
Infection-Induced Retrotransposon-Derived Noncoding RNAs Enhance Herpesviral Gene Expression via the NF-κB Pathway
Short interspersed nuclear elements (SINEs) are noncoding mobile genetic elements that are present at ~106 copies per mammalian genome, roughly comprising 10% of mammalian genomic real estate. SINEs are typically transcriptionally silenced, though in some cases viral infection can promote their expression, yet to an unknown functional outcome. Thus, SINE elements represent the largest class of infection-inducible noncoding RNAs that are functionally uncharacterized. Here, we reveal that SINE RNAs play a critical role in the host-pathogen interaction in that they are required for efficient murine gammaherpesvirus 68 (MHV68) replication and gene expression. We demonstrate that SINE RNAs, both exogenously expressed and infection-induced, are robust activators of the IKKβ kinase, a key signaling molecule in the innate immune response. Activation of the IKKβ kinase by SINE RNA is mediated through both MAVS-dependent and independent mechanisms. Moreover, we demonstrate the activation of the IKKβ via SINE RNA is required to drive the phosphorylation of MHV68 RTA, the main viral transcriptional activator, which enhances its transcriptional activating property. Collectively, we reveal the first example of a role for SINE RNAs in the host-pathogen interaction and identify them as a key immune signaling molecule early during infection. Though SINE RNAs activate the innate immune response, MHV68 has co-opted SINE-mediate innate immune activation to enhance the viral lifecycle.
Published in the journal:
. PLoS Pathog 11(11): e32767. doi:10.1371/journal.ppat.1005260
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005260
Summary
Short interspersed nuclear elements (SINEs) are noncoding mobile genetic elements that are present at ~106 copies per mammalian genome, roughly comprising 10% of mammalian genomic real estate. SINEs are typically transcriptionally silenced, though in some cases viral infection can promote their expression, yet to an unknown functional outcome. Thus, SINE elements represent the largest class of infection-inducible noncoding RNAs that are functionally uncharacterized. Here, we reveal that SINE RNAs play a critical role in the host-pathogen interaction in that they are required for efficient murine gammaherpesvirus 68 (MHV68) replication and gene expression. We demonstrate that SINE RNAs, both exogenously expressed and infection-induced, are robust activators of the IKKβ kinase, a key signaling molecule in the innate immune response. Activation of the IKKβ kinase by SINE RNA is mediated through both MAVS-dependent and independent mechanisms. Moreover, we demonstrate the activation of the IKKβ via SINE RNA is required to drive the phosphorylation of MHV68 RTA, the main viral transcriptional activator, which enhances its transcriptional activating property. Collectively, we reveal the first example of a role for SINE RNAs in the host-pathogen interaction and identify them as a key immune signaling molecule early during infection. Though SINE RNAs activate the innate immune response, MHV68 has co-opted SINE-mediate innate immune activation to enhance the viral lifecycle.
Introduction
While only ~1.5% of mammalian genomes consist of protein coding sequence, upwards of 75% of the genome is transcribed [1, 2]. A considerable amount of this transcription generates stable non-protein-coding RNAs (ncRNAs) of potential biological relevance. Similarly, transcription from the genomes of many large double stranded (ds) DNA viruses is pervasive and can generate an abundance of long and short ncRNAs, a number of which have key roles in viral replication and pathogenesis [3–11]. While there is an increasing appreciation that viruses have adopted ncRNAs as part of their gene regulatory repertoire, with the exception of some small ncRNAs such as microRNAs, how most other cellular ncRNAs may impact the gene expression landscape during infection remains unknown. Given that viruses have provided significant insight into mammalian gene regulation, they have the potential to reveal new features of ncRNA biology.
One of the largest potential sources of host-derived ncRNAs is a class of retrotransposons called short interspersed nuclear elements (SINEs), as these comprise greater than 10% of the human and mouse genomes [12–15]. SINEs are non-autonomous and require co-expression of protein products encoded within long interspersed nuclear elements (LINEs) for retrotransposition [16]. Alu elements are the predominant SINE family in humans, while the B1 and B2 SINEs are the major families in the murine genome. All SINE families are evolutionarily derived from endogenous RNA Polymerase III (Pol III) transcripts: Alu and B1 SINEs are derived from 7SL RNA, the RNA component of signal recognition particle, and B2 SINEs are derived from transfer RNA (tRNA) [17–20]. SINE elements contain internal Box A and Box B RNA Pol III promoter elements that drive transcription of a SINE ncRNA. In general, SINE elements are transcriptionally silenced in healthy somatic cells, although they can be activated by a variety of chemical and biological stresses [21]. In this regard, several viruses including herpes simplex virus 1 (HSV-1) [22, 23], adenovirus type 5 (Ad5) [24], and Minute virus of mice (MVM) have been shown to induce SINE RNA expression upon infection [25]. SINE elements are thus a robust source of inducible ncRNAs, whose expression could impact the gene expression environment during infection. Indeed, there is precedence for SINE RNA functioning in the regulation of gene expression during heat shock, where transcribed Alu and B2 SINE RNAs participate in transcriptional repression through direct interactions with RNA pol II [26–30].
Additional observations suggest that SINE ncRNA expression can also interface with components of the innate immune system, perhaps in a manner linked to their secondary structure. SINE RNAs are highly structured with multiple regions of long double stranded RNA (dsRNA) [31, 32], and the majority possess 5’-triphosphate moieties [33]. These ncRNAs thus have the potential to be recognized by cellular dsRNA sensors and could therefore serve as inducible immune signaling molecules. Early studies revealed that Alu RNA is efficiently bound by the double stranded RNA activated protein kinase (PKR) [34], and can function in either an inhibitory or activating capacity depending on the ncRNA concentration [35]. Furthermore, aberrant expression of Alu RNAs within retinal pigmented epithelium induces TLR-independent activation of the NLRP3 inflammasome, leading to geographic atrophy, a form of age-related macular degeneration (AMD) [36–38]. Thus, it is possible that mammalian cells have incorporated the regulated induction of SINE ncRNAs as a means to help control immune activation and gene expression, although aberrant or sustained SINE transcription is likely detrimental.
Here, we explored potential roles for SINE ncRNAs induced during viral infection using the murine gammaherpesvirus MHV68, which we found induces SINE ncRNA transcription in a rapid and sustained manner. MHV68 is a widely used model system for probing the in vivo biology and replication of gammaherpesviruses, a subfamily of large, nuclear replicating dsDNA viruses that includes the oncogenic human viruses Kaposi’s sarcoma-associated herpesvirus (KSHV) and Epstein-Barr virus (EBV). Unexpectedly, the induction of SINE ncRNAs boosts viral replication and gene expression, suggesting that herpesviruses have co-opted SINEs for proviral functions. The stimulatory effect on viral replication is linked to SINE RNA-based activation of the innate immune system, in particular the IKKβ component of the NF-κB signaling pathway. IKKβ is a known activator of the primary viral transcriptional transactivator protein RTA [39], and SINE expression enhances the IKKβ-dependent phosphorylation of RTA, thereby boosting its activity. We find that SINE RNAs activate NF-κB through both mitochondrial antiviral-signaling (MAVS) protein dependent and independent mechanisms. Collectively, our findings reveal that virus-induced retrotransposon expression contributes to activation of innate immune signaling during infection, but that herpesviruses exploit this pathway to bolster viral gene expression.
Results
MHV68 infection activates SINE transcription
In unstressed somatic cells, SINE loci are transcriptionally repressed and thus RNA Pol III-transcribed SINE RNAs are either undetectable or only weakly expressed. To determine whether gammaherpesvirus infection activated SINE RNA expression, NIH3T3 cells were infected with MHV68 and B1 and B2 SINE RNA levels were quantified by primer extension. Indeed, both B1 and B2 SINEs were induced specifically upon infection, with B2 RNA levels exceeding those of the highly abundant RNA Pol III-transcribed 7SK small nuclear RNA (snRNA) (Fig 1A). This increase in RNA Pol III transcriptional activity during MHV68 infection was specific for SINE loci, as infected cells displayed no alteration in the levels of multiple other RNA Pol III-derived transcripts, including tRNAVal, 5S ribosomal RNA (rRNA), and 7SL RNA as measured by Northern blotting (Fig 1B). Additionally, B1 and B2 SINE RNAs localized to both the cytoplasmic and nuclear compartments (Fig 1C).
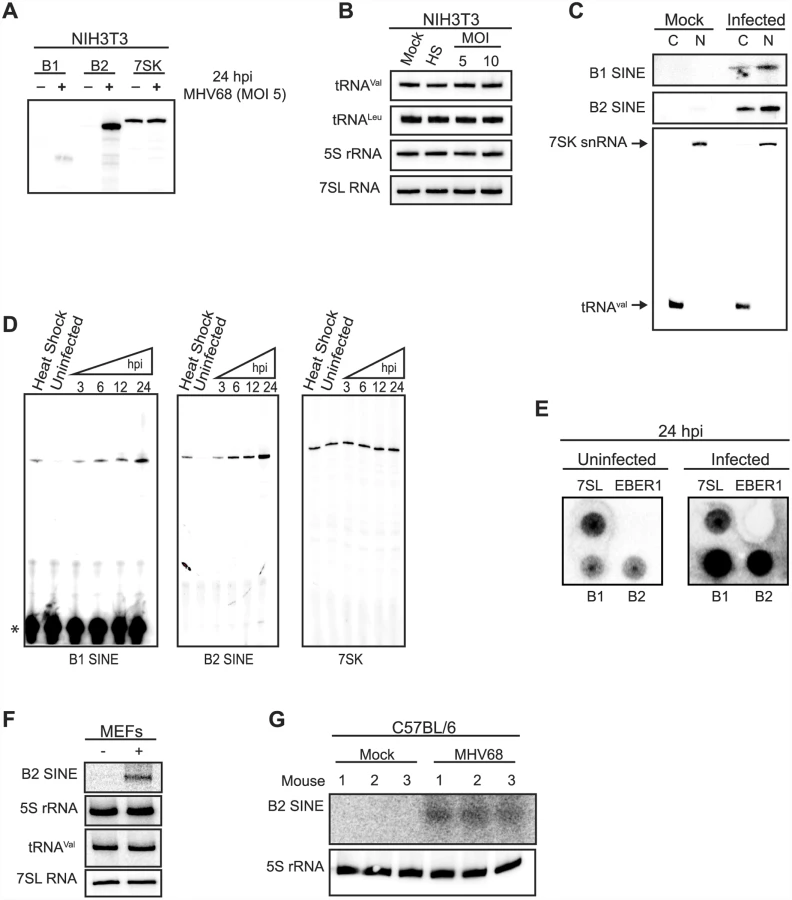
SINE RNA induction was both rapid and sustained, occurring by 3 h post infection (hpi) at levels similar to heat shock-driven induction and continually increasing throughout the 24 hpi time course (Fig 1D). During the same time course 7SK levels remained constant in both infected and uninfected cells (Fig 1D). To determine whether the increase in SINE RNAs occurred at the level of transcription, nuclear run-on assays were performed using nuclei from uninfected or MHV68-infected cells. A robust transcriptional increase was observed for both B1 and B2 in infected cells, whereas no transcriptional difference was observed for the RNA Pol III transcribed 7SL RNA (Fig 1E). No signal was detected for the negative control ncRNA EBER1 from Epstein-Barr virus, which is not expressed in these cells, confirming the specificity of the run-on signals (Fig 1E). Finally, the increase in SINE RNA was also observed upon infection of primary mouse embryonic fibroblasts (MEFs) and in vivo in the lung tissue of MHV68-infected C57BL/6 mice at 5 days post infection, indicating the induction was not restricted to established cell lines (Fig 1F and 1G). Together, these data demonstrate that MHV68 infection results in a rapid, sustained, and specific transcriptional activation of B1 and B2 SINE loci.
Viral early gene expression is required for sustained SINE induction
We next sought to identify aspect(s) of the MHV68 life cycle responsible for SINE RNA induction. After entry, the viral genome is delivered to the nucleus and transcription of immediate early and early genes occurs, which in turn enable viral DNA replication and subsequent expression of late viral genes necessary for progeny virion assembly. UV crosslinking of viral particles does not block viral attachment to cells, but inactivates the viral genome thereby inhibiting viral gene expression and replication (Fig 2A and 2B). NIH3T3 cells incubated with UV-inactivated virus still displayed the early SINE RNA induction at 3 hpi, but unlike incubation with infectious virus, B1 and B2 RNA levels rapidly subsided over the 24 hpi time course (Fig 2C). (It should be noted that the B1 gels in Fig 2 were exposed longer than the B2 gels to better visualize the faint B1 signal). This suggests that viral activation of SINEs is biphasic, with the initial induction occurring in response to viral attachment and/or entry, and sustained SINE transcription reliant on downstream aspects of the viral life cycle.

To determine whether sustained SINE activation required viral genome replication or late gene expression, cells were treated with the viral DNA polymerase inhibitor phosphonoacetic acid (PAA) prior to MHV68 infection (Fig 2D). SINE RNA induction was similar at 24 hpi in control - and PAA-treated cells (Fig 2E). These data suggest that viral early gene expression, but not DNA replication or late gene expression drives sustained transcriptional activation of SINE loci. Although it is possible that a specific viral gene product might be responsible for SINE activation, we have been unable to identify any MHV68 genes whose individual expression in NIH3T3 cells induced SINE transcription. Thus, SINE RNA induction is likely to occur as a consequence of the combined activity of multiple viral genes and/or as a more general cellular response to infection.
B2 SINE RNA enhances viral replication and gene expression
We hypothesized that the virus-induced noncoding SINE RNAs may play regulatory roles related to infection. To test whether SINE transcription impacts the MHV68 lifecycle, we measured viral replication upon specific knockdown of B1 or B2 SINE RNAs using 2′-O-methylated and phosphorothioate-substituted antisense oligonucleotides (ASO), which direct RNase H-based cleavage of target RNAs. Transfection of B1 and/or B2 ASOs 3 h prior to MHV68 infection of NIH3T3 cells significantly reduced the levels of SINE RNA at 24 hpi (Fig 3A). Control ASOs did not impact B1 or B2 levels, and ASO treatment of uninfected cells did not induce SINE expression (Fig 3A). Remarkably, B2 depletion resulted in delayed viral replication kinetics in a multistep growth curve and a ~15-fold decrease in progeny virion production (Fig 3B). We observed a similar (~10-fold) decrease in viral replication in a single-step growth curve upon treatment of NIH3T3 cells with the specific RNA Pol III inhibitor ML-60218 6 h prior to infection to block SINE transcription (S1 Fig). Although ML-60218 blocks transcription of all RNA Pol III genes, constitutive Pol III transcripts in general have long half-lives and thus, unlike B1 and B2 RNAs, their steady state levels are not appreciably affected by the treatment. Specific ASO-mediated depletion of B1 RNA, which accumulates to far lower levels than B2 RNA during MHV68 infection, had no effect on viral replication, and co-depletion of both B1 and B2 did not enhance the replication defect observed upon B2 knockdown (Fig 3B).

The above results indicated that induction of B2 SINE RNA by MHV68 is beneficial to the viral lifecycle. We therefore examined whether the SINE RNAs impacted viral gene expression by measuring the accumulation of viral mRNAs from different kinetic classes in cells treated with control or SINE-specific ASOs. Indeed, RT-qPCR analyses revealed that depletion of B2 RNA resulted in a significant reduction in the levels of all viral RNAs tested (Fig 3C). In agreement with the viral replication data, depletion of B1 RNA had no effect on viral gene expression. Interestingly, the effect of B2 RNA was primarily directed at viral genes, as we observed no decrease in the abundance of a set of 5 cellular mRNAs upon treatment with B1 or B2 ASOs (Fig 3C). However, we did note an increase in the levels of the GAPDH mRNA upon B2 depletion, in agreement with data showing that heat shock-induced B2 RNA can transcriptionally repress select promoters [26, 27]. We obtained similar results when SINE RNA transcription was inhibited by pre-treatment with ML-60218 (S1 Fig). Thus, virus-induced expression of B2 SINE RNA broadly enhances MHV68 mRNA abundance, and this stimulatory effect primarily impacts viral gene expression even though RNA Pol II transcribes both viral and cellular mRNAs.
B2 SINEs are not associated with viral promoters
Several long noncoding RNAs manipulate gene expression through their recruitment to specific cellular gene promoters [40]. In this regard, B2 SINE RNA has been shown to interact with RNA Pol II at select promoters to inhibit transcription [26, 27]. Although this explains the apparent suppression of GAPDH mRNA by B2, the stimulatory effect of B2 on MHV68 mRNA suggested that SINE RNAs might not function through their recruitment to promoters. Using chromatin isolation by RNA purification (ChIRP) we examined whether B2 RNA was present at viral or cellular promoters. ChIRP-qPCR analyses detected B2 RNA at the promoter of GAPDH and RPL13a (Fig 3D and 3E). In contrast to our findings with cellular promoters, we were unable to ChIRP B2 RNA to any viral promoters, suggesting that B2 SINE RNA modulates viral gene expression indirectly.
SINE RNAs activate the NF-κB signaling pathway
Our above data suggested that SINE RNAs have promoter-specific effects on cellular genes. We tested this hypothesis by analyzing the effect of B1 and B2 SINE RNA expression on a panel of promoter elements cloned upstream of a luciferase reporter (Fig 4A). To monitor effects specifically linked to SINE RNAs as opposed to secondary effects stemming from infection, we co-expressed each reporter with consensus B1 or B2 SINEs derived from RNA Pol III-driven SINE expression constructs in NIH3T3 cells. SINE RNAs modestly suppressed the AP1 promoter, had no impact on the p53 and ISRE promoters, but significantly activated both the NF-κB-driven and Sp1-driven luciferase reporters (Fig 4B). In each case, B2 RNA had a more potent effect than B1, and all changes in luciferase reporter levels required SINE RNA expression, as they were blocked upon treatment with the RNA Pol III inhibitor ML-60218 (Fig 4B).

MHV68 infection has been shown to activate the NF-κB pathway, components of which are used by the virus for robust induction of lytic gene expression [39, 41–43]. Though MHV68 blunts the NF-κB transcriptional response by subsequently inducing degradation of the RelA/p65 subunit of NF-κB during the first 4 hpi [41], the IKKβ kinase component of the pathway is co-opted to promote phosphorylation of the major viral lytic transactivator RTA. Our findings suggested that induction of SINE RNAs might contribute to NF-κB activation during infection, thereby potentiating viral replication. To test this hypothesis, we first examined whether SINE RNA expression was sufficient to activate endogenous components of the NF-κB signaling pathway. Indeed, transfection of plasmid-based SINEs in uninfected cells resulted in increased phosphorylation of the NF-κB p65 subunit, a marker of NF-κB activation (Fig 4C). Furthermore, inhibition of IKKβ by treatment with its specific inhibitor BAY 11–7082 significantly reduced the ability of SINE RNA to activate the NF-κB luciferase reporter, but did not impact SINE-driven activation of the Sp1 reporter (Fig 4D). Thus, SINE RNAs likely activate this pathway in the cytoplasm, upstream of IKKβ.
SINE RNAs signal through MAVS to activate IKKβ
IKKβ activation during MHV68 infection is at least partially mediated through activation of the MAVS adaptor protein [39]. However, the mechanism underlying MAVS activation during MHV68 infection remains unclear. MHV68 is a dsDNA virus, yet MAVS activation generally occurs upon recognition by upstream RIG-I-like receptors of nucleic acid features associated with RNA viruses, including double-stranded RNA with either a 5′ diphosphate or 5′ triphosphate [44]. We therefore explored whether the structured SINE RNAs might serve as the MAVS activation signal to stimulate IKKβ during MHV68 infection. Indeed, the ability of B1 or B2 SINE RNA expression to activate the NF-κB promoter was significantly, though not completely, impaired in MAVS-/- fibroblasts relative to WT fibroblasts (Fig 5A). This effect was specific for the NF-κB promoter, as no decrease in B1 - or B2-induced Sp1 promoter activation occurred in the cells lacking MAVS (Fig 5A).

We next examined the role of MAVS in endogenous, MHV68-induced SINE RNA-mediated phosphorylation of RelA/p65. Within 1 hpi, RelA/p65 is phosphorylated at Serine 468 in a MAVS-IKKβ-dependent manner, a mark that primes its degradation via the proteasome, thereby blunting the NF-κB response [41]. The ability of MHV68 to induce phosphorylation of RelA/p65 was significantly reduced in both MAVS-/- fibroblasts and in WT fibroblasts that had been depleted of B2 RNA using a B2-specific ASO (Fig 5B). As anticipated, no effect was observed upon depletion of the B1 RNA, in agreement with the fact that it is only weakly induced relative to B2 RNA by MHV68 (Fig 5B). Additionally, MHV68-induced RelA/p65 phosphorylation was further reduced to background levels when B2 RNA was depleted in the MAVS-/- cells, suggesting that at least a portion of SINE RNA-based IKKβ activation occurs in a MAVS-independent manner (Fig 5C). These results indicate that B2 RNA contributes to the blunting of the NF-κB response during MHV68 infection.
We next compared the relative importance of MAVS and B2 SINE RNA in productive MHV68 infection. MHV68 replication was impaired to a similar extent in fibroblasts lacking MAVS as in cells depleted of B2 SINE RNA, as measured in multi-step growth curves (Fig 5D). However, depletion of B2 RNA in a MAVS-/ - background caused a further reduction in viral replication, to levels ~30-fold lower than in untreated WT cells. Collectively, these data indicate that the B2 RNAs are major contributors to the IKKβ activation observed during MHV68 infection, and that they stimulate this pathway via both MAVS-dependent and -independent mechanisms to boost viral replication.
SINE RNAs enhance MHV68 RTA activity via IKKβ-mediated phosphorylation to increase viral gene expression
The broad enhancement of MHV68 gene expression by B2 RNA, coupled with the absence of B2 at viral promoters, suggested that SINE RNAs act at an early stage of the viral lifecycle to boost the subsequent gene expression cascade. Notably, the major viral lytic transactivator protein RTA was recently shown to be phosphorylated by IKKβ in a manner that increased RTA activity [39]. RTA is a viral protein expressed with immediate early kinetics, and its activity broadly impacts the viral gene expression cascade [45–47]. We therefore hypothesized that the pro-viral effects of the SINE RNAs might be mediated through an increase in RTA activity caused by B2-induced NF-κB pathway activation.
We tested this idea first by examining whether the activation of IKKβ by B1 and B2 RNA could potentiate RTA phosphorylation, as measured by in vivo P32 orthophosphate labeling. FLAG-RTA was expressed in NIH3T3 cells in the presence or absence of B1 or B2 expression plasmids in 32P orthophosphate containing growth media, and immunoprecipitated with anti-FLAG coupled beads to quantify its phosphorylation status. Expression of B1 and B2 promoted a significant increase in RTA phosphorylation relative to the control plasmid (Fig 6A). Furthermore, 32P orthophosphate labeling in MHV68-infected cells revealed that depletion of B2 RNA during infection reduced FLAG-RTA phosphorylation (Fig 6B).

Next, to determine whether this SINE-mediated phosphorylation change resulted in increased RTA activity, we examined the ability of RTA to transactivate several established RTA-responsive promoters in the presence or absence of B1 and B2 SINEs. Cells were co-transfected with RTA and luciferase reporter plasmids containing the RTA-responsive viral promoters from either RTA, ORF57, or M3 in the presence or absence of B1 and B2 SINE constructs (Fig 6C). Indeed, RTA mediated transcriptional activation of all three promoters was significantly enhanced upon co-expression of either B1 or B2 relative to the control plasmid. This ability of the SINE RNAs to enhance RTA activity was dependent on IKKβ phosphorylation of RTA, as an RTA mutant (RTA-TTS/A) [39] in which the IKKβ phosphorylation sites were mutated retained WT basal transcriptional activity but was unresponsive to the SINE RNAs (Fig 6C). Collectively, these data indicate that SINE RNA-mediated activation of IKKβ drives RTA phosphorylation, thereby increasing its activity on viral promoters and enhancing MHV68 gene expression.
Discussion
Here, we demonstrate that MHV68-induced activation of SINE RNA serves to regulate viral and cellular gene expression through distinct mechanisms (Fig 7). SINE RNAs reside in the nucleus and in the cytoplasm, and participate in gene regulation in both compartments. Nuclear SINE RNAs appear absent from viral promoters but associate with specific cellular promoters that are repressed upon SINE activation, while SINE RNAs in the cytoplasm drive activation of the antiviral NF-κB signaling pathway. Though this pathway is normally detrimental to viral replication, MHV68 co-opts the IKKβ component of the NF-κB cascade to boost the activity of the viral lytic transactivator RTA, thereby enhancing viral gene expression and replication [39]. Thus, beyond direct regulation of cellular gene expression, induction of this class of ncRNAs may be linked to early immune-based sensing of infection, a process that has been hijacked by gammaherpesviruses to enhance viral gene expression.

Activation of SINE expression following MHV68 infection is a biphasic response with an initial phase arising as a result of either viral attachment or entry in to cells, and a second response that requires progression of the infection past entry, and includes immediate early and early viral gene expression. While it is possible that the initial burst of SINE expression observed following infection with U.V. inactivated virus is mediated by protein components of the tegument, we disfavor this scenario as we have been unable to identify an individual MHV68 gene whose expression is sufficient to induce SINE expression. This suggests that the mechanism by which MHV68 induces SINE expression is distinct from that of HSV-1 and Ad5, in which specific viral proteins have been implicated in promoting SINE induction. For instance, in the context of Ad5 infection, loss of E1a, E1b, or E4 ORFs 3 and 6 results in a significant decrease in SINE expression in infected cells [24]. Additionally, HSV-1 ICP27 has been demonstrated to enhance the activity of the RNA Pol III general transcription factor TFIIIC leading to increased SINE RNA expression [48]. In the case of MHV68, we believe a more likely model is one that focuses on the cellular stresses imposed on the cell during infection, including disruption of plasma membrane homeostasis during viral attachment and entry, and triggering of the innate immune response. This suggests that at least the initial burst of SINE transcription should occur regardless of whether infection progresses to latency or lytic replication. However, whether a latent infection results in sustained SINE activation similar to a lytic infection is currently unknown. As expression of SINE RNA potentiates a normally antiviral NF-κB response, the coupling of viral entry receptors and/or plasma membrane homeostasis to the production of immunogenic RNAs would constitute a means to rapidly prime the innate immune response for a potential pathogen exposure.
SINE induction may also occur as an early component of innate immune sensing. Indeed, many pathogen recognition receptors (PRRs) such as the toll like receptors (TLRs), which are present at both the cell surface and endosome, are engaged by herpesviruses [49]. In this regard, cell surface associated TLR2 has been implicated in detecting MHV68, likely recognizing viral glycoproteins [50]. Alternatively or perhaps concomitantly, the incoming viral DNA may be sensed by nucleic acid sensing PRRs, for which both endosomal TLR9 and the cytoplasmic cGAS-STING sensing pathways have been implicated [51–54]. As both stimulation of PRRs and exogenous overexpression of SINEs can mediate NF-κB, the induction of SINEs in response to PRR stimulation could establish a feed forward or signal amplification mechanism. In support of a model in which SINE RNAs are generated in response to PRR stimulation are the recent findings that lipopolysaccharide (LPS) stimulation of TLR4 induces a general activation of RNA Pol III transcription [55]. Though we do not detect a general increase in all RNA Pol III synthesized RNAs, its possible that MHV68 has evolved ways to manipulate the RNA Pol III transcriptional program to selectively drive synthesis of SINE RNAs, which enhance the MHV68 lifecycle. Interestingly, stimulation of TLRs 3,4,5, and 9 promote MHV68 reactivation from latently infected B cells [56].
SINE RNAs are present in both the cytoplasm and nucleus, and our and others’ data argue that they regulate gene expression via multiple mechanisms, some of which are linked to their location within the cell. For MHV68 it is the cytoplasmic fraction that is likely important for enhancing viral gene expression through the activation of the NF-κB pathway. Multiple studies have established that the NF-κB pathway is crucial for gammaherpesvirus latent infection [57], although reports have been more varied as to its roles in the viral lytic cycle. For instance, suppression of NF-κB signaling via expression of the IκBα super suppressor did not impair viral replication [43], whereas overexpression of the RelA/p65 subunit of the transcriptionally active NF-κB dimer inhibited the MHV68 lytic cycle [58]. However, these apparently inconsistent findings have recently been clarified by Feng and colleagues, who revealed that MHV68 activates the NF-κB pathway immediately following infection but that the downstream NF-κB transcriptional response is blunted as RelA/p65 is robustly targeted for degradation early in infection [39, 41]. Furthermore, optimal MHV68 gene expression and replication require the role of the IKKβ component of the NF-κB pathway [39, 41], which operates upstream of the IκBα super suppressor. IKKβ promotes phosphorylation of RelA/p65, thereby priming it for proteasomal degradation, as well as phosphorylation of MHV68 RTA, which enhances its transcriptional activating properties.
Interestingly, MHV68 activates IKKβ in both MAVS-dependent and independent ways. While some of the MAVS-dependent activation likely comes from MHV68-induced RIG-I deamidation [59], we establish that B2 SINEs are necessary for robust IKKβ activation, and that this also occurs through both MAVS-dependent and independent mechanisms. Though the RNA ‘sensor’ for SINE RNA is not known, the antiviral dsRNA binding protein PKR has been shown to bind to SINE RNA [34]. Furthermore, PKR is capable of directly associating with MAVS [60, 61]. Additionally, many other RNA sensors including RIG-I preferentially recognize many features present in SINE RNA, including 5’-triphosphate moieties and double stranded regions. However, we have not detected B2 RNAs in RNA immunoprecipitations of RIG-I, suggesting RIG-I may not be responsible for ‘sensing’ B2 RNAs. Future studies aimed at characterizing the composition of viral-induced SINE ribonucleoprotein complexes may provide insight here.
Many viruses, including the related gammaherpesvirus EBV, up regulate host RNA Pol III transcription [62]. In the case of EBV, the up regulation of RNA Pol III transcription is more general, although whether this includes SINE elements is unknown. Interestingly, it was recently shown that EBV infection stimulates RNA Pol III dependent expression of vault RNAs, which can also activate NF-κB and enhance viral establishment [63]. These results suggest that multiple RNA Pol III ncRNAs engage components of the cytoplasmic innate immune system, and their transcriptional induction in response to viral infection could be one of the early stress responses of the cell. Additionally, the production of abundant dsRNAs following reactivation of latent KSHV in iSLK cells was recently reported [64]. Whether the dsRNAs detected are human SINE RNAs is unknown but worthy of investigation.
SINE RNA expression also manipulates host gene expression. For example, during the heat shock response, global RNA Pol II transcription is down regulated. This is mediated in part through the direct binding of B2 RNAs to RNA Pol II, which prevent it from establishing contacts with the promoter during closed complex formation [26–28]. Our knockdown data coupled with our ChIRP-qPCR analyses reveal that B2 RNA-mediated transcriptional repression can similarly operate during viral infection, perhaps also through an interaction with RNA Pol II. At the moment we are unaware of the extent to which this occurs globally during infection. However, data from our laboratory suggest that it is unlikely that B2 RNAs globally repress transcription during MHV68 infection, as both WT virus and a viral mutant unable to restrict host gene expression induce B2 RNAs to a similar extent, yet widespread transcriptional repression is only observed during a WT infection [65]. In this regard, future studies will be important to determine the basis for promoter specificity.
SINE RNAs are also abundantly expressed early during embryogenesis within stem cells, a cell type in which dsRNA sensing is inefficient. The function of these ncRNAs within this context is unclear, though a fraction of SINE RNAs are processed into endosiRNAs [66, 67]. Interestingly, it has recently been reported that that efficient induction of induced pluripotent stem cells (iPSCs) requires activation of the dsRNA sensor TLR3 [68]. Whether continued low-level activation of the innate immune system is required for iPSC maintenance is unclear, but it is possible that SINE RNA expression provides chronic low-level innate immune stimulation to promote stem cell maintenance.
The use of viruses to perturb host systems, such as described here, presents a valuable platform to probe SINE ncRNA biology. SINE ncRNAs are a critical component of the gammaherpesvirus lifecycle, but they are also activated upon infection with multiple other human and murine viruses. Whether in other systems SINE RNAs serve as anti-viral signaling components, as well as if and how they are co-opted by the diversity of viral and non-viral pathogens remain exciting avenues for future research.
Materials and Methods
Cells, viruses, and infections
NIH3T3 (ATCC), NIH3T12 (ATCC), Vero, MEF, and WT and MAVS-/- fibroblasts (kindly provided by Russell Vance, University of California Berkeley) were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen). The green fluorescent protein (GFP)-expressing MHV68 bacterial artificial chromosome (BAC) has been described elsewhere [69]. BAC-derived MHV68 virus was produced by transfecting BAC DNA into NIH3T3 cells using SuperFect (Qiagen). Virus was then amplified in NIH 3T12 cells and titered by plaque assay on NIH3T3 cells. Before infecting mice, the loxP-flanked BAC vector sequence was removed by passaging the virus through Vero cells expressing Cre recombinase (kindly provided by Dr. Samuel Speck, Emory University).
Nucleic acid isolation and measurement, and Luciferase assays
For analysis of gene expression by RT-qPCR, total or subcellular fractions of RNA were isolated with TRIzol (Invitrogen) in accordance with the manufacturer's instructions. cDNA was synthesized from 1 μg of RNA with random hexamers (Integrated DNA Technologies) and SuperScript II reverse transcriptase (Invitrogen). qPCR was performed using the DyNAmo ColorFlash SYBR green qPCR kit (Thermo Scientific) with appropriate primers.
For small RNA northern blot analysis total RNA was separated on 8% polyacrylamide–7M urea gels and electrotransferred at 4°C to Amersham Hybond-N+ membranes in 0.5X TBE buffer for 16h at 15V. Membranes were probed overnight using 32P-end labeled probes overnight at 55°C. Blots were washed three times in 0.1X SSC for 10 min each before exposed to phosphoimager screens overnight. For mRNA northern blot analysis total RNA was resolved on 1.2% agarose-formaldehyde gels and transferred to Hybond-N+ membranes by capillary action.
For primer extension, 15 μg of total RNA was incubated with 6 pmol 32P-labeled primer in 10 μl of Buffer A (250 mM KCl, 10 mM Tris, pH 7.5, and 1 mM EDTA) for 1 h at 55°C. Buffer B (40 μl) and 0.5 μl AMV reverse transcriptase (Promega) were added and incubated at 42°C for 1 h. Products were phenol-chloroform extracted, ethanol-precipitated, and resuspended in RNA loading dye before being resolved on 8% polyacrylamide–7M urea gels.
Nuclear run-on was performed as described previously with minor modifications [70]. Specifically, nuclei were isolated from two 10-cm plates of confluent mock - or MHV68 infected cells.
For Luciferase assays NIH3T3 cells were transfected in 6 well dishes with the indicated plasmids using TransIT-3T3 (Mirus Bio). 48 h post-transfection, lysates were prepared from approximately equal number of cells and luciferase activity was determined with the Promega luciferase assay system.
Chromatin isolation by RNA purification (ChIRP)
ChIRP was performed as previously described with minor modifications [71]. Briefly, ~ 100 million NIH3T3 cells were infected with MHV68 at an MOI 5. 24 hpi cells were cross-linked with 1.1% formaldehyde for 15 min at room temperature. Crosslinking was then quenched with 0.125 M glycine for 5 min. Cells were rinsed again with PBS, scraped into Falcon tubes, and pelleted at 1000 g for 5 min. The cell pellet was resuspended in 3 mL nuclei lysis buffer (50 mM Tris-HCl [pH 7.0], 10 mM EDTA, 1% SDS, protease cocktail inhibitor [Roche], and RNAse inhibitor [Fermentas]) and rotated for 10 min. at 4°C. Cells were dounced 10 times with a B type pestle and separated in to three 1 mL aliquots for sonication. Sonication was performed using a Covaris focused sonicator. After sonication chromatin aliquots were combined and 9 mL of hybridization buffer (750 mM NaCl, 1% SDS, 50 mM Tris 7.0, 1 mM EDTA, 15% Formamide, protease inhibitor cocktail, and RNAse inhibitor) was added. 50 pmol of five separate 3’-TEG biotinylated probes was added to the dilute chromatin and rotated over night for 16 h. Streptavidin-magnetic C1 beads (Life Technologies) were washed three times in nuclei lysis buffer, blocked with 500 ng/μl yeast total RNA, and 1mg/ml BSA for 1 hr at room temperature, and washed three times again in nuclear lysis buffer before being resuspended in its original volume. One hundred microliters washed/blocked C1 beads were added to the chromatin mixture and rotated for an additional 4 h at 37°C. Beads:biotin-probes:RNA:chromatin adducts were captured by magnets (Invitrogen) and washed five times with 10 mL wash buffer (2× SSC, 0.5% SDS). After the last wash complexes were eluted by resuspending the beads in 500 μL G50 buffer (20 mM Tris-HCl, 300 mM NaCl, 2 mM EDTA, 0.2% SDS) plus 50 μg/mL Proteinase K (Fermentas) and incubating at 60°C for 1 h. The G50 buffer was separated from the beads, phenol chloroform extracted, and ethanol precipitated. RNA was analyzed by small RNA northern blotting and DNA was analyzed by qPCR, as described above.
In vivo labeling/phosphorylation assay, cellular fractionation and western blotting
For in vivo kinase assays in the absence of infection, NIH3T3 cells were cotransfected with FLAG-RTA and either a B1 SINE, B2 SINE, or control expression plasmid using TransIT-3T3 (Mirus Bio). 24 h post-transfection media was replaced with phosphate‐free DMEM (Life Technologies) for 1 h and then incubated in the same medium containing [32P]-orthophosphate (0.5 mCi/ml final concentration, carrier free; PerkinElmer) for 6 h. After labeling cells were lysed in high salt RIPA buffer (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate) containing a phosphatase inhibitor cocktail (Sigma) and immunoprecipitated using anti-FLAG (M2) magnetic beads (Sigma) overnight at 4°C. Beads were washed extensively with high salt RIPA buffer and then eluted with FLAG peptide (Sigma). The eluate was resolved by SDS–PAGE, transferred to PVDF membrane (Immobilon; Millipore) and visualized by autoradiography. Additionally western blot was performed as described below using anti-FLAG (M2, Sigma).
For in vivo labeling during MHV68 infection, NIH3T3 cells were transfected with FLAG-RTA as above. 24 h post-transfection cells were transfected with ASO’s using RNAiMAX as previously described [72]. 3 h post-ASO treatment the media was replaced with phosphate‐free DMEM (Life Technologies) for 1 h and then incubated in the same medium containing [32P]-orthophosphate (0.5 mCi/ml final concentration, carrier free; PerkinElmer) and infected with MHV68 at an MOI 5 for 6 h. Immunoprecipitations and downstream analysis was performed as described above.
Subcellular fractionation was performed using the REAP method with the minor modification of using one 10-cm plate for each fractionation condition [73].
For western blot analysis, cell lysates were prepared in NET-2 buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5% Nonidet P-40), and protein concentrations were determined by Bradford assay. Equivalent quantities of each sample were fractionated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and incubated with the appropriate antibodies. Western blot assays were developed with HRP-conjugated secondary antibodies and ECL reagents (Pierce).
Growth curves and in vivo experiments
For multi-step growth curves, 1.5×105 NIH3T3, WT or MAVS-/- fibroblasts were infected with MHV68 at MOI of 0.05 and both supernatant and cells were harvested at 0, 1, 2, and 3 dpi and frozen. Samples were freeze-thawed once before titering by plaque assay on NIH3T3 cells.
Female C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and infected when 4–6 weeks old. Mice were anesthetized with isoflourane and inoculated intranasally with 5×104 plaque forming units (pfu) in 20 μl DMEM (Invitrogen). Lungs were harvested 5 dpi and homogenized with a tissue homogenizer in 5 mL Trizol and RNA was isolated as described above.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of California Berkeley (Permit Number: R292-0507). All animals were anesthetized prior to infection with isoflurane, and all efforts were made to minimize suffering.
Supporting Information
Zdroje
1. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–8. doi: 10.1038/nature11233 22955620
2. Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–63. 16141072
3. Tycowski KT, Guo YE, Lee N, Moss WN, Vallery TK, Xie M, et al. Viral noncoding RNAs: more surprises. Genes & development. 2015;29(6):567–84.
4. Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, et al. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS pathogens. 2014;10(1):e1003847. doi: 10.1371/journal.ppat.1003847 24453964
5. Canny SP, Reese TA, Johnson LS, Zhang X, Kambal A, Duan E, et al. Pervasive transcription of a herpesvirus genome generates functionally important RNAs. MBio. 2014;5(2):e01033–13. doi: 10.1128/mBio.01033-13 24618256
6. O'Grady T, Cao S, Strong MJ, Concha M, Wang X, Splinter Bondurant S, et al. Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. Journal of virology. 2014;88(3):1604–16. doi: 10.1128/JVI.02989-13 24257595
7. Lin YT, Kincaid RP, Arasappan D, Dowd SE, Hunicke-Smith SP, Sullivan CS. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. Rna. 2010;16(8):1540–58. doi: 10.1261/rna.1967910 20566670
8. Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, et al. Decoding human cytomegalovirus. Science. 2012;338(6110):1088–93. doi: 10.1126/science.1227919 23180859
9. Juranic Lisnic V, Babic Cac M, Lisnic B, Trsan T, Mefferd A, Das Mukhopadhyay C, et al. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS pathogens. 2013;9(9):e1003611. doi: 10.1371/journal.ppat.1003611 24086132
10. Johnson LS, Willert EK, Virgin HW. Redefining the genetics of murine gammaherpesvirus 68 via transcriptome-based annotation. Cell Host Microbe. 2010;7(6):516–26. doi: 10.1016/j.chom.2010.05.005 20542255
11. Chandriani S, Xu Y, Ganem D. The lytic transcriptome of Kaposi's sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. Journal of virology. 2010;84(16):7934–42. doi: 10.1128/JVI.00645-10 20534856
12. Deininger PL, Batzer MA. Mammalian retroelements. Genome research. 2002;12(10):1455–65. 12368238
13. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. 11237011
14. Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. 12466850
15. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304–51. 11181995
16. Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nature genetics. 2003;35(1):41–8. 12897783
17. Daniels GR, Deininger PL. Repeat sequence families derived from mammalian tRNA genes. Nature. 1985;317(6040):819–22. 3851163
18. Kriegs JO, Churakov G, Jurka J, Brosius J, Schmitz J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 2007;23(4):158–61. 17307271
19. Ullu E, Tschudi C. Alu sequences are processed 7SL RNA genes. Nature. 1984;312(5990):171–2. 6209580
20. Weiner AM. An abundant cytoplasmic 7S RNA is complementary to the dominant interspersed middle repetitive DNA sequence family in the human genome. Cell. 1980;22(1 Pt 1):209–18. 6159101
21. Liu WM, Chu WM, Choudary PV, Schmid CW. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic acids research. 1995;23(10):1758–65. 7784180
22. Panning B, Smiley JR. Regulation of cellular genes transduced by herpes simplex virus. Journal of virology. 1989;63(5):1929–37. 2539495
23. Jang KL, Latchman DS. HSV infection induces increased transcription of Alu repeated sequences by RNA polymerase III. FEBS letters. 1989;258(2):255–8. 2557237
24. Panning B, Smiley JR. Activation of RNA polymerase III transcription of human Alu repetitive elements by adenovirus type 5: requirement for the E1b 58-kilodalton protein and the products of E4 open reading frames 3 and 6. Molecular and cellular biology. 1993;13(6):3231–44. 7684492
25. Williams WP, Tamburic L, Astell CR. Increased levels of B1 and B2 SINE transcripts in mouse fibroblast cells due to minute virus of mice infection. Virology. 2004;327(2):233–41. 15351211
26. Allen TA, Von Kaenel S, Goodrich JA, Kugel JF. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nature structural & molecular biology. 2004;11(9):816–21.
27. Mariner PD, Walters RD, Espinoza CA, Drullinger LF, Wagner SD, Kugel JF, et al. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Molecular cell. 2008;29(4):499–509. doi: 10.1016/j.molcel.2007.12.013 18313387
28. Yakovchuk P, Goodrich JA, Kugel JF. B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5569–74. doi: 10.1073/pnas.0810738106 19307572
29. Kassube SA, Fang J, Grob P, Yakovchuk P, Goodrich JA, Nogales E. Structural insights into transcriptional repression by noncoding RNAs that bind to human Pol II. Journal of molecular biology. 2013;425(19):3639–48. doi: 10.1016/j.jmb.2012.08.024 22954660
30. Ponicsan SL, Houel S, Old WM, Ahn NG, Goodrich JA, Kugel JF. The non-coding B2 RNA binds to the DNA cleft and active-site region of RNA polymerase II. Journal of molecular biology. 2013;425(19):3625–38. doi: 10.1016/j.jmb.2013.01.035 23416138
31. Espinoza CA, Goodrich JA, Kugel JF. Characterization of the structure, function, and mechanism of B2 RNA, an ncRNA repressor of RNA polymerase II transcription. Rna. 2007;13(4):583–96. 17307818
32. Sinnett D, Richer C, Deragon JM, Labuda D. Alu RNA secondary structure consists of two independent 7 SL RNA-like folding units. The Journal of biological chemistry. 1991;266(14):8675–8. 1709156
33. Shumyatsky GP, Tillib SV, Kramerov DA. B2 RNA and 7SK RNA, RNA polymerase III transcripts, have a cap-like structure at their 5' end. Nucleic acids research. 1990;18(21):6347–51. 1700854
34. Chu WM, Ballard R, Carpick BW, Williams BR, Schmid CW. Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR. Molecular and cellular biology. 1998;18(1):58–68. 9418853
35. Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18(45):6112–20. 10557102
36. Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471(7338):325–30. doi: 10.1038/nature09830 21297615
37. Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–59. doi: 10.1016/j.cell.2012.03.036 22541070
38. Kerur N, Hirano Y, Tarallo V, Fowler BJ, Bastos-Carvalho A, Yasuma T, et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(12):7395–401. doi: 10.1167/iovs.13-12500 24114535
39. Dong X, Feng H, Sun Q, Li H, Wu TT, Sun R, et al. Murine gamma-herpesvirus 68 hijacks MAVS and IKKbeta to initiate lytic replication. PLoS pathogens. 2010;6(7):e1001001. doi: 10.1371/journal.ppat.1001001 20686657
40. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81 : 145–66. doi: 10.1146/annurev-biochem-051410-092902 22663078
41. Dong X, Feng P. Murine gamma herpesvirus 68 hijacks MAVS and IKKbeta to abrogate NFkappaB activation and antiviral cytokine production. PLoS pathogens. 2011;7(11):e1002336. doi: 10.1371/journal.ppat.1002336 22110409
42. Krug LT, Collins CM, Gargano LM, Speck SH. NF-kappaB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. Journal of virology. 2009;83(10):4732–48. doi: 10.1128/JVI.00111-09 19264770
43. Krug LT, Moser JM, Dickerson SM, Speck SH. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS pathogens. 2007;3(1):e11. 17257062
44. Vazquez C, Horner SM. MAVS Coordination of Antiviral Innate Immunity. Journal of virology. 2015;89(14):6974–7. doi: 10.1128/JVI.01918-14 25948741
45. Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, et al. Transcription program of murine gammaherpesvirus 68. Journal of virology. 2003;77(19):10488–503. 12970434
46. Wu TT, Tong L, Rickabaugh T, Speck S, Sun R. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. Journal of virology. 2001;75(19):9262–73. 11533188
47. Wu TT, Usherwood EJ, Stewart JP, Nash AA, Sun R. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. Journal of virology. 2000;74(8):3659–67. 10729142
48. Jang KL, Latchman DS (1992) The herpes simplex virus immediate-early protein ICP27 stimulates the transcription of cellular Alu repeated sequences by increasing the activity of transcription factor TFIIIC. Biochem J 284 (Pt 3): 667–73 1320373
49. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11(2):143–54. doi: 10.1038/nri2937 21267015
50. Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PloS one. 2010;5(10):e13742. doi: 10.1371/journal.pone.0013742 21060793
51. Pezda AC, Penn A, Barton GM, Coscoy L. Suppression of TLR9 immunostimulatory motifs in the genome of a gammaherpesvirus. J Immunol. 2011;187(2):887–96. doi: 10.4049/jimmunol.1003737 21666062
52. Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J Immunol. 2008;180(1):438–43. 18097045
53. Sun C, Schattgen SA, Pisitkun P, Jorgensen JP, Hilterbrand AT, Wang LJ, et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J Immunol. 2015;194(4):1819–31. doi: 10.4049/jimmunol.1402495 25595793
54. Yang K, Wang J, Wu M, Li M, Wang Y, Huang X. Mesenchymal stem cells detect and defend against gammaherpesvirus infection via the cGAS-STING pathway. Sci Rep. 2015;5 : 7820. doi: 10.1038/srep07820 25592282
55. Graczyk D, White RJ, Ryan KM. Involvement of RNA Polymerase III in Immune Responses. Molecular and cellular biology. 2015;35(10):1848–59. doi: 10.1128/MCB.00990-14 25776554
56. Gargano LM, Forrest JC, Speck SH. Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. Journal of virology. 2009;83(3):1474–82. doi: 10.1128/JVI.01717-08 19019960
57. Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene. 2006;25(51):6844–67. 17072332
58. Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. Journal of virology. 2003;77(15):8532–40. 12857922
59. He S, Zhao J, Song S, He X, Minassian A, Zhou Y, et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Molecular cell. 2015;58(1):134–46. doi: 10.1016/j.molcel.2015.01.036 25752576
60. Zhang P, Li Y, Xia J, He J, Pu J, Xie J, et al. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J Cell Sci. 2014;127(Pt 11):2471–82. doi: 10.1242/jcs.139626 24659800
61. Arnaud N, Dabo S, Akazawa D, Fukasawa M, Shinkai-Ouchi F, Hugon J, et al. Hepatitis C virus reveals a novel early control in acute immune response. PLoS pathogens. 2011;7(10):e1002289. doi: 10.1371/journal.ppat.1002289 22022264
62. Felton-Edkins ZA, Kondrashov A, Karali D, Fairley JA, Dawson CW, Arrand JR, et al. Epstein-Barr virus induces cellular transcription factors to allow active expression of EBER genes by RNA polymerase III. The Journal of biological chemistry. 2006;281(45):33871–80. 16956891
63. Amort M, Nachbauer B, Tuzlak S, Kieser A, Schepers A, Villunger A, et al. Expression of the vault RNA protects cells from undergoing apoptosis. Nat Commun. 2015;6 : 7030. doi: 10.1038/ncomms8030 25952297
64. West JA, Wicks M, Gregory SM, Chugh P, Jacobs SR, Zhang Z, et al. An important role for mitochondrial antiviral signaling protein in the Kaposi's sarcoma-associated herpesvirus life cycle. Journal of virology. 2014;88(10):5778–87. doi: 10.1128/JVI.03226-13 24623417
65. Abernathy E, Gilbertson S, Alla R, Glaunsinger B. Viral Nucleases Induce an mRNA Degradation-Transcription Feedback Loop in Mammalian Cells. Cell Host Microbe. 2015.
66. Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes & development. 2008;22(20):2773–85.
67. Hu Q, Tanasa B, Trabucchi M, Li W, Zhang J, Ohgi KA, et al. DICER - and AGO3-dependent generation of retinoic acid-induced DR2 Alu RNAs regulates human stem cell proliferation. Nature structural & molecular biology. 2012;19(11):1168–75.
68. Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151(3):547–58. doi: 10.1016/j.cell.2012.09.034 23101625
69. Adler H, Messerle M, Wagner M, Koszinowski UH. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. Journal of virology. 2000;74(15):6964–74. 10888635
70. Ashe HL, Monks J, Wijgerde M, Fraser P, Proudfoot NJ. Intergenic transcription and transinduction of the human beta-globin locus. Genes & development. 1997;11(19):2494–509.
71. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Molecular cell. 2011;44(4):667–78. doi: 10.1016/j.molcel.2011.08.027 21963238
72. Zong X, Huang L, Tripathi V, Peralta R, Freier SM, Guo S, et al. Knockdown of nuclear-retained long noncoding RNAs using modified DNA antisense oligonucleotides. Methods in molecular biology. 2015;1262 : 321–31. doi: 10.1007/978-1-4939-2253-6_20 25555591
73. Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K. REAP: A two minute cell fractionation method. BMC research notes. 2010;3 : 294. doi: 10.1186/1756-0500-3-294 21067583
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 11
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins
- On the Discovery of TOR As the Target of Rapamycin
- Parasite Glycobiology: A Bittersweet Symphony
- Broadening of Virus-Specific CD8 T-Cell Responses Is Indicative of Residual Viral Replication in Aviremic SIV Controllers