
-Sialidase in Complex with a Neutralizing Antibody: Structure/Function Studies towards the Rational Design of Inhibitors
Trans-sialidase (TS), a virulence factor from Trypanosoma cruzi, is an enzyme playing key roles in the biology of this protozoan parasite. Absent from the mammalian host, it constitutes a potential target for the development of novel chemotherapeutic drugs, an urgent need to combat Chagas' disease. TS is involved in host cell invasion and parasite survival in the bloodstream. However, TS is also actively shed by the parasite to the bloodstream, inducing systemic effects readily detected during the acute phase of the disease, in particular, hematological alterations and triggering of immune cells apoptosis, until specific neutralizing antibodies are elicited. These antibodies constitute the only known submicromolar inhibitor of TS's catalytic activity. We now report the identification and detailed characterization of a neutralizing mouse monoclonal antibody (mAb 13G9), recognizing T. cruzi TS with high specificity and subnanomolar affinity. This mAb displays undetectable association with the T. cruzi superfamily of TS-like proteins or yet with the TS-related enzymes from Trypanosoma brucei or Trypanosoma rangeli. In immunofluorescence assays, mAb 13G9 labeled 100% of the parasites from the infective trypomastigote stage. This mAb also reduces parasite invasion of cultured cells and strongly inhibits parasite surface sialylation. The crystal structure of the mAb 13G9 antigen-binding fragment in complex with the globular region of T. cruzi TS was determined, revealing detailed molecular insights of the inhibition mechanism. Not occluding the enzyme's catalytic site, the antibody performs a subtle action by inhibiting the movement of an assisting tyrosine (Y119), whose mobility is known to play a key role in the trans-glycosidase mechanism. As an example of enzymatic inhibition involving non-catalytic residues that occupy sites distal from the substrate-binding pocket, this first near atomic characterization of a high affinity inhibitory molecule for TS provides a rational framework for novel strategies in the design of chemotherapeutic compounds.
Published in the journal:
. PLoS Pathog 8(1): e32767. doi:10.1371/journal.ppat.1002474
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002474
Summary
Trans-sialidase (TS), a virulence factor from Trypanosoma cruzi, is an enzyme playing key roles in the biology of this protozoan parasite. Absent from the mammalian host, it constitutes a potential target for the development of novel chemotherapeutic drugs, an urgent need to combat Chagas' disease. TS is involved in host cell invasion and parasite survival in the bloodstream. However, TS is also actively shed by the parasite to the bloodstream, inducing systemic effects readily detected during the acute phase of the disease, in particular, hematological alterations and triggering of immune cells apoptosis, until specific neutralizing antibodies are elicited. These antibodies constitute the only known submicromolar inhibitor of TS's catalytic activity. We now report the identification and detailed characterization of a neutralizing mouse monoclonal antibody (mAb 13G9), recognizing T. cruzi TS with high specificity and subnanomolar affinity. This mAb displays undetectable association with the T. cruzi superfamily of TS-like proteins or yet with the TS-related enzymes from Trypanosoma brucei or Trypanosoma rangeli. In immunofluorescence assays, mAb 13G9 labeled 100% of the parasites from the infective trypomastigote stage. This mAb also reduces parasite invasion of cultured cells and strongly inhibits parasite surface sialylation. The crystal structure of the mAb 13G9 antigen-binding fragment in complex with the globular region of T. cruzi TS was determined, revealing detailed molecular insights of the inhibition mechanism. Not occluding the enzyme's catalytic site, the antibody performs a subtle action by inhibiting the movement of an assisting tyrosine (Y119), whose mobility is known to play a key role in the trans-glycosidase mechanism. As an example of enzymatic inhibition involving non-catalytic residues that occupy sites distal from the substrate-binding pocket, this first near atomic characterization of a high affinity inhibitory molecule for TS provides a rational framework for novel strategies in the design of chemotherapeutic compounds.
Introduction
Chagas' disease, the American trypanosomiasis, is a chronic disabling parasitic disease caused by the flagellate protozoon Trypanosoma cruzi. With an estimated global burden of 100 million people at risk, 8 million already infected, and approximately 40,000 new cases/year, Chagas' disease represents a major health and economic problem in Latin America [1]. The infection is naturally transmitted by triatomine vectors (“kissing bugs”), from the south of the USA to the southern region of South America, although chagasic patients are in fact dispersed worldwide due to migrations. Patients can also transmit the disease either by in utero infection leading to the congenitally acquired disease or by accidental transmission through contaminated blood. The acute infection is characterized by patent parasite burden. During this initial stage, T. cruzi induces several alterations in the infected mammal including intense polyclonal activation of lymphocytes [2], transient thymic aplasia [3], [4] and other clinical hematological findings [5], [6]. The majority of the patients control the parasitemia, survive the acute phase, and enter into an indeterminate form of the disease that may last for many years or even indefinitely [1]. Up to 20 years after the infection, ∼35% of patients develop different pathologies, such as cardiomyopathy, peripheral nervous system damage, and/or dysfunction of the digestive tract [1].
Sialic acids have proven to be crucial during the parasite's life cycle and survival in the mammalian host [7]–[10]. However, T. cruzi is unable to perform de novo synthesis of sialic acids [11]. This family of nine-carbon carbohydrates, is actually scavenged from the host's glycoconjugates, through a glycosyl-transfer reaction mediated by trans-sialidase (TS), a modified sialidase expressed by the parasite. In this way, the surface of the parasite becomes rapidly sialylated, with mucins being the main sialyl acceptors, in a process that allows the parasite to evade its destruction by serum factors [9], [10]. TS activity is also involved in host cell attachment and invasion [7], [8], as well as in parasite escape from the parasitophorous vacuole into the cytoplasm, where the parasite replicates [12].
In the trypomastigote stage, TS is a glycosylphosphatidylinositol-anchored non-integral membrane protein [13], actively released to the extracellular milieu, leading to a systemic distribution of the enzyme through the bloodstream. Its half-life in blood is significantly extended due to the presence of a C-terminal repetitive domain named SAPA [14]. TS activity is detectable in the bloodstream of infected humans and mice, until antibodies able to neutralize its catalytic activity are elicited [15]. The systemic distribution of TS is associated with several pathologies observed during the early steps of infection including depletion of thymocytes [16], absence of germinal centers in secondary organs [17] and thrombocytopenia and erythropenia [5], [6], all alterations that can be prevented by the passive transfer of TS-neutralizing antibodies [17], [18]. In fact, administration of the enzyme in mice before T. cruzi challenge, leads to more severe evolution of the infection [19]. These finding are also consistent with the fact that increased shedding of the enzyme correlates with increased virulence of the corresponding parasite strains [20].
TS has thus been identified as a potential target for drug discovery and design. Added to its key roles in host response evasion, cell invasion and pathogenesis, TS is not present in the mammalian host. The development of suitable drugs to treat/prevent Chagas' disease is urgently needed [21]. Only two compounds, benznidazol and nifurtimox, are currently available for treating both acute and chronic infections. These drugs are far from being optimal: fairly toxic, they trigger serious side effects, while also showing suboptimal efficacy in a high proportion of patients. The emergence of resistant parasite strains adds a concerning issue [22]. Several attempts to obtain suitable TS inhibitors have been made, especially once its 3D structure became available [23], [24]. However, only low affinity molecules have been obtained so far [25], [26], some of them toxic in in vivo assays [27], ultimately suggesting that further and more active efforts must be pursued.
We have obtained a TS-neutralizing mouse monoclonal antibody (mAb 13G9) that displays very high affinity and specificity towards the T. cruzi enzyme. This mAb is able to prevent immune system and hematological abnormalities, even when assaying highly virulent parasites under lethal infection conditions [5], [17]. We now report an extensive functional characterization of mAb 13G9, as well as the crystal structure of the 13G9-TS binary complex. The molecular features of the inhibitory mechanism are unveiled, providing novel insight for the development of TS inhibitors, which might also be relevant for related neuraminidases in other pathogens.
Results
Biochemical Characterization of the TS-neutralizing Monoclonal Antibody
Mice were immunized with a TS recombinant protein (Δ1443TS), identical to the wt except it includes a deletion of a non-neutralizing epitope. Δ1443TS retains full enzymatic activity, while avoiding the otherwise typical delay in eliciting TS-neutralizing antibodies [28], [29]. Hybridomas were screened by TS-inhibition assay [30] and the 13G9 clone secreting a TS-neutralizing mAb (IgG2aκ) was obtained. The specificity of this mAb was confirmed by the absence of reactivity against the closely related sialidase from Trypanosoma rangeli and the TS from Trypanosoma brucei (data not shown). As depicted in Figure 1A, this mAb showed high affinity for the T. cruzi TS (KD ∼7.2×10−10 M) as calculated from the kinetic constants determined by surface plasmon resonance. In agreement, isothermal titration calorimetry assays indicated an equilibrium dissociation constant lower than 10−9 M (raw data not shown).

The mAb was purified by Protein A-affinity chromatography from filtered hybridoma supernatants. This purified material was further subjected to anionic chromatography (Figure S1). The mAb eluted as a single peak as evaluated both by TS-neutralizing activity (not shown) as well as by TS recognition in dot-blot assays (Figure S1). The same sequence was found in several mRNAs encoding for the antibody (not shown), in support of a clonal nature of the hybridoma. Purified mAb was proteolized with papain to generate the Fab fragment. Inhibitory activity of the fragment was determined and compared with that from the whole IgG protein (Figure 1, panels B and C). Although the full-length mAb appears to have a higher inhibitory activity (half maximal inhibitory concentration IC50 5.6×10−11 M), its Fab fragment still retains a nanomolar IC50 (1.6×10−9 M), clearly conserving its antigen-binding mechanism. These high inhibitory potencies are consistent with the apparent dissociation constant determined by surface plasmon resonance (see above), even though IC50 figures cannot be compared with affinity constants in absolute terms at this point (allosteric effects, or yet mixed inhibition mechanisms, may flaw a linear relationship). The purified Fab proved to be fairly unstable when non-complexed to TS, requiring immediate use for biochemical characterizations. This may be one of the main reasons for the observed inhibitory potency decrease compared to the entire immunoglobulin molecule. The Fab's instability precluded its use for further in vivo and in vitro biologic assays.
T. cruzi TS belongs in fact to a huge superfamily of genes, among which at least four families can be discriminated [31]. TSs are only included in one of these families, which encodes for a number of enzymatically active and inactive members [32]. These two forms of TS can be distinguished by the single Tyr342His mutation [33]: only the active TSs have the Tyr342 residue acting as the enzyme's nucleophile during the ping-pong reaction [34]. TS-mAb competition assays performed with the inactive TS showed that both proteins reacted similarly with the mAb. An equimolar mixture of inactive and active TSs, displayed ∼50% reduction of the neutralizing reactivity (Figure 1D). In a separate set of assays, heat-inactivated TS was not recognized by the mAb 13G9 (Figure S1), consistent with the hypothesis that the neutralizing epitope is conformational [35]. In the infective trypomastigote stages, all TSs include the SAPA C-terminal extension [31], which is absent in all the other TS-related families allowing for clear-cut discrimination. To address whether the mAb 13G9 was specific only for TS proteins, extracts from biotinylated trypomastigotes were reacted with the antibody (Figure 1E). Pulled-down material was subjected to Western blot and developed in parallel with anti-SAPA (for TS) and streptavidin for all the biotinylated parasite surface components. Strong signals were readily observed in both lanes, matching the TS expected protein sizes. No differential pattern was detected whatsoever, confirming the very high specificity of 13G9 antibody only towards proteins belonging to the TS family.
mAb 13G9 Reduces Cell Invasion and Inhibits the Sialylation of the Parasite
The reactivity of mAb 13G9 with whole parasites was assayed by immunofluorescence showing surface labeling consistent with the expected cellular membrane localization of TS (Figure 2A). The ability of the mAb to inhibit TS-mediated transfer of sialic acid from the surrounding environment to the parasite's surface molecules was then tested. To reduce the basal sialylation of parasites, sialyl residue donors were largely depleted replacing fetal bovine serum (FBS) by bovine serum albumin (BSA) in the infected tissue cultures; only host cells remained as the unique source of the sugar. Trypomastigotes were then collected and incubated with α(2,3)sialyllactose as sialic acid donor and TS, in the presence of mAb 13G9. The amount of transferred sialic acid was determined by the thiobarbituric acid method [36]. As shown in Figure 2B, mAb 13G9 very efficiently inhibited the parasites' sialylation, demonstrating its biologic relevance as a TS-inhibitory molecule. The sialylation observed in the treated parasites corresponds to the sugar acquired before the addition of the mAb. These quantitative results are in agreement with the Western blot assays we have recently reported for sialyl-transfer inhibition by mAb 13G9 using azido-modified sialic acids [37].
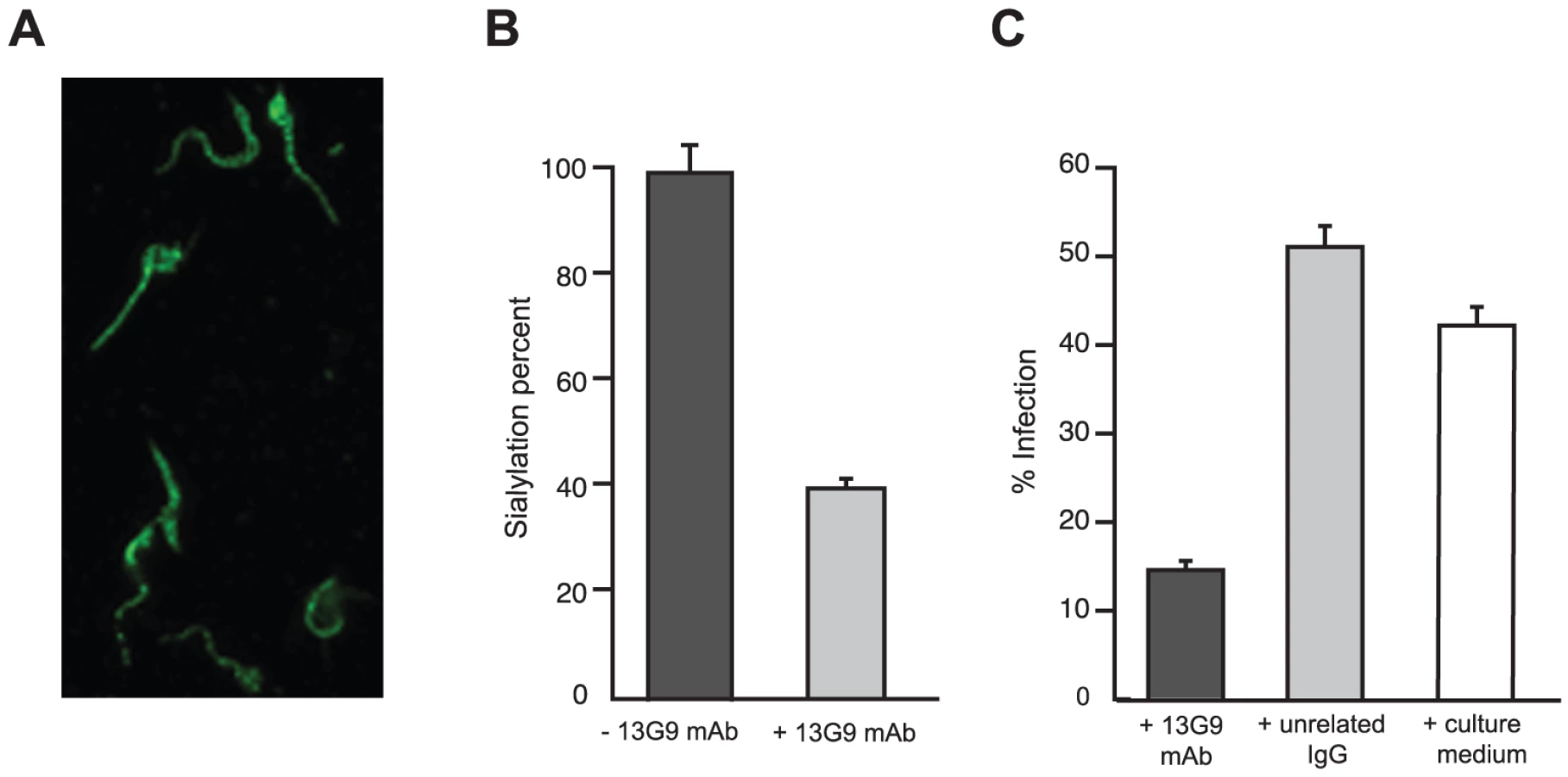
TS is involved in cell invasion [8], [12] given that sialic acid is required for competent interplay with the host cells. The ability of mAb 13G9 to interfere with the invasion process was therefore studied. The addition of the mAb (Figure 2C) strongly reduced the number of infected cells, highlighting its biologic activity and contributing direct evidence that TS is a valid target for drug discovery.
3D Structure of the Immunocomplex Fab/TS
To gain atomic insight into the antigen-antibody interactions allowing mAb 13G9 to neutralize the TS catalytic activity with extremely high efficiency, we solved the structure of the immunocomplex by X ray crystallography.
Crystallogenesis screenings were performed under a sitting-drop vapor diffusion setup with a Honeybee963 robotic station, using standard 96-well plates. Several initial hits were obtained. Further manual optimization eventually allowed to grow crystals (0.7×0.05×0.05 mm) in polyethylene glycol (PEG) 20,000 plus dioxane, suitable for X ray diffraction data to be collected (Table 1). Limiting resolution was 3.4Å on a Cu rotating anode generator, and indexing was straightforward, indicating a primitive cell in the trigonal/hexagonal system. Cell parameters (a = b = 178.1Å, c = 140.7Å) suggested the presence of as many as 3 binary complexes per asymmetric unit, raising as well the hypothesis that its weak diffraction could respond to limiting X ray beam intensity in the context of a fairly large unit cell (low number of scattering cells per crystal unit volume). To rule out this possibility, several crystals were tested at the ALS (Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA) beamline 5.0.2 (8×1011 photons/s with 1.5 mrad divergence at 12.4 keV), with no detectable improvement in resolution as judged by standard quantitative statistics, strongly suggesting that crystal disorder linked to high solvent content (66% as determined after full refinement) is the major cause for maximum resolution sphere limitation.

No 6-fold peaks were found in self-rotation function maps, and the κ = 180° section revealed significantly weaker signals than the 3-fold axis (data not shown) consistent with point group 3. Systematic extinctions were observed in the reciprocal 00l axis, strongly suggesting space groups P31 or P32. The structure was solved by molecular replacement confirming SG P31. Two search probes were used to calculate rotation and translation functions: Protein Data Base (PDB) 3CLF (mouse IgG Fab fragment, chosen according to sequence similarity to mAb 13G9) and 2AH2 (high resolution T. cruzi TS model). Iterative cycles of maximum likelihood refinement [38] were interspersed with manual rebuilding [39]. The high resolution of the molecular replacement search models resulted in excellent maps and straightforward rebuilding, mostly adding missing side chains on the immunoglobulin heavy and light chains. Tight non-crystallographic symmetry restraints were kept only in the first refinement cycles, thereafter allowing for automatic local NCS detection, with variable weights according to evolving rms deviations, as implemented in the program Buster/TNT [40]. Model refinement statistics are summarized in Table 1. Interestingly, the PISA server (European Bioinformatics Institute, Hinxton) predicts that the TS-Fab 13G9 complex would not be stable in solution, contradicting our experimental results. This discrepancy reveals the still challenging task of predicting energetic and thermodynamic properties of protein/protein associations, based on the analysis of crystal structures of partners and derived complexes, despite the fact that prediction algorithms are complex and attempt integrating enthalpic and entropic effects, as well as solvent accessible surface burial and geometric complementarity [41].
Indeed, three binary Fab-TS complexes are located in the asymmetric unit, all very similar at the level of precision of our data. Refined models of immunocomplex 2 (IC2, composed by TS chain B, and chains I and M of the Fab molecule) and IC3 (TS chain C, complexed to Fab J and N) were superposed sequentially onto complex IC1 (TS chain A with H "heavy" and L "light" chains from the Fab molecule) minimizing root mean squared deviations (rmsd) of atomic coordinates. Such structural alignments resulted in 0.84Å rmsd between IC1 and IC2, and 0.82Å between IC1 and IC3. Regions of highest variation correspond to intrinsically mobile segments, as reflected by detailed analysis of atomic displacement parameters (isotropic B factors). The mean B factor for all atoms is relatively high (59.9 Å2), consistent with the low resolution to which these crystals diffract X rays. Crystal packing is indeed loose, leading to high bulk solvent content and corresponding protein flexibility. TS molecules display lower B factors then the Fab dimers to which they are bound. A global tendency is also maintained among the independent complexes, IC3 showing greater mobility than IC2, which in turn is more flexible than complex IC1 (59>53>48 Å2), probably due to the different packing environments. In the case of the immunoglobulin heterodimers, chains also display a clear difference among variable domains, more rigid, compared to the constant domains, which show a reproducible flexibility on the distal half, away from the interdomain hinge.
Given the overall structural similarity among the three complexes and the fact that complex IC1 resulted in a model with lower B factors, subsequent analyses will be referred only to this complex. Figure 3 shows the immunocomplex IC1 highlighting that the variable regions of the Fab light chain are interacting with TS loops located closer to the entrance of the enzyme's catalytic pocket, while the heavy chain associates to an adjacent, more distal patch.
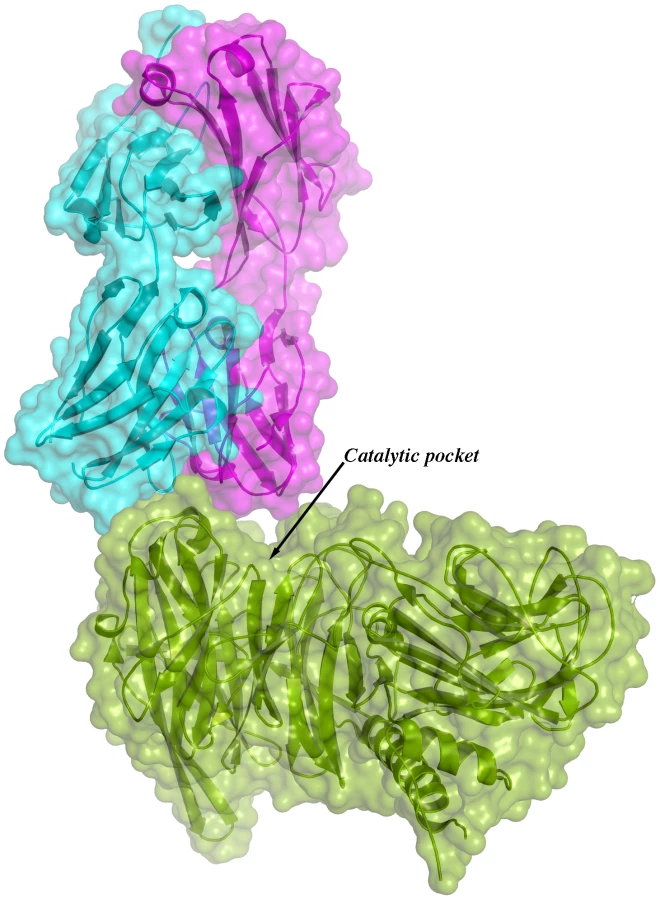
The solvent accessible surface that becomes buried due to the enzyme-antibody interaction corresponds to 1810.2 Å2 (916.5 Å2 on the TS and 893.7 Å2 on the Fab, adding 506 Å2 from the heavy chain, and 387.7 Å2 from the light chain), within the typical range of antibodies reacting with protein antigens. On this interface, 15 hydrogen bonds and one salt bridge can be distinguished, as well as a number of residues that establish contact interactions (van der Waals forces), as listed on Table 2. The resolution limit of the diffraction data allowed for the identification of very few water molecules, none of which are directly involved in the accessible nor the buried surfaces engaged in interaction. The shape complementarity statistics [42] correspond to 0.673 and 0.645, after analysis of the interface areas with the light and the heavy chains, respectively. These figures are within the typical range (0.64–0.74) of specific protein:protein interfaces. The epitope (Figure 4) consists of residues H171, Y248, R311–W312, and loops 199–201 (KKK) and 116–128 (SRSYWTSHGDARD - W120 and A126 do not interact directly).


The structural bases of the catalytic inhibitory effect that this mAb elicits, can start to be elucidated by modeling the entrance of the sialylated substrate into the TS reactional center in the context of the TS-Fab complex (Figure 5). Superimposing TS PDB models 1S0I and 1S0J, onto our structure, allowed to define the positions of the substrates N-acetyl-neuraminyl-lactose (α(2,3)sialyllactose) and 4-methylumbelliferyl-N-acetyl-neuraminic acid (MU-NANA), respectively (Figure 5). The most readily observable feature is the steric hindrance that TS residue Y119 imposes, blocking the entrance of the sialyl residue in the reactional pocket.

The free mobility of the phenolic side chain of Y119 is limited by the juxtaposed residue S30 from the Fab's light chain (Figure 5). This restraint seems to play a central role in precluding the entrance of sialylated substrates into the catalytic pocket, entrance that absolutely requires the movement of Y119 [23]. A second effect could not be excluded, namely the spatial constraint exerted by the overall architecture of the associated complex. Residues S26–S28 (within the light chain complementarily determining region CDRL1) and S66–G67 on the same Fab chain, establish direct contact with TS residues R311 and W312. This interaction is located just on top of the catalytic pocket entrance, functioning as a ‘roof’ (SG/RW roof), where the catalytic center itself would be the floor. As shown in Figure 5B, when sialyllactose is located in position, the substrate pocket appears to be too small, predicting direct clashes of the glucosyl residue with the SG/RW roof (particularly residues Ser66–Gly67 of the Fab light chain). This scenario of course implies that Y119 could eventually be forced to move out of the sialic acid binding site, an unlikely event. The light chain loop 29–31 is also prone to interfere with the saccharide, if rearrangements are to be considered during its accommodation (data not shown). In order to obtain further experimental data evaluating the relative effects of Y119-mobility hindrance and/or the spatial constraints exerted by the SG/RW roof onto the catalytic pocket cavity volume, MU-NANA was assayed in TS-catalyzed sialidase reactions. MU-NANA is an artificial substrate that allows for TS-catalyzed hydrolytic and trans-glycosidase activities [43], and given its smaller volume, could better accommodate, avoiding steric clashes with the SG/RW roof structure (Figure 5B). TS-mediated MU-NANA hydrolysis was efficiently inhibited by mAb 13G9 (Figure 6), suggesting that the immobilization of Y119 does play a central role. The spatial confinement in the pocket, partly due to the SG/RW roof structure, might impose secondary constraints precluding torsional accommodation, even in the case of smaller compounds.

Discussion
This report describes an extensive biochemical and structural characterization of the mouse mAb 13G9, which is herein demonstrated to act as a powerful inhibitor of the T. cruzi TS catalytic activity, displaying high specificity and affinity for the enzyme. T. cruzi TS is a virulence factor required for the survival of the parasite in the mammalian host. Several different biologic activities of the enzyme can be discriminated. The parasite uses TS activity to sialylate its own surface molecules, allowing it to evade lysis by serum factors [9], [10]. In this context, it should be noted that the addition of mAb 13G9 inhibited this sialylation process (Figure 1) in agreement with our previous findings with azido-modified sugars [37]. As well, TS is not only directly involved in the parasite/host cell interaction through the generation of a required sialylated epitope [7], [8] but also in escaping from the parasitophorous vacuole to the cytoplasm [12]. In concert with these findings, here we report that mAb 13G9 significantly reduces parasite infection of cell cultures (Figure 1). Passive transfer of neutralizing mAb 13G9 to heavily infected mice, protects them against TS-induced deleterious effects on the immune system and platelets [5], [17]. In this sense, it is well known that antibodies against neuraminidases are also effective in preventing other diseases such as Influenza [44]. These protective effects are very much promising to delineate a therapeutic tool. The high molecular weight of antibodies constitutes a main drawback in their use, due to eventual hindrance for effective diffusion into infected tissues, where high concentrations of locally produced TS are expected to be found. On the other hand, Fab fragments, small recombinant antibody-derived molecules (e.g. scFv), or yet antibody-mimetic engineered molecules [45], can be cleared exceedingly fast from the bloodstream [46], resulting in poor pharmacokinetic figures. PEGylation, and other modifications to improve bioavailability of these smaller protein scaffolds, constitute interesting approaches to be tested using mAb 13G9 as starting lead [47].
As a second interesting avenue to explore for therapeutic derivatives, the high affinity and specificity of this mAb, prompted us to elucidate its neutralizing mechanism, as an attempt to thereafter conceive low molecular weight inhibitors, suitable as chemotherapy leads. Some information can be gathered in this respect from previous studies of the neuraminidase from Influenza virus, a protein orthologous to TS. The overall geometry of the antibody/TS association that we are now reporting, is reminiscent of the one described for a Fab/Influenza-N2 neuraminidase complex (PDB 2AEP; [48]), which shows interaction with enzyme's loops on the same side of the reaction pocket, opposite to the patch where most other anti-neuraminidase antibodies have been reported (such as the ones involving avian N9 neuraminidase with antibodies NC41 and NC10, PDBs 1NCA and 1NMB, respectively; among others) [49]–[51]. The interaction surfaces of TS-13G9 mAb (this report) and N2NA-Mem5Fab (2AEP) are largely overlapping, although the antibodies are bound in inverted configurations with respect to the location of the heavy and light chains. Well defined escape mutations in Influenza (loops including positions 198–199 and 220–221, following N2 Influenza numbering scheme) identify epidemiologically important antigenic sites of neuraminidase, revealing antigenic drift in human viruses seemingly under natural antibody selection of enzyme variants [52]. These loops, connecting β2–β3 within the second blade of the six-bladed β-propeller domain, and β4 of this blade with β1 of the next one, are not structurally conserved between T. cruzi and Influenza enzymes, being longer in the former. Nevertheless, it is clear that the equivalent loops in T. cruzi TS do play a critical role in the 13G9 Fab association that we are now reporting.
One of the specific mAb loops that interact in a proximal position to the catalytic pocket of the enzyme, was observed precluding the displacement of Y119, a critical residue that has already been shown to be flexible in TS [24], [53]. Indeed, the mobility of Y119 plays a key role in the trans-glycosidase mechanism of TS. The determination of the three-dimensional coordinates of the paratope, including these features that lead to spatial constraints, uncovers relevant information. This is to be used as a precise guide, not only to undertake peptidomimetic syntheses, but most importantly, to use as a working template for the synthesis of non-peptidic molecules including critical pharmacophores [54].
Materials and Methods
Ethics Statement
The protocol of this study was approved by the Committee on the Ethics of Animal Experiments of the Universidad Nacional de San Martín, which also approved protocol development under the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Recombinant Enzymes
Recombinant T. cruzi TSs (constructs 1N1, 2Vo, Δ1443TS and 3.2) [24], [28], [33], T. rangeli sialidase [23] and T. brucei TS [55] were used. The 1N1 and 2Vo clones correspond to the full-length (including the SAPA repeats [33]) wild type genes that encode for enzymatically active and inactive molecules, respectively. The Δ1443TS recombinant TS was used for immunization procedures. Δ1443TS is an engineered variant where the deletion of a non-neutralizing epitope in the globular domain was done [28]. The TS 3.2 construct [24] is engineered to express the enzymatically competent globular domain only, containing seven mutations of surface-located residues that allow for protein crystallization. All TSs were expressed in Escherichia coli BL21 and immediately used after purification, avoiding >3 weeks storage at 4°C. Recombinant proteins were purified to homogeneity as described elsewhere [56], briefly, TS was subjected to immobilized metal affinity chromatography (Ni++-charged, Hi-Trap Chelating HP) followed by MonoQ anionic exchange chromatography (both from GE-Healthcare).
Mice, Immunization Procedures and Neutralizing Titer Determination
C3H/HeJ male animals (60 day old) were used. Mice received three intramuscular doses of Δ1443TS recombinant enzyme [28], 10 µg each with 100 µg of thiophosphodiester backbone CpG-ODN 1826 oligonucleotide (5′-TCCATGACGTTCCTGACGTT-3′, CpG motifs underlined) (Sigma-Genosys) as adjuvant [57]. TS-inhibition assay was performed as previously described [30], preincubating sera with TS and then testing for remnant activity using α(2,3)sialyllactose (Sigma) and [D-glucose-1-14C]-lactose (GE-Healthcare) as donor and acceptor substrates, respectively. Best responders were selected for cell fusion procedures.
Hybridoma Screening and mAb Production
Splenocyte suspensions were mixed with Sp2/0-Ag14 cells (ATCC) and fusions performed with polyethylene glycol (GIBCO) following standard procedures [58]. Cells were seeded on 96-well flat-bottom plates at a density of 1×105 cells/well in RPMI 1640 with 2 mM Na Piruvate, 10% FBS, 1X hypoxanthine-aminopterin-thymidine (HAT) solution (all from Invitrogen) and supplemented with 2% supernatant of Sp2/0–Ag14 cultures. One-week later, plates were observed under microscopy and the supernatant of those wells containing hybridomas were taken and refilled with fresh medium. ELISA was performed with these samples in search for TS-specific antibody production. To preserve discontinuos epitopes, the recombinant TS 1N1 containing the C-terminus repetitive extension (SAPA) was linked to the plate (MaxiSorb, NUNC) by Protein A-Sepharose (HiTrap, GE-Healthcare)-purified rabbit IgG anti-SAPA, a procedure that safely retained the enzymatic activity (not shown). Those culture wells where anti-TS antibodies were detected were further assayed by TS-inhibition assay [30]. Hybridomas secreting neutralizing antibodies were cloned twice by cell dilution. From four inhibitory antibody-secreting hybridomas detected, only one (named 13G9) was successfully recloned twice by the dilution method and then expanded. The mAb 13G9 was typed as IgG2aκ using the Mouse Antibody Isotyping Kit (GIBCO).
mAb Production and Purification
The 13G9 hybridoma was cultured in RPMI 1640 plus 2 mM Na Piruvate and 10% FBS. Supernatants were clarified and subjected to Protein A-Sepharose (GE-Healthcare) affinity chromatography. The mAb was eluted with 150 mM NaCl, 0.1 M Glycine-HCl pH 3.5 and aliquots were received on 0.1 M Tris-ClH pH 7.6 and dialyzed against 50 mM NaCl, 20 mM Tris-HCl, pH 7.6. Fractions were then loaded into an ion-exchange column (MonoQ, GE-Healthcare) and eluted with a 50–500 mM NaCl gradient in the same buffer (Figure S1). Purified 13G9 mAb was tested by TS-inhibition assay [30] and by reactivity to native and denatured TS-SAPA molecules spotted on nitrocellulose (Figure S1).
Sequence Analysis
cDNA was obtained from 13G9 hybridoma cultures from total RNA using the SuperScript II retrotranscriptase (Invitrogen). cDNA quality control was performed by GAPDH amplification. To amplify the immunoglobulin Fab chains, oligonucleotide primer sets Fwh1 (5′-GTCAGGAGTTGAGCTGGTAAG-3′), Fwh2 (5′-CCTGGGACTTCAGTGAAGATG-3′) and Rvh (5′-TGGAGGACAGGGCTTGATTG-3′) were used for the heavy chain, and Fwl1 (5′-AACAATCATGTGTGCATCTATA-3′), Fwl2 (5′-GAGGAGATCACCCTAACCTG-3′) and Rvl (5′-TCAGGATGTGGTTGCAACAC-3′), for the light chain. Pfu DNA polymerase (Promega) was used and amplicons cloned and sequenced.
Determination of Kinetic Parameters of mAb 13G19 Reactivity
The association/dissociation kinetic constants (kon/koff) were determined with a BIAcore 2000 (BIAcore AB, Uppsala, Sweden). Purified mAb was dialyzed against 20 mM sodium acetate pH 5.6 and immobilized to sensor chips CM5 by using the amine-coupling kit (BIAcore AB). Chips were quenched with 1 M ethanolamine/HCl. After equilibration with 150 mM NaCl, 0.05% P20 surfactant, 10 mM HEPES pH 7.4 (HBS-EP), different concentrations of TS (from 1 nM to 10 µM) were injected at 50 µl/min. After each recording cycle, chips were regenerated with an injection of 2 mM HCl for 30 sec. A free surface of the chip was used as control throughout the experiments. Kinetic constants were evaluated using the program BIAevaluation 3.01 (BIAcore AB). Isothermal titration calorimetry assays were performed in the laboratory of Dr. Alan Cooper (Department of Chemistry, Joseph Black Building University of Glasgow, UK).
Inhibition constants of TS activity were determined for mAb 13G9 and its derived Fab fragment (see below for digestion details) by testing increasing amounts of inhibiting antibody with 2 ng of TS in 30 µl of 150 mM NaCl, Tris-HCl pH 7.6. After 5 min at room temperature (RT), 1 mM sialyllactose and 0.4 nmol (about 40,000 cpm) of [D-glucose-1-14C]-lactose (54.3 mCi/mmol, GE-Healthcare) were added. Remnant TS activity was evaluated [30] after 30 min incubation at RT.
Specificity of mAb 13G9 Reactivity
Trypomastigotes (120×106) were purified from supernatants of infected Vero cell cultures, biotinylated (Sulfo-NHS-LC-Biotin kit form Pierce, Rockford, IL) washed and lysed in the presence of protease inhibitors and centrifuged at 16,000 g. Supernatant was precleared with Protein A-Sepharose (GE-Healthcare) and then reacted with 50 µl of mAb 13G9 hybridoma supernatant for 30 min. Then, Protein A-Sepharose was added and beads extensively washed before SDS-PAGE sample buffer addition and boiling. SDS-PAGE was performed with two parallel aliquots that were then transferred to polyvinylidene fluoride (PVDF) membrane (GE-Healthcare) and developed with either rabbit IgG anti-SAPA followed by horseradish peroxidase (HRP)-labeled secondary antibody or HRP-streptavidin and Super Signal West Pico Chemiluminescent substrate (Pierce).
Inhibition of Parasite Cell Invasion
T. cruzi trypomastigotes (CL-Brenner strain) obtained from Vero cell cultures (Minimum Essential Medium (Invitrogen) supplemented with 0.2% BSA instead of FBS to reduce sialic acid donors) were exhaustively washed with PBS. Parasites were tested by infection of Vero and HeLa cell cultures in the same medium at a multiplicity of infection of 30 in the presence of 0.1 mg/ml of mAb 13G9. After 3 h, cells were washed and medium plus 10% FBS was added. Cells were fixed and stained 24 h later for counting infected cells under microscopy. IgG purified from naïve mouse was used as control.
Inhibition of Parasite Sialylation
Parasites obtained under low sialic acid conditions as above were incubated with 1 mM sialyllactose (Sigma) as sialyl residue donor substrate and TS (2 µg/ml) with or without mAb 13G9 (0.1 mg/ml). After washings with PBS, sialyl residue content was determined by the thiobarbituric HPLC assay after hydrolysis in 0.1 M HCl for 1 h at 80°C [36]. IgG purified from naïve mouse was used as control.
Immunofluorescence
Cell culture-derived trypomastigotes were washed with PBS and incubated with mAb 13G9 (0.05 mg/ml) for 15 min, washed, fixed with 1% paraformaldehyde for 10 min on ice, washed again and blocked for 1 h with 2% BSA plus 5% swine serum in PBS. After that, the parasites were adhered to glass slides via Poly-L-Lysine (Sigma), blocked again, developed with a FITC-conjugated secondary antibody (DAKO, Denmark) and observed by epifluorescence microscopy.
Inhibition of Sialidase Activity
The sialidase activity of TS was determined by measuring the fluorescence of 4-methylumbelliferone released by the hydrolysis of 0.2 mM MU-NANA (Sigma). To 50 ng of TS, different amounts of hybridoma culture supernatant (0–10 µl) or RPMI plus 10% FBS (control) were added. The assay was performed in 50 µl of 150 mM NaCl, 20 mM Tris-ClH pH 6.8. After 10 min at RT, 200 µM of MU-NANA was added and incubation continued for 30 min. The reaction was stopped by dilution in 0.2 M NaHCO3 pH 10, and fluorescence was measured with a DYNA Quant TM 200 fluorometer (GE-Healthcare). Fluorescence values were referred to each RPMI control.
Generation of Antibody Fragments and Immunocomplex
Purified mAb was dialyzed against 2 mM EDTA, 0.1 M Tris-HCl pH 7.6. Before papain digestion 1 mM dithiothreitol (DTT) was added. Papain-agarose beads (Sigma) were washed with the same buffer and activated by addition of 1 mM DTT for 15 min at 37°C. The Fab fragment was generated by digestion for 5 h at 37°C with papain-agarose beads (3U papain/mg mAb; 30 mg of beads for 14 mg of mAb) with gentle end-over-end agitation [58]. After centrifugation at 3,000 rpm, 10 µM trans-epoxysuccinyl-L-leucylamido(4-guanidino)butane (E-64) was added. Undigested antibody and Fc fragment were depleted by Protein A-Sepharose (GE-Healthcare) chromatography and Fab digestion and purity was assayed by SDS-PAGE.
To generate the immunocomplex, pure TS (3.2 clone) was immediately added after the depletion of papain-beads and E-64 addition step before subjecting the mixture to Protein A-Sepharose chromatography as above (Figure S1). The immunocomplex was brought to 25 mM NaCl and concentrated on a BIOMAX 30 K (Millipore) to 14 mg/ml and the buffer changed to 25 mM NaCl, 20 mM Tris-HCl pH 7.6. The purified immunocomplex was essentially free from contaminating proteins and only traces of TS activity remained (see Figure S1). Before crystallization trials, the immunocomplex was repurified by size exclusion chromatography (Superdex200 10/300, GE Healthcare) in an AKTA Purifier, (GE Healthcare) with isocractic elution in 100 mM NaCl, 20 mM Tris-HCl pH 7.6. The resulting single symmetric peak was pooled and concentrated to 7.5 mg/ml by ultrafiltration (Vivaspin, Sartorius-Stedim Biotech; 30 kDa-cutoff membrane) in buffer 25 mM NaCl, 20 mM Tris-HCl pH 7.6.
Immunocomplex Crystallization
Crystallogenesis conditions were screened with a HoneyBee 963 robot (Digilab), using the vapor diffusion method in sitting-drops and reservoirs filled with 150 µl mother liquors (kits JCSG Core Suites I, II, III and IV, Qiagen), rendering 396 different conditions in 96-well plates (3-drop round bottom, Greiner). Protein drops were dispensed mixing equal parts of protein and reservoir solutions (300 nl + 300 nl). Plates were immediately sealed and incubated at 20°C. Hits were obtained in several conditions, one of them was chosen for manual optimization in 24-well plates (VDX, Hampton Research). Final optimized conditions consisted in 2+2 µl hanging-drops, 0.1 M bicine pH 8.5, 10% PEG 20,000, 4% 1,4-dioxane as mother liquor. To obtain larger crystals suitable for single crystal X ray diffraction experiments, repeated macroseeding cycles proved to be essential. Each cycle included selection of best crystal seeds that were transferred to protein-free drops of mother liquor and crystals etched for 30 sec (this washing procedure was repeated three times). Finally, the seed was added to a fresh hanging-drop containing 2 µl protein + 2 µl mother liquor, over 1 ml pure mother liquor. Single needles grew in 5–10 days, cryoprotected with mother liquor containing 12% PEG 20,000 and 30% glycerol and flash frozen in liquid nitrogen until data collection.
Crystal Structure Determination
Single crystal X ray diffraction experiments were performed with a rotating copper anode (Micromax007-HF, Rigaku), multilayer mirrors (Varimax HF, Rigaku) and an image plate detector (Mar345 dtb, Mar Research). Crystals were mounted to collect data under cryogenic temperature (108°K, Cryostream Series 700, Oxford Cryosystems). To attempt improving diffraction resolution, similar crystals were subjected to X ray diffraction using synchrotron radiation at beamline 5.0.2 ALS, equipped with a wiggler inserted device. All data sets were processed with MOSFLM [59], SCALA and TRUNCATE [60].
The structure was solved by molecular replacement with the program Phaser [61], using the models 3CLF (mouse IgG Fab) and 2AH2 (T. cruzi TS in complex with 3-flourosialic acid) as search probes. The Fab probe was previously modified using Chainsaw [60], keeping only the conserved side chains, the rest pruned to alanine or glycine.
The model was refined to the highest collected resolution (3.4 Å) with the program Buster/TNT [38], using a maximum likelihood target function and non-crystallographic restraints throughout the entire process. A TLS model was used to refine correlated anisotropic atomic displacement parameters in large rigid-body domains. Reciprocal space refinement cycles were iterated with manual model rebuilding [39]. Validation tools within Coot were inspected regularly during the refinement process. Last validation steps were done with MolProbity [62].
Accession Numbers
The atomic coordinates and structure factors of the Fab-TS immunocomplex that we have solved in this report are accessible in the PDB with accession code 3OPZ. The models used to solve the phase problem have PDB accession codes 3CLF (mouse IgG Fab fragment) and 2AH2 (T. cruzi TS). A certain number of sialidase and trans-sialidase structures solved previously by us or by other groups, are mentioned in the Discussion section and can be accessed in the PDB with codes: 2AEP (Fab/Influenza-N2 neuraminidase complex); 1NCA (avian N9 neuraminidase complexed with antibody NC41); 1NMB (avian N9 neuraminidase complexed with antibody NC10); 2AEP (N2NA-Mem5Fab); 1S0I (T. cruzi TS in complex with sialyllactose) and 1S0J (T. cruzi TS in complex with MUNANA). Sequence of T. cruzi trans-sialidase can be accessed from the GenBank with the code L26499.
Supporting Information
Zdroje
1. RassiAJrRassiAMarin-NetoJA 2010 Chagas disease. Lancet 375 1388 1402
2. MinoprioPItoharaSHeusserCTonegawaSCoutinhoA 1989 Immunobiology of murine T. cruzi infection: the predominance of parasite-nonspecific responses and the activation of TCRI T cells. Immunol Rev 112 183 207
3. TaliaferroWHPizziT 1955 Connective tissue reactions in normal and immunized mice to a reticulotropic strain of Trypanosoma cruzi. J Infect Dis 96 199 226
4. SavinoW 2006 The thymus is a common target organ in infectious diseases. PLoS Pathog 2 e62
5. TribulattiMVMucciJVan RooijenNLeguizamónMSCampetellaO 2005 The trans-sialidase from Trypanosoma cruzi induces thrombocytopenia during acute Chagas' disease by reducing the platelet sialic acid contents. Infect Immun 73 201 207
6. de TittoEHAraujoFG 1988 Serum neuraminidase activity and hematological alterations in acute human Chagas' disease. Clin Immunol Immunopathol 46 157 161
7. SchenkmanRPVandekerckhoveFSchenkmanS 1993 Mammalian cell sialic acid enhances invasion by Trypanosoma cruzi. Infect Immun 61 898 902
8. SchenkmanSJiangMSHartGWNussenzweigV 1991 A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell 65 1117 1125
9. TomlinsonSPontes de CarvalhoLCVandekerckhoveFNussenzweigV 1994 Role of sialic acid in the resistance of Trypanosoma cruzi trypomastigotes to complement. J Immunol 153 3141 3147
10. Pereira-ChioccolaVLAcosta-SerranoACorreia de AlmeidaIFergusonMASouto-PadronT 2000 Mucin-like molecules form a negatively charged coat that protects Trypanosoma cruzi trypomastigotes from killing by human anti-alpha-galactosyl antibodies. J Cell Sci 113 1299 1307
11. PreviatoJOAndradeAFPessolaniMCMendonca-PreviatoL 1985 Incorporation of sialic acid into Trypanosoma cruzi macromolecules. A proposal for a new metabolic route. Mol Biochem Parasitol 16 85 96
12. Rubin-de-CelisSSUemuraHYoshidaNSchenkmanS 2006 Expression of trypomastigote trans-sialidase in metacyclic forms of Trypanosoma cruzi increases parasite escape from its parasitophorous vacuole. Cell Microbiol 8 1888 1898
13. AgustiRCoutoASCampetellaOEFraschACde LederkremerRM 1997 The trans-sialidase of Trypanosoma cruzi is anchored by two different lipids. Glycobiology 7 731 735
14. AlvarezPBuscagliaCACampetellaO 2004 Improving protein pharmacokinetics by genetic fusion to simple amino acid sequences. J Biol Chem 279 3375 3381
15. LeguizamónMSCampetellaORussomandoGAlmironMGuillenI 1994 Antibodies inhibiting Trypanosoma cruzi trans-sialidase activity in sera from human infections. J Infect Dis 170 1570 1574
16. LeguizamónMSMocettiEGarcia RivelloHArgibayPCampetellaO 1999 trans-sialidase from Trypanosoma cruzi induces apoptosis in cells from the immune system in vivo. J Infect Dis 180 1398 1402
17. RissoMGPitcovskyTACaccuriRLCampetellaOLeguizamónMS 2007 Immune system pathogenesis is prevented by the neutralization of the systemic trans-sialidase from Trypanosoma cruzi during severe infections. Parasitology 134 503 510
18. MucciJHidalgoAMocettiEArgibayPFLeguizamónMS 2002 Thymocyte depletion in Trypanosoma cruzi infection is mediated by trans-sialidase-induced apoptosis on nurse cells complex. Proc Natl Acad Sci U S A 99 3896 3901
19. ChuenkovaMPereiraME 1995 Trypanosoma cruzi trans-sialidase: enhancement of virulence in a murine model of Chagas' disease. J Exp Med 181 1693 1703
20. RissoMGGarbarinoGBMocettiECampetellaOGonzález CappaSM 2004 Differential expression of a virulence factor, the trans-sialidase, by the main Trypanosoma cruzi phylogenetic lineages. J Infect Dis 189 2250 2259
21. MunozMJMurciaLSegoviaM 2011 The urgent need to develop new drugs and tools for the treatment of Chagas disease. Expert Rev Anti Infect Ther 9 5 7
22. WilkinsonSRTaylorMCHornDKellyJMCheesemanI 2008 A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci U S A 105 5022 5027
23. BuschiazzoATavaresGACampetellaOSpinelliSCremonaML 2000 Structural basis of sialyltransferase activity in trypanosomal sialidases. Embo J 19 16 24
24. BuschiazzoAAmayaMFCremonaMLFraschACAlzariPM 2002 The crystal structure and mode of action of trans-sialidase, a key enzyme in Trypanosoma cruzi pathogenesis. Mol Cell 10 757 768
25. NeresJBrewerMLRatierLBottiHBuschiazzoA 2009 Discovery of novel inhibitors of Trypanosoma cruzi trans-sialidase from in silico screening. Bioorg Med Chem Lett 19 589 596
26. NeresJBryceRADouglasKT 2008 Rational drug design in parasitology: trans-sialidase as a case study for Chagas disease. Drug Discov Today 13 110 117
27. BuchiniSBuschiazzoAWithersSG 2008 A new generation of specific Trypanosoma cruzi trans-sialidase inhibitors. Angew Chem Int Ed Engl 47 2700 2703
28. PitcovskyTABuscagliaCAMucciJCampetellaO 2002 A functional network of intramolecular cross-reacting epitopes delays the elicitation of neutralizing antibodies to Trypanosoma cruzi trans-sialidase. J Infect Dis 186 397 404
29. PitcovskyTAMucciJAlvarezPLeguizamónMSBurroneO 2001 Epitope mapping of trans-sialidase from Trypanosoma cruzi reveals the presence of several cross-reactive determinants. Infect Immun 69 1869 1875
30. LeguizamónMSRussomandoGLuquettiARassiAAlmironM 1997 Long-lasting antibodies detected by a trans-sialidase inhibition assay of sera from parasite-free, serologically cured chagasic patients. J Infect Dis 175 1272 1275
31. CampetellaOSánchezDOCazzuloJJFraschACC 1992 A superfamily of Trypanosoma cruzi surface antigens. Parasitol Today 8 378 381
32. CremonaMLCampetellaOSanchezDOFraschAC 1999 Enzymically inactive members of the trans-sialidase family from Trypanosoma cruzi display beta-galactose binding activity. Glycobiology 9 581 587
33. CremonaMLSanchezDOFraschACCampetellaO 1995 A single tyrosine differentiates active and inactive Trypanosoma cruzi trans-sialidases. Gene 160 123 128
34. WattsAGDamagerIAmayaMLBuschiazzoAAlzariP 2003 Trypanosoma cruzi trans-sialidase operates through a covalent sialyl-enzyme intermediate: tyrosine is the catalytic nucleophile. J Am Chem Soc 125 7532 7533
35. BuscagliaCACampetellaOLeguizamónMSFraschAC 1998 The repetitive domain of Trypanosoma cruzi trans-sialidase enhances the immune response against the catalytic domain. J Infect Dis 177 431 436
36. PowellLDHartGW 1986 Quantitation of picomole levels of N-acetyl - and N-glycolylneuraminic acids by a HPLC-adaptation of the thiobarbituric acid assay. Anal Biochem 157 179 185
37. MuiáRPYuHPrescherJAHellmanUChenX 2010 Identification of glycoproteins targeted by Trypanosoma cruzi trans-sialidase, a virulence factor that disturbs lymphocyte glycosylation. Glycobiology 20 833 842
38. BlancERoversiPVonrheinCFlensburgCLeaSM 2004 Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60 2210 2221
39. EmsleyPCowtanK 2004 Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 2126 2132
40. SmartOSBrandlMFlensburgCKellerPPaciorekW 2008 Refinement with Local Structure Similarity Restraints (LSSR) Enables Exploitation of Information from Related Structures and Facilitates use of NCS. Abstr Annu Meet Am Crystallogr Assoc Abstract TP139
41. DaviesDRPadlanEASheriffS 1990 Antibody-antigen complexes. Annu Rev Biochem 59 439 473
42. LawrenceMCColmanPM 1993 Shape complementarity at protein/protein interfaces. J Mol Biol 234 946 950
43. MucciJRissoMGLeguizamónMSFraschACCampetellaO 2006 The trans-sialidase from Trypanosoma cruzi triggers apoptosis by target cell sialylation. Cell Microbiol 8 1086 1095
44. WebsterRGReayPALaverWG 1988 Protection against lethal influenza with neuraminidase. Virology 164 230 237
45. StumppMTBinzHKAmstutzP 2008 DARPins: a new generation of protein therapeutics. Drug Discov Today 13 695 701
46. HolligerPHudsonPJ 2005 Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23 1126 1136
47. NatarajanADeNardoSJ 2010 PEGylation of Antibody Fragments to Improve Pharmacodynamics and Pharmacokinetics. KontermannRDübelS Antibody Engineering 2 ed. Heidelberg Springer
48. VenkatramaniLBochkarevaELeeJTGulatiUGraeme LaverW 2006 An epidemiologically significant epitope of a 1998 human influenza virus neuraminidase forms a highly hydrated interface in the NA-antibody complex. J Mol Biol 356 651 663
49. TulipWRVargheseJNLaverWGWebsterRGColmanPM 1992 Refined crystal structure of the influenza virus N9 neuraminidase-NC41 Fab complex. J Mol Biol 227 122 148
50. TulipWRVargheseJNWebsterRGLaverWGColmanPM 1992 Crystal structures of two mutant neuraminidase-antibody complexes with amino acid substitutions in the interface. J Mol Biol 227 149 159
51. MalbyRLTulipWRHarleyVRMcKimm-BreschkinJLLaverWG 1994 The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 2 733 746
52. GulatiUHwangCCVenkatramaniLGulatiSStraySJ 2002 Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98). J Virol 76 12274 12280
53. AmayaMFBuschiazzoANguyenTAlzariPM 2003 The high resolution structures of free and inhibitor-bound Trypanosoma rangeli sialidase and its comparison with T. cruzi trans-sialidase. J Mol Biol 325 773 784
54. CohenNC 2007 Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R&D pipeline challenge. Chem Biol Drug Des 70 557 565
55. MontagnaGCremonaMLParisGAmayaMFBuschiazzoA 2002 The trans-sialidase from the african trypanosome Trypanosoma brucei. Eur J Biochem 269 2941 2950
56. BuschiazzoAFraschACCampetellaO 1996 Medium scale production and purification to homogeneity of a recombinant trans-sialidase from Trypanosoma cruzi. Cell Mol Biol (Noisy-le-grand) 42 703 710
57. FrankFMPetrayPBCazorlaSIMunozMCCorralRS 2003 Use of a purified Trypanosoma cruzi antigen and CpG oligodeoxynucleotides for immunoprotection against a lethal challenge with trypomastigotes. Vaccine 22 77 86
58. GodingJW 1996 Monoclonal antibodies: principles and practices. London Academic Press
59. LeslieAGW 1990 Molecular data processing. MorasDPodjarnyADThierryJC Crystallographic computing New York Oxford University Press
60. Collaborative Computational Project N 1994 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50 760 763
61. McCoyAJGrosse-KunstleveRWAdamsPDWinnMDStoroniLC 2007 Phaser crystallographic software. J Appl Crystallogr 40 658 674
62. ChenVBArendallWB3rdHeaddJJKeedyDAImmorminoRM 2010 MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66 12 21
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Type 1 Interferons and Antiviral CD8 T-Cell Responses
- Sequence Divergent RXLR Effectors Share a Structural Fold Conserved across Plant Pathogenic Oomycete Species
- Temporal Expression of Bacterial Proteins Instructs Host CD4 T Cell Expansion and Th17 Development
- Sexual Development in : Lessons from Functional Analyses