuclein, and ß-amyloid demonstrate that less stable fibrils have a higher propensity to undergo breakage, thereby creating new seeds for conversion [33], [34], [41], [42], [43], [44], [45].
The PrPSc conformational stability data presented here suggest that conformationally stable PrPSc may also be more susceptible to fragmentation. SDS, like Gdn-HCl, can increase the susceptibility of PrPSc to PK digestion and inactivate the agent [27], [46], [47]. Since treatment of PrPSc that is enriched using detergent extraction and ultracentrifugation with SDS results in the disaggregation of PrPSc and the production of smaller PrPSc particles, SDS can affect the aggregation state of PrPSc [32], [48]. Therefore, the higher concentration of SDS required to increase the susceptibility of PrPSc to PK digestion of short incubation period strains may be due to increased PrPSc particle size compared to long incubation period strains.
Short incubation period strains have more efficient PrPSc amplification compared to long incubation period strains. We used PMCA to determine the relative efficiency of PrPSc conversion between hamster strains. We have previously shown that PMCA of HY and DY TME recapitulates the strain-specific properties of PrPSc and faithfully replicates the HY and DY TME agents [49]. In examining the eight hamster strains we found that the efficiency of PrPSc amplification correlated with the strains respective incubation periods, as the strains with more efficiently replicating PrPSc had a shorter incubation period compared to long incubation period strains (Table 1). This is consistent with cell-free conversion experiments that demonstrated a faster rate of HY PrPSc synthesis compared to the rate of DY PrPSc synthesis [50]. The data presented here also indicate that conformationally more stable PrPSc amplifies more efficiently compared to less stable PrPSc. Interestingly, the short incubation period strain HaCWD has conformationally less stable PrPSc in SDS compared to 263K and HY PrPSc which corresponded with a lower amplification efficiency compared to the two other short incubation period strains. A possible explanation for the increased amplification efficiency of PrPSc from the short incubation period strains is that this PrPSc is more likely to fragment due to its large PrPSc particle size compared to the longer incubation period strains used in this study. Alternatively, a minor subpopulation of PrPSc that is conformationally less stable may be responsible for the highly efficient PrPSc replication that was observed. This conformationally less stable subpopulation may be masked by an excess of conformationally more stable PrPSc that replicates with lower efficiency [32], [51], [52].
Strain and cell-specific variations in the proteolytic processing of PrPSc have been observed in both brain tissue and cultured cells [36], [53], [54], [55], [56]. The results presented here are consistent with these findings and additionally suggest a relationship between the extent of truncation of PrPSc within the soma of neurons and the strains respective incubation periods. The short incubation period strains, HY TME, 263K, and HaCWD, contained a longer portion of C-terminal protein intact and a large punctate deposition of PrPSc within the soma of neurons, compared to the longer incubation period strains suggesting a strain-specific clearance of PrPSc (Figures 8, Figure 9). Furthermore, the low immunoreactivity of PrPSc in ME7H infected animals observed with all six anti-PrP antibodies and within all three cell types examined (Figure 8, Table S1) may represent the more efficient clearance of PrPSc in both neurons and glia for this particular strain and account for its significantly longer incubation period. However, we cannot exclude the possibility that the inability to detect intense PrPSc immunoreactivity in the soma of neurons from animals inoculated with the long incubation period strains is due to a failure of PrPSc transport to the soma. This strain-specific truncation pattern was only observed in neurons, as the same N-terminally truncated PrPSc species was detected in astrocytes and microglia for all strains examined, with the lone exception of the loss of the POM3 epitope from DY PrPSc within astrocytes (Figure 8, Figure 9). These data support the hypothesis that direct infection of neurons leads to more rapid death of neurons resulting in shorter incubation periods, compared to indirect neuronal death via infection of astrocytes and microglia [37], [38], [57].


The results presented here suggest the strain-encoded relationship between PrPSc replication, stability and processing in neurons is predictive of the incubation period of disease (Figure 9). Here we show that strains with a short incubation period have conformationally stable PrPSc that replicates efficiently. The fast replication and stable PrPSc may be responsible for the accumulation of PrPSc in the soma of neurons resulting in a shorter incubation period. The long incubation period strains displayed relatively less efficient PrPSc replication and less stable PrPSc. In these strains, the combination of a slower replicating agent and PrPSc that is less stable may result in neurons to be able to more effectively cleared of PrPSc resulting in longer incubation periods.
Materials and Methods
Ethics statement
All procedures involving animals were approved by the Creighton University Institutional Animal Care and Use Committee and were in compliance with the Guide for the Care and Use of Laboratory Animals.
Animal inoculations
Sciatic nerve and intracerebral inoculations of the HY or DY TME agents were performed on male Syrian golden hamsters (Harlan-Sprague-Dawley, Indianapolis, IN) as previously described [21]. Groups of five hamsters were inoculated in the sciatic nerve or intracerebrally with 1 or 25 µl, respectively, of a 1% (wt/vol) brain homogenate from animals at the terminal stage of disease infected with either the HY TME, DY TME, 263K, HaCWD, 22AH, 22CH, 139H, or ME7H agents. Hamsters were observed three times per week for the onset of clinical signs as described previously [58]. Incubation period was calculated as the number of days between inoculation and onset of clinical signs.
Tissue collection
Tissue from infected and mock-infected hamsters was collected for either immunohistochemistry (IHC) or Western blot analysis. For IHC analysis animals were anesthetized with isoflurane and perfused transcardially with 50 ml of 0.01 M Dulbecco's phosphate-buffered saline followed by 75 ml of McLean's paraformaldehyde-lysine-periodate (PLP) fixative as previously described [21], [59]. Brain was immediately removed and placed in PLP for 5 to 7 h at room temperature prior to paraffin processing. For Western blot analysis, animals were sacrificed by CO2 asphyxiation, and the brain was rapidly removed and flash frozen and stored at −80°C.
Western blot analysis
Brain tissue and spinal cord tissue were homogenized to 10% w/v in Dulbecco's Phosphate Buffered Saline (DPBS) without Ca++ or Mg++ (Mediatech, Herndon, VA) containing protease inhibitors (Roche Diagnostics Corporation, Indianapolis, IN) by passing the tissue through a 26 g needle, followed by a 30 second incubation in a cup horn sonicator (Fisher Scientific, Atlanta, GA). The tissue was diluted to 5% w/v in DPBS containing proteinase K (PK) at a final concentration of 1 U/ml (Roche Diagnostics Corporation, Indianapolis, IN) and incubated at 37°C for 1 hour with constant agitation. The PK digestion was terminated by incubating the samples at 100°C for 10 minutes. SDS-PAGE and Western blot analysis were performed as described previously [49] using the anti-PrP antibody 3F4 (1∶600; Chemicon; Billerica, MA). The blot was developed with Pierce Supersignal West Femto Maximum Sensitivity Substrate according to manufactures instructions (Pierce, Rockford, IL) and imaged in the linear range of detection on a Kodak 4000R Imaging Station (Kodak, Rochester, NY) and analysis was performed using Kodak Molecular Imaging Software v.5.0.1.27 (New Haven, CT) as described previously [49].
Protein misfolding cyclic amplification
Protein misfolding cyclic amplification (PMCA) was performed as previously described [1], [49]. Briefly, uninfected brain was homogenized to 10% (w/v) in ice-cold conversion buffer [phosphate buffer saline (pH 7.4) containing 5 mM EDTA, 1% v/v Triton X-100, and complete protease inhibitor tablet (Roche Diagnostics, Mannheim, Germany)] using a Tenbroeck tissue grinder (Vineland, NJ). The brain homogenate was centrifuged at 500× g for 30 seconds and the supernatant was stored at −80°C. PMCA was performed with a Misonix 3000 sonicator (Farmingdale, NY) with the sonicator output set to level 6 with an average output of 156 watts for each sonication cycle. All PMCA reactions were replicated in triplicate. One round of PMCA consisted of 144 cycles of a five-second sonication followed by a ten-minute incubation at 37°C. Before each PMCA round, an aliquot was placed at −80°C as an unsonicated control. Samples seeded with prion-infected brain homogenate were replicated using a minimum of three individual hamster brains to control for variation between animals. Samples containing uninfected brain homogenate in conversion buffer alone were included in every round of PMCA as a negative control. The amplification efficiency was calculated as the reciprocal of the µg equivalent of last dilution of prion-infected brain homogenate that resulted in detectable amplified PrPSc following one round of PMCA.
Conformation stability assay
Brain homogenates [7.5% (w/v)] were diluted in either SDS (Fischer Scientific, Atlanta GA) to a final concentration of 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, or 2% (w/v) or in Gdn-HCl (Sigma-Aldrich, St. Louis, MO) to a final concentration of 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, or 2 molar and were immediately heated at 70°C for 10 min. Proteinase K was added to 0.0625 U/ml (Roche Diagnostics, Indianapolis, IN) and the samples were incubated at 37°C for 15 min while shaking. All samples were brought to 200 µl in DPBS and the concentration of PrPSc was determined using a 96-well immunoassay as described previously [60]. Gdn-HCL and SDS treated samples were performed in quintuplicate. Serial two-fold dilutions of brain homogenate each strain were performed in triplicate to ensure that the PrPSc levels of the SDS or Gdn-HCl treated samples were in the linear range of PrPSc detection. Denaturation curves were generated by dividing the intensity of all samples by the average intensity of the 0% SDS or Gdn-HCl samples. A sigmoidal dose response curve with variable slope was fitted to the standardized values (Prism statistical software, GraphPad, La Jolla, CA). The [SDS]1/2 and [Gdn-HCl]1/2 values is the percentage of SDS or molarity of Gdn-HCl required for a 50% reduction in the PrPSc signal intensity.
PrPSc immunohistochemistry
PrPSc IHC was performed as previously described [21], [59]. Briefly, 7 µm tissue sections were deparaffinized and incubated in 95% formic acid (Sigma-Aldrich, St.Louis, MO) followed by blocking of endogenous peroxidases by immersion in 0.3% H2O2 in methanol. Following blocking of non-specific staining with 10% horse serum, sections were incubated overnight with an anti-PrP antibody (Table 2) at 4°C. The sections were then incubated with a biotinylated horse anti-mouse immunoglobulin G conjugate and subsequent incubation with the ABC-horseradish peroxidase elite (Vector Laboratories, Burlingame, CA) staining kit. Sections were developed using 0.05% w/v 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO) in tris-buffered saline containing 0.0015% H2O2 and counterstained with hematoxylin (Richard Allen Scientific, Kalamazoo, MI). Microscopy was performed using a Nikon i80 microscope (Nikon, Melville, NY) and images were captured using DigiFire camera and ImageSys digital imaging software (Soft Imaging Systems, GmbH) and processed using Adobe Photoshop CS2 v9.0.1 (Adobe Systems Inc., San Jose, CA).
For double immunofluorescence, tissue sections were deparaffinized and treated with 95% formic acid as described above. The tissue sections were blocked with 10% goat serum in tris-buffered saline for 30 minutes at room temperature, followed by overnight incubation at 4°C with the same panel of anti-PrP monoclonal antibodies (Table 2) and anti-glial fibrillary acidic protein (GFAP; 1∶16,000; Dako; Carpinteria, CA) or anti-ionized calcium binding adaptor molecule 1 (Iba-1; 1∶500; Abcam; Cambridge, MA). Sections were then incubated with both Alexa Fluor goat anti-mouse 546 and Alexa Fluor goat anti-rabbit 488 (1∶500; Invitrogen; Carlsbad, CA) secondary antibodies for one hour at room temperature. Slides were cover slipped using ProLong Gold antifade reagent with DAPI (Invitrogen; Carlsbad, CA).
Confocal laser scanning microscopy
Fluorescent images were captured on a Zeiss LSM 510 META NLO confocal scanning system (Carl Zeiss Jena; Jena, Germany) using a Plan Neo 40× 1.3-NA DIC oil objective. Excitation of the Alexa Fluor antibodies and DAPI was achieved using an Argon laser at 488 nm, a Helium Neon laser at 543 nm, and a Coherent Chameleon near infrared tunable Ti:Sapphire laser. To increase the signal to noise ratio, each line was scanned 4 times and averaged. The pinhole aperture for each channel was adjusted so that an optical slice of 1.0 µm was imaged. In the profile view for each image, the line tool was used to draw an arbitrary line, and the relative fluorescent intensities along that line were compared to determine intracellular staining.
Semi-quantitative measurement of PrPSc immunoreactivity
Semi-quantitative measurements of PrPSc immunoreactivity was performed as previously described [61]. Briefly, captured images were randomized and the relative magnitude of neuropil, intraneuronal, intra-astrocytic, and intra-migroglial PrPSc immunoreactivity was classified as absent (0), slight (1), moderate (2), or striking (3) from a minimum of 6 observations by three independent observers. The PrPSc immunoreactivity scores were compared between strains and cell types and analyzed by two-way analysis of variance and Bonferroni post-tests for statistical significance (p<0.05). These tests were performed using the Prism 4.0 (for Macintosh) software program (GraphPad Software, Inc., San Diego, CA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CastillaJ
SaaP
HetzC
SotoC
2005
In vitro generation of infectious scrapie prions.
Cell
121
195
206
2. DeleaultNR
HarrisBT
ReesJR
SupattaponeS
2007
Formation of native prions from minimal components in vitro.
Proc Natl Acad Sci U S A
104
9741
9746
3. PrusinerSB
1982
Novel proteinaceous infectious particles cause scrapie.
Science
216
136
144
4. WangF
WangX
YuanCG
MaJ
2010
Generating a prion with bacterially expressed recombinant prion protein.
Science
327
1132
1135
5. MakaravaN
KovacsGG
BocharovaO
SavtchenkoR
AlexeevaI
2010
Recombinant prion protein induces a new transmissible prion disease in wild-type animals.
Acta Neuropathol
119
177
187
6. SigurdsonCJ
NilssonKP
HornemannS
HeikenwalderM
MancoG
2009
De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis.
Proc Natl Acad Sci U S A
106
304
309
7. HoriuchiM
PriolaSA
ChabryJ
CaugheyB
2000
Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers.
Proc Natl Acad Sci U S A
97
5836
5841
8. PrusinerSB
ScottM
FosterD
PanKM
GrothD
1990
Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication.
Cell
63
673
686
9. WeissmannC
1991
A ‘unified theory’ of prion propagation [see comments].
Nature
352
679
683
10. CaugheyBW
DongA
BhatKS
ErnstD
HayesSF
1991
Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy [published erratum appears in Biochemistry 1991 Oct 29;30(43):10600].
Biochemistry
30
7672
7680
11. PanKM
BaldwinM
NguyenJ
GassetM
SerbanA
1993
Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins.
Proc Natl Acad Sci U S A
90
10962
10966
12. FraserH
DickinsonAG
1968
The sequential development of the brain lesion of scrapie in three strains of mice.
J Comp Pathol
78
301
311
13. CollingeJ
ClarkeAR
2007
A general model of prion strains and their pathogenicity.
Science
318
930
936
14. BartzJC
DejoiaC
TuckerT
KincaidAE
BessenRA
2005
Extraneural prion neuroinvasion without lymphoreticular system infection.
J Virol
79
11858
11863
15. BessenRA
MarshRF
1994
Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy.
J Virol
68
7859
7868
16. DeArmondSJ
SanchezH
YehielyF
QiuY
Ninchak-CaseyA
1997
Selective neuronal targeting in prion disease.
Neuron
19
1337
1348
17. MahalSP
BakerCA
DemczykCA
SmithEW
JuliusC
2007
Prion strain discrimination in cell culture: the cell panel assay.
Proc Natl Acad Sci U S A
104
20908
20913
18. GreilCS
VorbergIM
WardAE
Meade-WhiteKD
HarrisDA
2008
Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent.
Virology
379
284
293
19. MagalhaesAC
BaronGS
LeeKS
Steele-MortimerO
DorwardD
2005
Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells.
J Neurol Sci
25
5207
5216
20. KarapetyanYE
SaaP
MahalSP
SferrazzaGF
ShermanA
2009
Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay.
PLoS One
4
e5730
21. AyersJI
KincaidAE
BartzJC
2009
Prion strain targeting independent of strain-specific neuronal tropism.
J Virol
83
81
87
22. CaugheyB
RaymondGJ
BessenRA
1998
Strain-dependent differences in beta-sheet conformations of abnormal prion protein.
J Biol Chem
273
32230
32235
23. KascsakRJ
RubensteinR
MerzPA
CarpRI
RobakisNK
1986
Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains.
J Virol
59
676
683
24. LegnameG
NguyenHO
PeretzD
CohenFE
DeArmondSJ
2006
Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes.
Proc Natl Acad Sci U S A
103
19105
19110
25. SafarJ
WilleH
ItriV
GrothD
SerbanH
1998
Eight prion strains have PrP(Sc) molecules with different conformations [see comments].
Nature Med
4
1157
1165
26. TellingGC
ParchiP
DeArmondSJ
CortelliP
MontagnaP
1996
Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity [see comments].
Science
274
2079
2082
27. PeretzD
ScottMR
GrothD
WilliamsonRA
BurtonDR
2001
Strain-specified relative conformational stability of the scrapie prion protein.
Prot Sci
10
854
863
28. TixadorP
HerzogL
ReineF
JaumainE
ChapuisJ
2010
The physical relationship between infectivity and prion protein aggregates is strain-dependent.
PLoS Pathog
6
e1000859
29. ColbyDW
GilesK
LegnameG
WilleH
BaskakovIV
2009
Design and construction of diverse mammalian prion strains.
Proc Natl Acad Sci U S A
106
20417
20422
30. WilleH
BianW
McDonaldM
KendallA
ColbyDW
2009
Natural and synthetic prion structure from X-ray fiber diffraction.
Proc Natl Acad Sci U S A
106
16990
16995
31. MaselJ
JansenVA
NowakMA
1999
Quantifying the kinetic parameters of prion replication.
Biophys Chem
77
139
152
32. SilveiraJR
RaymondGJ
HughsonAG
RaceRE
SimVL
2005
The most infectious prion protein particles.
Nature
437
257
261
33. TanakaM
CollinsSR
ToyamaBH
WeissmanJS
2006
The physical basis of how prion conformations determine strain phenotypes.
Nature
442
585
589
34. KryndushkinDS
AlexandrovIM
Ter-AvanesyanMD
KushnirovVV
2003
Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104.
J Biol Chem
278
49636
49643
35. GonzalezL
MartinS
Begara-McGorumI
HunterN
HoustonF
2002
Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie.
J Comp Pathol
126
17
29
36. JeffreyM
MartinS
GonzâalezL
2003
Cell-associated variants of disease-specific prion protein immunolabelling are found in different sources of sheep transmissible spongiform encephalopathy.
J Gen Virol
84
1033
1045
37. JeffreyM
GoodsirCM
RaceRE
ChesebroB
2004
Scrapie-specific neuronal lesions are independent of neuronal PrP expression.
Ann Neurol
55
781
792
38. MallucciG
DickinsonA
LinehanJ
KlohnPC
BrandnerS
2003
Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis.
Science
302
871
874
39. MallucciGR
WhiteMD
FarmerM
DickinsonA
KhatunH
2007
Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice.
Neuron
53
325
335
40. GreenKM
CastillaJ
SewardTS
NapierDL
JewellJE
2008
Accelerated high fidelity prion amplification within and across prion species barriers.
PLoS Pathog
4
e1000139
41. LeeS
FernandezEJ
GoodTA
2007
Role of aggregation conditions in structure, stability, and toxicity of intermediates in the Abeta fibril formation pathway.
Prot Sci
16
723
732
42. SunY
MakaravaN
LeeCI
LaksanalamaiP
RobbFT
2008
Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size.
J Mol Biol
376
1155
1167
43. XueWF
HellewellAL
GosalWS
HomansSW
HewittEW
2009
Fibril fragmentation enhances amyloid cytotoxicity.
J Biol Chem
284
34272
34282
44. YonetaniM
NonakaT
MasudaM
InukaiY
OikawaT
2009
Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant.
J Biol Chem
284
7940
7950
45. ZhouZ
FanJB
ZhuHL
ShewmakerF
YanX
2009
Crowded cell-like environment accelerates the nucleation step of amyloidogenic protein misfolding.
J Biol Chem
284
30148
30158
46. PeretzD
SupattaponeS
GilesK
VergaraJ
FreymanY
2006
Inactivation of prions by acidic sodium dodecyl sulfate.
J Virol
80
322
331
47. PrusinerSB
GrothD
SerbanA
StahlN
GabizonR
1993
Attempts to restore scrapie prion infectivity after exposure to protein denaturants.
Proc Natl Acad Sci U S A
90
2793
2797
48. RiesnerD
KellingsK
PostK
WilleH
SerbanH
1996
Disruption of prion rods generates 10-nm spherical particles having high alpha-helical content and lacking scrapie infectivity.
J Virol
70
1714
1722
49. ShikiyaRA
AyersJI
SchuttCR
KincaidAE
BartzJC
2010
Co-infecting prion strains compete for a limiting cellular resource.
J Virol
84
5706
5714
50. MulcahyER
BessenRA
2004
Strain-specific kinetics of prion protein formation in vitro and in vivo.
J Biol Chem
279
1643
1649
51. GhaemmaghamiS
AhnM
LessardP
GilesK
LegnameG
2009
Continuous quinacrine treatment results in the formation of drug-resistant prions.
PLoS Pathog
5
e1000673
52. LiJ
BrowningS
MahalSP
OelschlegelAM
WeissmannC
2009
Darwinian evolution of prions in cell culture.
Science
327
869
872
53. ChenSG
TeplowDB
ParchiP
TellerJK
GambettiP
1995
Truncated forms of the human prion protein in normal brain and in prion diseases.
J Biol Chem
270
19173
19180
54. DronM
MoudjouM
ChapuisJ
SalamatMK
BernardJ
2010
Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell - and tissue-dependent.
J Biol Chem
285
10252
10264
55. Jimenez-HueteA
LievensPM
VidalR
PiccardoP
GhettiB
1998
Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues.
Am J Pathol
153
1561
1572
56. YadavalliR
GuttmannRP
SewardT
CentersAP
WilliamsonRA
2004
Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation.
J Biol Chem
279
21948
21956
57. KercherL
FavaraC
ChanCC
RaceR
ChesebroB
2004
Differences in scrapie-induced pathology of the retina and brain in transgenic mice that express hamster prion protein in neurons, astrocytes, or multiple cell types.
Am J Pathol
165
2055
2067
58. BessenRA
MarshRF
1992
Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters.
J Gen Virol
73
329
334
59. WilsonMI
McBrideP
2000
Technical aspects of tracking scrapie infection in orally dosed rodents.
J Cell Pathol
5
17
22
60. KramerML
BartzJC
2009
Rapid, high-throughput detection of PrP(Sc) by 96-well immunoassay.
Prion
3
44
48
61. GonzalezL
MartinS
JeffreyM
2003
Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie - and BSE-infected sheep: implications for differential cell targeting and PrP processing.
J Gen Virol
84
1339
1350
62. KimberlinRH
WalkerC
1977
Characteristics of a short incubation model of scrapie in the golden hamster.
J Gen Virol
34
295
304
63. BartzJC
MarshRF
McKenzieDI
AikenJM
1998
The host range of chronic wasting disease is altered on passage in ferrets.
Virology
251
297
301
64. KimberlinRH
WalkerCA
FraserH
1989
The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice.
J Gen Virol
70
2017
2025
65. YuS
YinS
LiC
WongP
ChangB
2007
Aggregation of prion protein with insertion mutations is proportional to the number of inserts.
Biochem J
403
343
351
66. ManserJ
MassonW
JonesP
BostockC
2002
TSE Resource Centre and generation of monoclonal antibodies to recombinant ovine PrP (residues Met 23–230) in 129/Ola PrP null mice [abstract P3.16].
In: International Conference on Transmissible Spongiform Encephalopathies; 15–18 September 2002; Edinburgh, United Kingdom
67. PolymenidouM
MoosR
ScottM
SigurdsonC
ShiYZ
2008
The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes.
PLoS ONE
3
e3872
68. KascsakRJ
RubensteinR
MerzPA
Tonna-DeMasiM
FerskoR
1987
Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins.
J Virol
61
3688
3693
69. KorthC
StierliB
StreitP
MoserM
SchallerO
1997
Prion (PrPSc)-specific epitope defined by a monoclonal antibody.
Nature
390
74
77
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 3
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- A Toxin that Hijacks the Host Ubiquitin Proteolytic System
- Invasive Extravillous Trophoblasts Restrict Intracellular Growth and Spread of
- Blood Meal-Derived Heme Decreases ROS Levels in the Midgut of and Allows Proliferation of Intestinal Microbiota
- Metabolite Cross-Feeding Enhances Virulence in a Model Polymicrobial Infection