
Identification of Genes Required for Neural-Specific Glycosylation Using Functional Genomics
Glycosylation plays crucial regulatory roles in various biological processes such as development, immunity, and neural functions. For example, α1,3-fucosylation, the addition of a fucose moiety abundant in Drosophila neural cells, is essential for neural development, function, and behavior. However, it remains largely unknown how neural-specific α1,3-fucosylation is regulated. In the present study, we searched for genes involved in the glycosylation of a neural-specific protein using a Drosophila RNAi library. We obtained 109 genes affecting glycosylation that clustered into nine functional groups. Among them, members of the RNA regulation group were enriched by a secondary screen that identified genes specifically regulating α1,3-fucosylation. Further analyses revealed that an RNA–binding protein, second mitotic wave missing (Swm), upregulates expression of the neural-specific glycosyltransferase FucTA and facilitates its mRNA export from the nucleus. This first large-scale genetic screen for glycosylation-related genes has revealed novel regulation of fucTA mRNA in neural cells.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001254
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001254
Summary
Glycosylation plays crucial regulatory roles in various biological processes such as development, immunity, and neural functions. For example, α1,3-fucosylation, the addition of a fucose moiety abundant in Drosophila neural cells, is essential for neural development, function, and behavior. However, it remains largely unknown how neural-specific α1,3-fucosylation is regulated. In the present study, we searched for genes involved in the glycosylation of a neural-specific protein using a Drosophila RNAi library. We obtained 109 genes affecting glycosylation that clustered into nine functional groups. Among them, members of the RNA regulation group were enriched by a secondary screen that identified genes specifically regulating α1,3-fucosylation. Further analyses revealed that an RNA–binding protein, second mitotic wave missing (Swm), upregulates expression of the neural-specific glycosyltransferase FucTA and facilitates its mRNA export from the nucleus. This first large-scale genetic screen for glycosylation-related genes has revealed novel regulation of fucTA mRNA in neural cells.
Introduction
Neural cells require correct glycosylation patterns for their development, function, and viability. An example of this is the attachment of an α1,3-fucose moiety to an N-glycan core via α1,3-linkage [1], a process which is prevalent in neural cells in Drosophila [2]. This α1,3-fucose moiety can be detected with an anti-horse radish peroxidase (HRP) antibody and therefore has also previously been referred to as an HRP epitope. The α1,3-fucose is thought to be essential for neural development, function, and behavior because a nac (neurally altered carbohydrate) Drosophila mutant that lacks this α1,3-fucose moiety exhibits deformation of the eyes [3], the misrouting of wing sensory neurons [4], and abnormal grooming behavior [5]. However, as it remains unclear that the nac mutation impairs only α1,3-fucosylation, the necessity of α1,3-fucosylation for neural development and/or function in Drosophila has not been conclusively demonstrated.
The enzyme α1,3-fucosyltransferase (FucTA) [6], which is mainly expressed in neural cells, directly catalyzes α1,3-fucosylation. In addition to FucTA, other glycosylation-related proteins such as UDP-GlcNAc: α-3-D-mannoside-β -1,2-N-acetylglucosaminyltransferase I (Mgat1) [7], GDP-mannose 4,6-dehydratase (Gmd) [8], and a GDP-fucose transporter (Gfr) [9], [10] are required for α1,3-fucosylation. Whereas Mgat1 provides a preferred substrate for FucTA by adding N-acetylglucosamine to the nonreducing end of an N-glycan, Gmd and Gfr are responsible for the synthesis and transport, respectively, of GDP-fucose, another substrate for FucTA. These genes, in contrast to the gene encoding FucTA, are widely expressed in various tissues and also utilized for other glycosylation processes such as O-fucosylation of Notch, α1,6-fucosylation of N-glycans, and formation of complex type N-glycans. Hence, the neural-specific expression of FucTA appears to account for the neural-specific regulation of α1,3-fucosylation. However, the mechanisms regulating FucTA expression have remained largely unknown.
Forward genetic approaches have proven to be powerful methods of elucidating novel mechanisms. For example, the study of Drosophila genetics has yielded important contributions to our understanding of the developmental significance of proteoglycans [11], [12] and Fringe-dependent Notch glycosylation [13]. Genetic screens for mutations affecting morphogenesis and growth factor signaling have now identified a number of genes involved in Notch glycosylation and/or proteoglycan formation. Most of these genes are conserved in mammals, suggesting that Drosophila is a useful model system for the study of glycosylation in metazoans. However, although previously performed screens of this nature have identified glycosyl enzymes and nucleotide sugar transporters, to date they have not been used to uncover regulators of these molecules.
To elucidate novel regulatory mechanisms underlying neural-specific glycosylation, we performed a genetic screen in Drosophila and identified 109 genes required for glycosylation of a retinal neural cell-specific protein. These included 95 genes that are newly implicated in this process and 9 functional groups. Furthermore, 17 genes were identified to be specifically required for α1,3-fucosylation. Among these genes, we further analyzed the function of second mitotic wave missing (Swm), which contains an RNA-binding motif. Here, we show that Swm directly binds to fucTA mRNA, upregulates fucTA mRNA and protein levels, and facilitates the nuclear export of fucTA mRNA in neural cells. These results indicate that Swm is involved in neural-specific glycosylation in addition to the cell cycle, in which its involvement has been previously reported [14]. This report, the first large-scale screen for glycosylation in a multicellular organism, has thus identified a number of new genes directly or indirectly involved in glycosylation and unveiled a novel regulatory mechanism of neural-specific glycosylation.
Results
Biological significance of α1,3-fucosylation
As α1,3-fucosylation has not proven to be essential for neural development and/or function in Drosophila, we first aimed to determine the neural function of α1,3-fucosylation. A phenotype of a piggyBac insertional mutant for the fucTA gene, fucTAf03774, was examined. To validate this mutant, its central nervous system (CNS) was stained using anti-HRP antibody. The mutant CNS was negative for anti-HRP staining, suggesting that α1,3-fucosylation was compromised (data not shown). CNS morphology was then compared between wild-type and fucTA mutant third instar larvae. The longitudinal length-to-width ratio of the ventral nerve cord (VNC) was significantly lower in fucTA mutants than in wild-type larvae (Figure 1A and 1B), indicating that α1,3-fucosylation plays an important role in neural development. However, the staining patterns of antibodies such as 22C10, BP102, anti-FasI, anti-FasII, and anti-FasIII were not noticeably different in the fucTA mutant (data not shown).

Screening strategy
To reveal novel regulatory mechanisms underlying neural-specific glycosylation, a genetic screen was performed in adult Drosophila using the RNAi-mediated tissue-specific gene knockdown method. The procedure is summarized in Figure 2A. We chose eye-specific knockdown because eyes bear the neural-specific glycan α1,3-fucose. Additionally, the ablation of genes essential for development and/or cell viability in eyes might not cause knockdown fly lethality, whereas their ablation in the central and/or peripheral nervous systems would be lethal. DsRNAs targeting different genes were expressed in Drosophila eyes by crossing the eye-specific Gal4 driver, GMR-Gal4, with the corresponding strains. These strains harbored inverted repeats of a portion of the corresponding cDNA downstream of the yeast upstream activating sequence (UAS), which is activated by the Gal4 protein (http://www.shigen.nig.ac.jp/fly/nigfly/about/aboutRnai.jsp).

In our screen, the glycosylation of a model eye-specific glycoprotein was examined, as this allowed us to accurately measure the number of glycans added. We chose Chaoptin (Chp) [15] as our model glycoprotein based on the following advantages: (i) Chp is modified by neural-specific α1,3-fucosylation; (ii) detailed information is available on both the structures of the N-glycans and their attachment sites on Chp (described below in detail) [16]; and (iii) simple protein affinity purification is possible using an anti-Chp antibody (24B10). As shown in Figure 2B, Chp protein could be purified as a single band detected by Coomassie Brilliant Blue (CBB) staining.
Chp is a Drosophila glycoprotein essential for rhabdomere formation in adult photoreceptor cells, which is mediated through cell adhesion activity [15], [17]. N-glycosylation of Chp was recently shown to play a pivotal role in its stability, transport to the plasma membrane, and cell adhesion activity [18]. Chp N-glycan structures are classified as high-mannose type, pauci-mannose type, or complex type [16]. Glycan structures added to Chp are summarized in Figure S1. Interestingly, α1,3-fucose is added only at N1012, suggesting that Chp has a single difucosylated glycan at N1012. To detect these glycan structures, the binding of glycan probes such as lectins and anti-HRP antibody was examined. Immunopurified Chp was subjected to lectin blot or immunoblot analysis using ABA, ConA, DBA, DSA, LCA, Lotus, PHA-L4, PNA, SBA, UEA, and WGA lectins and anti-HRP antibody. The specificity of these lectins and anti-HRP antibody is summarized in Table S1. The binding abilities of these probes were quantified relative to the amount of Chp protein (Figure 2C). These analyses suggested that the WGA, ConA, LCA, and DSA lectins and anti-HRP antibody would be useful as glycan probes in our assay system. As represented in Figure S1, WGA and ConA detect N-acetylglucosamine (GlcNAc) and mannose moieties of N-glycan core regions, respectively. LCA preferentially binds to N-glycans with α1,6-fucose and even weakly recognizes the core regions of N-glycans. Anti-HRP antibody binds to the α1,3-fucose moieties attached to the core [1], [19]. DSA recognizes GlcNAc at the non-reducing end of N-glycans.
Chp protein purified from knockdown flies was subjected to lectin and immuno-dot blot analyses using the same glycan probes described above and an anti-Chp antibody. An example of the dot blot analysis is shown in Figure S2. The glycan moieties detected by these probes were quantified relative to Chp protein, and the z-scores for each knockdown experiment were thus calculated. For verification of this procedure, a dsRNA that targets FucTA was expressed. The resulting knockdown of fucTA decreased the affinity of Chp to the anti-HRP antibody (66.2±11.8% of the control, Figure 2D). Given the results of our previous mass spectrometry study showing that α1,3-fucose is added to the Chp N1012 site [16], Chp was purified from fucTA knockdown eyes, and mass spectrometry analysis was performed. The glycan structures attached to N1012 are summarized in Figure S3. The number of glycans with two fucoses present at N1012 (M3F2Gn2 and M3F2Gn3) selectively decreased in fucTA knockdown eyes (Figure 2E). This finding is consistent with those of previous studies reporting that most single - or double-fucosylated N-glycans bear α1,6-fucose alone, or both α1,6 - and α1,3-fucose, respectively, in embryos [20] and adults [6].
Identification of glycosylation-related genes
A large-scale screen was performed in the current study using an RNAi library that was previously constructed in National Institute of Genetics, Japan (NIG, http://www.shigen.nig.ac.jp/fly/nigfly/about/aboutRnai.jsp). With this library, the expression of 6923 Drosophila genes can be suppressed. From the primary screen followed by tests for reproducibility, 171 genes were identified as candidate genes, knockdown of which compromised glycosylation (Table S2). Since these candidate genes likely included false-positives due to the off-target effects of RNAi [21], the results were validated by repeating the knockdown experiments using secondary sets of dsRNAs. During the construction of these secondary sets of dsRNAs, we ensured that the target regions did not overlap.
Of the 171 primary candidate genes, only 80 could be tested because, in most cases, the induction of the secondary sets of dsRNA resulted in lethal or severe eye malformation or cDNAs were too short to permit the design of secondary target regions. Of the 80 genes tested successfully, 57 were verified to be involved in glycosylation. Sources of the RNAi strains used for the verification of the 57 genes are listed in Table S2 (see “Line ID or transformant ID used for validation”). When the validation results were compared with the calculated off-target probability scores (OTPS) provided by dsCheck software [22], 95.6% of the validated genes had scores of less than 3, whereas the scores of 60.7% of the genes yet to be validated were greater than 2. Thus, the genes with OTPS less than 3 were rescreened by testing whether knockdown of their suspected off-target genes led to glycosylation defects. When knockdown of the suspected off-target genes did not induce any glycosylation defects, the corresponding genes were classified as glycosylation-related genes (rank 2) along with the genes that were validated by secondary dsRNAs (rank 1) (Table S4).
To successfully identify additional glycosylation genes, we searched for genes that interacted with our newly identified genes in the yeast two-hybrid database BIOGRID (http://www.thebiogrid.org/) and in the genetic interaction data listed in FlyBase (http://flybase.bio.indiana.edu/). The RNAi fly strains harboring candidate genes that did not already exist in our library were obtained from the Vienna Drosophila RNAi Library Center (VDRC) [23] (Table S3). Among the 186 genes tested, 10 genes showed glycosylation defects. Further validation experiments identified 1 rank-1 and 5 rank-2 glycosylation-related genes among them. In total, 109 genes were eventually isolated and identified as glycosylation-related genes (Table S4). Significantly, although 14 of the 109 identified genes have already been shown to be involved in glycosylation, the remaining 95 genes were newly assigned to this category by our analysis.
Clustering of glycosylation genes
Glycosylation-related genes were classified based on their domain structures and presumptive functions using the InterPro (http://www.ebi.ac.uk/interpro/), FlyBase (http://flybase.org/), and Panther (http://www.pantherdb.org/) databases. The gene functional groups obtained included glycosylation reactions, transcription, RNA regulation, translation, intracellular trafficking, cytoskeletal regulation, signal transduction, protein degradation, mitochondrial function, and other functions (Figure 3A, Table S4).
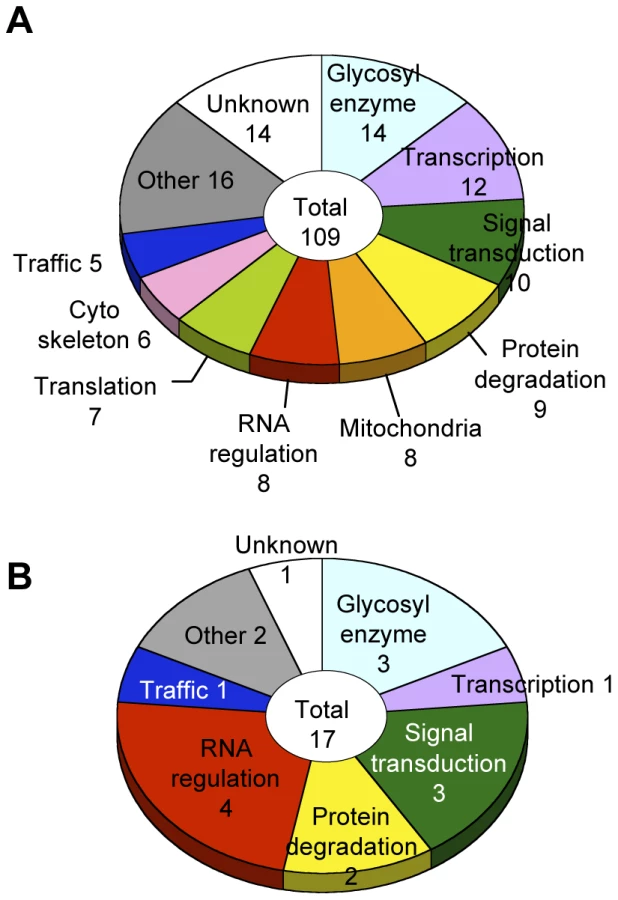
Genes regulating neural-specific glycosylation
To identify genes specifically involved in neural-specific glycosylation, we selected genes whose knockdown induced only abnormal anti-HRP binding activity from among the 109 genes listed in Table S4. From this analysis, 17 genes showing α1,3-fucosylation-specific defects were identified belonging to seven functional groups (Figure 3B). In the RNA regulation group, swm (CG10084) [14], Pabp2 (CG2163) [24], and Hel25E (CG7269) are suggested to interact with each other in the Drosophila interaction database STRING (http://string.embl.de/) and literature [25].
Swm and Pabp2 are required for α1,3-fucosylation
Since Swm and Pabp2 have been reported to physically interact with each other in BIOGRID, the roles of these genes were further analyzed in the context of glycosylation. To analyze the glycosylation defects induced by the knockdown of these genes in further detail, Chp glycans in the swm and Pabp2 knockdown lines were subjected to mass spectrometric analysis. The ratio of the α1,3-fucosylated form (M3F2Gn2) at N1012 was markedly decreased in both knockdown lines, whereas the other form (M3F2Gn3) was only slightly affected by swm or Pabp2 knockdown (Figure 4A). These results suggest that Swm and Pabp2 are required for α1,3-fucosylation and that the M3F2Gn2 form might be more sensitive than the M3F2Gn3 form to decreases in Swm and Pabp2.

Since swm and Pabp2 mutants have been previously isolated [26] (Flybase), glycosylation defects were also examined in the amorphic and hypomorphic alleles of swm, swmF14 and swmF15, respectively, and the hypomorphic allele of Pabp2, Pabp2KG02359. All cells homozygous for swm mutations were generated in whole Drosophila eyes using a modified FLP/FRT system [27]. Chp was isolated from these mutant eyes and detected by immunoblotting with anti-Chp and anti-HRP antibodies to quantify the amount of α1,3-fucose added to Chp. The α1,3-fucose added in swmF14, swmF15, and Pabp2KG02359 cells decreased to 22.7% ± 4.8%, 28.3% ± 7.3%, and 59.5% ± 8.8% of wild-type levels, respectively (Figure 4B). These results are further evidence from knockdown experiments that Swm and Pabp2 are somehow required for α1,3-fucosylation.
Although Pabp2 is known to participate in polyA addition, the function of Swm is largely unknown. Thus, we focused on the role of Swm in glycosylation. Modification of α1,3-fucose is widely utilized for Drosophila neural proteins. Thus, we first examined whether swm depletion affects not only Chp but also a wide range of other proteins. Homogenates of adult eyes heterozygous or homozygous for swmF14 and swmF15 were analyzed by immunoblot analysis with an anti-HRP antibody (Figure 5A). Several positive bands were detected, including a high-molecular-weight band corresponding to Chp (∼150 kDa). Quantitative comparison between heterozygotes and homozygotes of the same swm alleles revealed that anti-HRP antibody affinity was reduced in most bands in the homozygotes, with the exception of an approximately 30-kDa product (Figure 5A and 5B). The decrease was much larger in swmF14 than in swmF15 mutants, consistent with the severity of these mutations. Taken together, our data indicate that Swm is required for the α1,3-fucosylation of most proteins that are usually modified by α1,3-fucose.
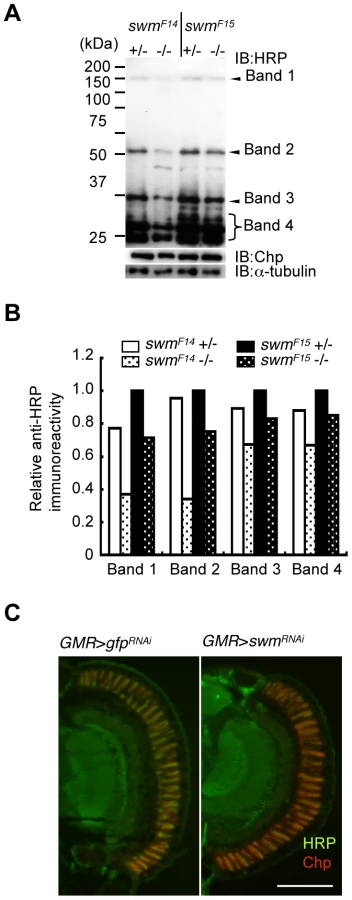
The observed requirement of Swm for most α1,3-fucosylation was also supported by the immunostaining of eyes with anti-HRP antibody. Adult photoreceptor cells from swm knockdown and control flies were simultaneously stained with anti-HRP and anti-Chp antibodies (Figure 5C). Control photoreceptor cells appeared yellow in color because the intensities of green signal (anti-HRP) and red signal (anti-Chp) were nearly equal in this condition. In contrast, swm knockdown cells appeared relatively red in color, suggesting that the intensity of green signal was reduced compared to the red signal. These data clearly indicate that the signal strength of anti-HRP relative to anti-Chp was reduced in the knockdown photoreceptor cells compared to control cells.
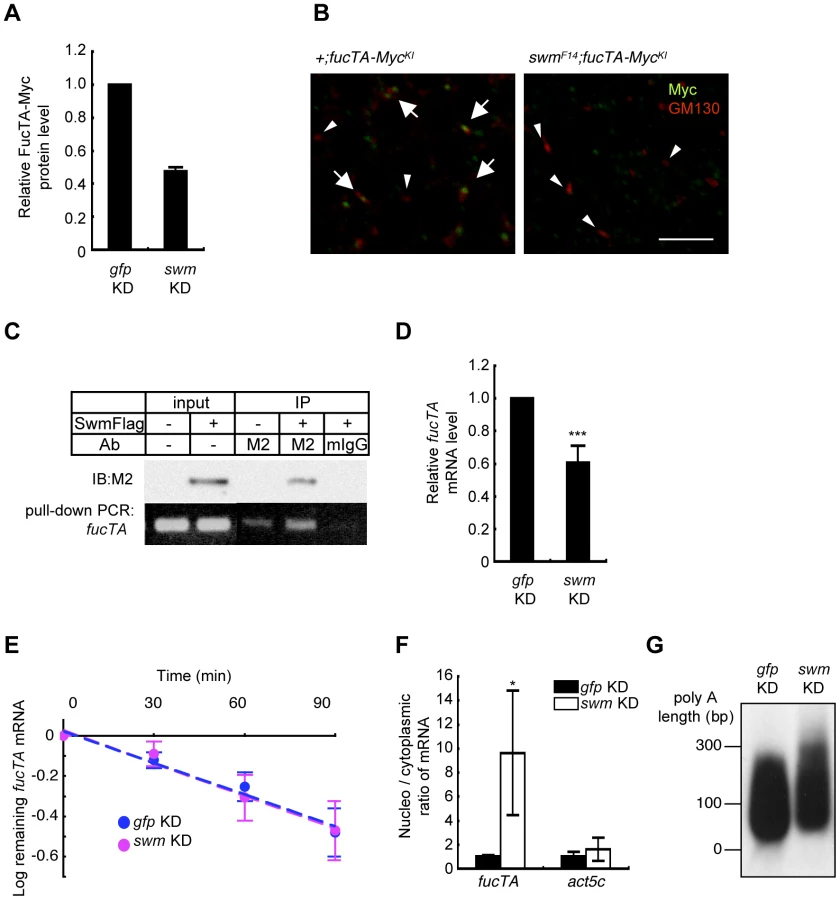
Since α1,3-fucosylation is directly catalyzed by FucTA, the Swm-dependence of FucTA protein expression was examined. Because no anti-FucTA antibody was available, a knock-in fly was generated in which the fucTA locus was replaced by a Myc-tagged fucTA gene (Figure S4A and S4B). When swm dsRNA was expressed by GMR-Gal4, FucTA-Myc protein levels were reduced to 47.5±2.4% of control values as revealed by immunoblot analysis (n = 2) (Figure 6A and Figure S4C). The reduction of FucTA-Myc levels in swm mutant tissue was also examined. We costained the CNS of first instar larvae with anti-Myc and anti-GM130, a Golgi marker. The number of Golgi harboring FucTA-Myc protein was markedly decreased in swmF14 mutants (2.0% of total Golgi) compared to wild-type larvae (43.1% of total Golgi, Figure 6B). The different ratios of FucTA-Myc reduction observed in the biochemical and immunostaining experiments may be due to different levels of Swm depletion in knockdown and mutant flies.
Functional analyses of Swm
Although Swm has an RNA recognition motif (RNP-1) that is present in a wide variety of other RNA binding proteins [26], it remained undetermined whether Swm directly bound to mRNA. Binding of Swm to fucTA mRNA was thus examined by expressing Flag-tagged Swm in the Drosophila neural cell line BG2-c6 [28] and by performing immunoprecipitation with an anti-Flag antibody. The quantity of endogenous fucTA mRNA that coprecipitated with Swm-Flag was higher in Swm-Flag–expressing cells than in controls in which Swm-Flag was not expressed or immunoprecipitation was performed using a control IgG (Figure 6C). These results strongly suggest that Swm binds to fucTA mRNA in vivo.
The amount of fucTA mRNA under the control of Swm was next examined. dsRNA targeting swm was introduced into BG2-c6 cells, and the amount of endogenous fucTA mRNA was measured. In addition to a decrease in swm mRNA, to 27.3 ± 1.9% of the control level (n = 3), the amount of fucTA mRNA also decreased to 60.6 ± 10.5% of the control level (Figure 6D). Furthermore, the stability of fucTA mRNA was examined in swm knockdown cells. The amount of fucTA mRNA was measured at 30, 60, and 90 min after total transcription was inhibited by addition of actinomycin D. Decay rates of fucTA were indistinguishable between swm knockdown and control cells (swm, t1/2 = 97.72; control, t1/2 = 100.48, Figure 6E). These data suggest that Swm is involved in the expression of fucTA mRNA but not in its stability.
Swm was recently reported to incorporate into the complex that regulates nuclear export of mRNAs [29]. The necessity of Swm for the nuclear export of fucTA mRNA was therefore examined in BG2-c6 cells. Following separation of cell lysates into nuclear and cytoplasmic fractions, fucTA mRNA levels were measured in each fraction of swm knockdown and control cells. The amount of fucTA mRNA in the nucleus increased by 9.6-fold in swm knockdown cells compared to control gfp knockdown cells (Figure 6F). The nuclear export of actin (act5c) mRNA was also examined in swm knockdown cells. The amount of actin mRNA in the nucleus increased by 1.7-fold in swm knockdown cells compared to control cells. However, the fold change in nuclear accumulation of actin mRNA was significantly lower than that of fucTA mRNA in swm knockdown cells (Figure 6F). These results suggest that relatively more Swm is required for nuclear export of fucTA mRNA than for nuclear export of actin mRNA.
Since nuclear export defects have been reported to result in an abnormally longer polyA length of mRNAs [29], the polyA length of fucTA mRNA was examined in swm knockdown cells. The polyA length of fucTA mRNA was clearly longer in swm knockdown cells than in control cells (Figure 6G). Collectively, these results indicate that Swm positively regulates α1,3-fucosylation by promoting nuclear export of fucTA mRNA in neural cells.
Discussion
The current literature on glycosylation regulatory mechanisms is remarkably limited, and our current study is in fact the first report of a large-scale screen for glycosylation genes using a multicellular organism. We identified 109 genes in our screen, including 95 genes that have not been previously associated with glycosylation. In silico analyses using the available Drosophila databases suggested that these gene products participate in a variety of cellular processes (Figure 3), among which RNA regulation is, for the first time, shown to play an important role in the context of glycosylation. Moreover, the candidate genes belonging to the RNA regulation group were enriched by rescreening for α1,3-fucosylation-specific genes.
Biological significance of α1,3-fucosylation
In the current study, the fucTA mutant showed an imbalanced VNC. The VNC longitudinal length-to-width ratio was significantly shorter in fucTA mutants compared to wild-type larvae. To our knowledge, this is the first report of such shortened VNC in the loss-of-function mutant, although elongated VNC has already been reported in mutants for integrin [30], collagen IV [31], DHR3 [32], worniu [33] and C1GalTA [34]. Interestingly, C1GalTA encodes a core 1 galactosyltransferase that adds galactose to O-GalNAc attached to Ser or Thr protein residues. Taken together with our results, glycosylation patterns might affect the VNC shape. Although how α1,3-fucose affects VNC formation remains to be revealed, we think that two possible defects may result in a shortened VNC in fucTA mutants. (1) The aberrantly elongated VNC in the C1GalTA mutant is proposed to result from deglycosylation of laminin, a component of the extracellular matrix. In addition, overexpression of FucTA has recently been reported to enhance cell migration in the embryonic nervous system [35]. By analogy with the case of C1GalTA, cell migration in the larval VNC may be impaired in fucTA mutants because of aberrant interactions between migrating cells and extracellular matrix. (2) Another possibility is that neural activity may be impaired in fucTA mutants because neural activity has been reported to be necessary for VNC condensation in embryos [31]. This possible defect is consistent with the finding that one of the major α1,3-fucosylated proteins is a β-subunit of Na+/K+ ATPase essential for the maintenance of neural cell membrane potentials [36]. It will be interesting to examine whether neural activity is controlled by glycosylation in the future.
Insight into the genes identified in this screen
Three types of genes were identified in this screen: (1) genes that specifically regulate glycosylation such as glycosyltransferases, (2) genes with multiple functions including regulation of expression, activity and/or localization of glycosyltransferases, and (3) genes with primary functions other than glycosylation that secondarily affect glycosylation. For example, knockdown of cytoskeletal regulation and mitochondrial function genes may secondarily affect glycosylation as a result of damage to cell structures and viability.
For several reasons, swm might be the second type of gene. Although swm has been previously reported to show involvement in the cell cycle [14], no glycosylation defects were observed with the knockdown of other known cell cycle regulators such as cdc2 (CG5363) or cdk4 (CG5072) (Table S2 and S3). In addition, no cell cycle defects could be detected in the fucTA mutant. These data suggest that cell cycle and glycosylation are regulated independently. Therefore, swm would play at least two independent roles in cell cycle and glycosylation.
Traffic genes identified in this screen may be another example of the second type of genes. The conserved oligomeric Golgi (COG) complex has been reported to participate in the Golgi localization of glycosyltransferases and membrane traffic [37], but no involvement of the COG complex in glycosylation has been reported in Drosophila. Our screen identified Cog3 as a gene whose knockdown impaired α1,3-fucosylation. Thus, localization of FucTA protein was examined in cog3 knockdown photoreceptor cells. Expressed FucTA was hardly localized to the Golgi complex in knockdown photoreceptor cells (data not shown). Moreover, Golgi localization of FucTA was also preliminarily found compromised by knockdown of vps2 and vps20, other traffic genes (data not shown). These data raise the possibility that some traffic genes may be involved in glycosylation by localizing glycosyltransferases to appropriate Golgi and/or endoplasmic reticulum sites.
Preferential requirement of Swm in glycoforms
Our detailed analysis of glycan structures using mass spectrometry revealed that one (M3F2Gn2) of the two forms bearing an α1,3-fucose (M3F2Gn3 and M3F2Gn2) is more sensitive to the decrease of swm and Pabp2 than the other (M3F2Gn3). Two reasons may account for this difference, which are not mutually exclusive. (1) FucTA productivity differs between these two glycoforms. The structural difference between the glycoforms consists of a GlcNAc moiety attached to the nonreducing end of N-glycans. Given that this GlcNAc moiety has been shown to increase the reactivity of FucTA to the core of N-glycans [6], we assume that M3FGn3 is preferentially modified by FucTA to form M3F2Gn3. Therefore, under the condition of reduction, not complete loss, of FucTA by swm and Pabp2 knockdown, M3F2Gn2 might be preferentially lost. (2) The other possibility is that Swm or Pabp2 regulate Mgat1 or Fused lobes (Fdl) [38], the enzymes that add and remove the GlcNAc moiety, respectively, from the nonreducing end. In fact, the amount of mRNA encoding Mgat1, but not fdl, was slightly but significantly increased in swm knockdown cells (Figure S5). This finding is consistent with the selective decrease of M3F2Gn2 induced by swm knockdown. However, it remains undetermined whether the preferential decrease of glycoforms is caused by an increase in Mgat1, the preferential productivity of FucTA, or both.
Regulation of fucTA mRNA by Swm
Expression of mRNA is generally regulated at transcription, splicing, 5′ - or 3′-end processing, and nuclear export, and by the stability of the mRNA. The stability of fucTA mRNA was not affected by swm knockdown (Figure 6E). Given that 5′-end processing is essential for mRNA stability, the 5′-end processing of fucTA mRNA is not regulated by Swm. In addition, splicing of fucTA mRNA does not appear to depend on Swm activity since the misspliced form of fucTA mRNA in the swm knockdown cells could not be detected when we amplified fucTA mRNA using PCR primers that targeted different exons (data not shown). Furthermore, both transcription and nuclear export of mRNAs are regulated by the THO/TREX complex [39] and Sus1 [40]; thus, Swm may be a factor participating in both processes, with its depletion resulting in reduced expression and defective nuclear export of fucTA mRNA. We assume that abnormal extension of polyA in swm knockdown cells would be a secondary effect of nuclear export defects, as observed in mutants of genes required for nuclear export [29]. Moreover, Pabp2, Hel25E/UAP56 [41], and Nup358 [42], which participate in the nuclear export of mRNA, were also identified in our screen (Table S4), suggesting that nuclear export might be essential for glycosylation regulation.
Nuclear accumulation of polyA(+) RNA in swm knockdown S2 cells was confirmed in our in situ hybridization experiment, as previously reported [43]. However, in neural BG2-c6 cells, weak polyA(+) RNA signals in nuclei were comparable between control gfp knockdown and swm knockdown cells (data not shown), suggesting that only a small portion of mRNA might be exported by Swm. In addition, the ratio of nuclear to cytoplasmic fucTA mRNA was relatively higher than that of actin mRNA in swm knockdown BG2-c6 cells (Figure 6F). Collectively, Swm may have preferred mRNAs for nuclear export. Moreover, the export efficiency and/or target RNA preference of Swm may differ between S2 and BG2-c6 cells.
Mammalian homologs of genes identified in this screen
Most of the genes identified in our present study have mammalian homologs (Table S5). Among these, Cog3 has been reported to be involved in glycosylation in organisms ranging from yeast to humans [37]. Interestingly, mutations of some COG components are causative for human diseases including congenital disorders of glycosylation (CDG) [44]. Further investigation of the genes identified here will likely provide additional insights into novel glycosylation regulatory mechanisms conserved in organisms ranging from Drosophila to humans and, possibly, into diseases involving glycosylation pathways.
Materials and Methods
Fly stocks and clone analysis
The following fly strains were used: Canton-S as the wild-type strain, GMR-Gal4 (Bloomington Drosophila Stock Center (BDSC)), fucTA mutant Pbac{WH}FucTAf03774 (BDSC), swm mutants swmF14 and swmF15(BDSC), and Pabp2 mutant P{SUPor-P}Pabp2{KG02359} (BDSC). RNAi lines were mainly supplied by the National Institute of Genetics (Japan), with the remainder purchased from the Vienna Drosophila RNAi Center (VDRC). The transgenic flies harboring UAS-dicer2 were also purchased from VDRC.
The swm and Pabp2 mutant mosaic clones were generated using the FLP-FRT system [27]. Adult heads from a cross of w[*]; P{w[+t*] ry[+t*] = white-un1}30C swm[F14] P{ry[+t7.2] = neoFRT}40A/CyO, P{Wee-P.ph0}2 or w[*]; P{w[+t*] ry[+t*] = white-un1}30C swm[F15] P{ry[+t7.2] = neoFRT}40A/CyO, P{Wee-P.ph0}2 with y[1]w[*]; P{w[+mC] = GMR-hid}G1 P{ry[+t7.2] = neoFRT}40A, l(2)CL-L[1]/CyO; P{w[+m*] = GAL4-ey.H}SS5, P{w[+mC] = UAS-FLP1.D}JD2 (BDSC), or y[1]; P{y[+mDint2] w[BR.E.BR] = SUPor-P}Pabp2[KG02359] P{neoFRT}42D/SM1 with y[1], w[*]; P{ry[+t7.2] = neoFRT}42D P{y[+t7.7] ry[+t7.2] = Car20y}44B, P{w[+mC] = GMR-hid}SS2, l(2)CL-R[1]/CyO; and P{w[+m*] = GAL4-ey.H}SS5, P{w[+mC] = UAS-FLP1.D}JD2 (BDSC) were dissected and used for glycosylation analyses as described below.
To investigate the localization of FucTA in RNAi-mutant flies, inducible Myc-tagged FucTA-expressing flies (UAS-fucTA-Myc) were generated. fucTA cDNA was a gift from S. Nishihara. A DNA fragment encoding the 3Myc sequence was ligated to the 3′end of the coding region of fucTA and inserted into the pUAST vector. The vector was injected into w1118 embryos for the production of transgenic flies.
To generate fucTA-Myc knock-in flies (fucTA-MycKI), the procedure was adopted from the ends-out knock in method [45]. Details are shown in Figure S4.
Antibodies and lectins
Commercial sources provided biotin-conjugated Lotus; HRP-conjugated ConA, WGA, DSA, LCA, UEA, PHAL4, PNA, SBA, and DBA lectins (Seikagaku Kogyo); and HRP-conjugated ABA (Sigma). Anti-HRP antibody was obtained from Jackson Immunoresearch. In addition, the antibodies used in this study are as follows: streptavidin-POD (Roche); anti-Myc A14 and anti-Myc 9E10 (Santa Cruz); anti-Flag M2 (Sigma); HRP-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG, and normal mouse IgG (Jackson Immunoresearch); Alexa 488-conjugated anti-mouse IgG, Alexa 488-conjugated anti-rabbit IgG, and TOPRO3 (Invitrogen); and Cy3-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG (Kirkegaard & Perry Laboratories, Inc.). Anti-Chp 24B10, anti-α-tubulin, 22C10, BP102, anti-FasI, anti-FasII, and anti-FasIII were obtained from the Developmental Studies Hybridoma Bank. Anti-Chp antibody N8A [18] and anti-GM130 [46] were generated as described previously.
Primers
The primer sets used for the generation of transgenic flies are listed in Table S6. The primer sets used for analysis in cultured cells are listed in Table S7.
Quantification of ventral nerve cord shape
The central nervous systems were manually isolated from CS and fucTAf03774 third instar larvae and fixed in 4% paraformaldehyde/PBS at 4°C overnight. After several washes with PBS, brain hemispheres were manually removed and placed flatly onto slide glasses. The images were captured using an AxioCam-equipped Zeiss AxioImager M1, and the length and width of VNCs were measured using AxioVision Rel. 4.7 software.
First screen
The first screen was carried out using a collection of transgenic fly lines carrying inducible dsRNA constructs. To drive expression of dsRNA for each gene in the eye, males from each RNAi fly line were crossed with GMR-Gal4 virgin females. Mating to inducible RNAi males and egg laying were carried out at 25°C for two days, and the resulting embryos were then incubated at 28°C. After 2 weeks, the hatched knockdown flies were collected and cultured for an additional 14 days at 28°C to enhance the effects of dsRNAs. More than 100 heads per line were isolated and homogenized in lysis buffer consisting of 10 mM sodium phosphate (pH 7.0), 100 mM NaCl, 0.5% Triton X-100, and complete protease inhibitor (Roche). The lysate was incubated with anti-Chp (24B10) covalently coupled with protein G-Sepharose (GE Healthcare) for 2 h. The Chp-bound Sepharose was subsequently washed with lysis buffer, and Chp was eluted with 50 mM triethylamine-acetic acid (pH 3.5).
Purified Chp was spotted onto Hybond ECL nitrocellulose membranes (GE Healthcare) using a Bio Dot slot format (Bio-Rad). After the membrane was blocked with 2% bovine serum albumin, immuno - or lectin blot analysis was performed with anti-Chp (N8A), anti-HRP, or HRP-conjugated ConA, LCA, DSA, and WGA. HRP-conjugated anti-mouse or HRP-conjugated anti-rabbit IgG was used as the secondary antibody. Detection was performed using Supersignal West Pico Chemiluminescent Substrate (Thermo), and quantification performed using a LAS-1000 Luminescent Image Analysis System (FujiFilm). An example of the dot blot is shown in Figure S2. Lectin affinity values per Chp values were converted to log values for normalization. On the basis of standard deviations from the values of GMR>GFPRNAi (as a control), genes with a z-score >3 were considered primary candidates. In the primary screen, we performed a single assay round. For the primary candidates, the lectin analysis was repeated (n = 3). Two-sided Student's t-tests were applied to the reproducibility test data between lectin values of knockdown and control flies. Genes with p<0.005 were considered to be reproducible findings.
Validation
Collection of the second transgenic fly stock lines was designed in such a way that there was no overlap between the primary construct in the original library and the new dsRNA sequence, and that the new dsRNA sequence had a low off-target probability score calculated by dsCheck [22]. Using the second lines, lectin analysis was independently repeated (n = 2), and the values between the knockdown and control (GMR>GFPRNAi) were tested by two-sided Student's t-tests. Genes with p<0.05 were considered glycosylation-related genes (Rank 1). When no change in lectin values was observed solely using GMR-Gal4, the examination was repeated using GMR-Gal4; UAS-dicer2 (VDRC). RNAi flies from VDRC whose target sequence had no overlap with the primary construct were also used as second lines.
For candidate genes that were not evaluated with the second lines, first the number of 19mers with a perfect match to each Drosophila transcript was calculated using dsCheck. The highest number of 19mers with a perfect match to the off-target gene was defined as the “off-target probability score”. The genes with a score below 3 were tested to determine whether knockdown of their suspected off-target genes led to glycosylation defects. When the knockdown of the suspected off-target genes did not lead to any evidence of glycosylation defects, the corresponding genes were added to the list of genes required for glycosylation (Rank 2).
GO analysis
Our final glycosylation hit list was annotated to include information on protein domain, molecular function, and protein family by cross-referencing InterPro (version 20.0 and 21.0) [47], Flybase, and Panther [48]. On the basis of the resulting annotation, genes were manually classified into 9 categories. These annotations are shown in Table S4.
Mass spectrometry (MS) analysis of glycopeptides and glycan structure
MS analysis of glycopeptides was performed as previously described [16]. Purified Chp was digested with modified trypsin (Promega). Glycopeptides and peptide mixtures were separated by reversed-phase high-performance liquid chromatography (RP-HPLC) using an Inertsil C18 column (GL Science). The matrices for peptide analysis by matrix-assisted laser desorption/ionization time-of-flight MS (MALDI-TOF-MS) spectra were acquired using a Voyager mass spectrometer (Applied Biosystems). MS/MS measurements were performed using an Ultraflex TOF/TOF mass spectrometer with the LIFT-MS/MS facility (Bruker Daltonics GmbH).
Immunostaining
To investigate the amount of α1,3-fucosylation in vivo, frozen sections of adult eye demonstrating swm or gfp knockdown were prepared. Immunostaining was performed as previously described [49]. Fluorescent images were acquired with a laser scanning confocal microscope, LSM700 (Zeiss).
Cell culture, plasmid construction, transfection, and siRNA treatment
BG2-c6 cells were cultured in M3 medium (Sigma) containing 10% FBS and 10 µg/mL insulin (Invitrogen) at 25°C. Transfection was performed using a calcium phosphate transfection kit (Invitrogen).
To construct an expression plasmid for C-terminal Flag-tagged Swm (Swm-Flag), cDNA containing the swm coding region was obtained from Canton-S total RNA by RT-PCR. A DNA fragment encoding the 3Flag sequence was ligated to the 3′end of the swm coding region and inserted into the expression vector pRmHa.
For the transient expression of Swm-Flag, 1.5 µg plasmid was added to each well of a 24-well plate, in which 5×105 BG2-c6 cells had been seeded. Plasmid expression was induced by incubating cells in the presence of 0.1–0.5 mM CuSO4 for periods from 3 h to overnight.
For knockdown experiments, dsRNA for swm or gfp was produced using in vitro transcription T7 kit (Takara). A 3-µg aliquot of dsRNA was added to each well of a 24-well plate in which 5×105 cells had been seeded. After 5 days, cells were harvested for the following assays.
Immunoblot
For protein analysis, equal amounts of protein extracted from fly eyes were subjected to SDS-PAGE. After transfer to PVDF membranes (Millipore), membranes were blocked in PBS containing 0.05% Tween 20 and 5% skim milk and incubated overnight with the primary antibody, followed by the secondary antibody. A monoclonal anti-α-tubulin antibody was used for normalization. Detection was performed using the Supersignal West Pico Chemiluminescent Substrate.
RNP-IP assay
Immunoprecipitation of protein–RNA complexes was performed according to a protocol by Niranjanakumari et al. [50]. BG2-c6 cells transfected with swm-Flag were harvested and cross-linked in PBS containing 0.2% PFA. Following sonication in RIPA buffer (50 mM Tris-HCl pH 7.5, 1% NP-40, 0.05% SDS, 1 mM EDTA, 150 mM NaCl) with protease inhibitor (Nakarai), each supernatant was mixed with a monoclonal anti-Flag M2 antibody or normal mouse IgG and then incubated with protein-G beads (GE Healthcare) at 4°C for 6 h. Sepharose beads were washed 5 times with RIPA buffer and incubated in 50 mM Tris (pH 7.0), 5 mM EDTA, 10 mM DTT, and 1% SDS at 70°C for 45 min to reverse cross-linking. RNA was extracted from the immunoprecipitates using Sepasol reagent (Nakarai) followed by DNase I treatment. The extracted RNA was used as a template to synthesize cDNA using Superscript III (Invitrogen) according to the manufacturer's protocol.
Real-time PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen) or Sepasol reagent (Nakarai). Complementary DNAs were synthesized using Superscript III according to the manufacturer's protocol. Real-time PCR was carried out using a 7900HT Fast Real Time PCR system (Applied Biosystems) with Power Syber Green. Samples were normalized with Drosophila rpl32.
RNA degradation analysis
BG2-c6 cells transfected with gfp or swm dsRNA were grown for 5 days, and then actinomycin D (Nakarai) was added to a final concentration of 5 µg/mL to arrest de novo RNA synthesis. At 30, 60, and 90 min after actinomycin D treatment, the cells were harvested, and fucTA and rpl32 (control) mRNA was quantified by real-time PCR as described above.
PAT assay
PolyA measurement of fucTA mRNA was carried out as described [51]. PCR fragments from the PAT assay were separated on 2% agarose gels and visualized by Southern blot using DIG DNA Labeling and Detection kit (Roche).
Subcellular fractionation
Fractionation and RNA isolation were performed as described [52]. The cytoplasmic fraction was extracted by hypotonic buffer (10 mM Tris (pH 7.5), 10 mM NaCl, 3 mM MgCl2, 0.5% (v/v) Nonident P-40, 40 units/mL RNase inhibitor, 1 mM DTT) and then extracted by EDTA buffer (10 mM Tris (pH 7.5), 10 mM NaCl, 40 mM EDTA, 0.5% (v/v) Nonident P-40, 40 units/mL RNase inhibitor, 1 mM DTT). The remaining pellet was used as the nuclear fraction. Sepasol solution was used to purify mRNA from each fraction as described above.
Human homologs
The InParanoid database (version 7.0) was searched for mammalian homologs [53].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KurosakaA
YanoA
ItohN
KurodaY
NakagawaT
1991 The structure of a neural specific carbohydrate epitope of horseradish peroxidase recognized by anti-horseradish peroxidase antiserum. J Biol Chem 266 4168 4172
2. JanLY
JanYN
1982 Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc Natl Acad Sci U S A 79 2700 2704
3. KatzF
MoatsW
JanYN
1988 A carbohydrate epitope expressed uniquely on the cell surface of Drosophila neurons is altered in the mutant nac (neurally altered carbohydrate). EMBO J 7 3471 3477
4. WhitlockKE
1993 Development of Drosophila wing sensory neurons in mutants with missing or modified cell surface molecules. Development 117 1251 1260
5. PhillisRW
BramlageAT
WotusC
WhittakerA
GramatesLS
1993 Isolation of mutations affecting neural circuitry required for grooming behavior in Drosophila melanogaster. Genetics 133 581 592
6. FabiniG
FreilingerA
AltmannF
WilsonIB
2001 Identification of core alpha 1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster. Potential basis of the neural anti-horseadish peroxidase epitope. J Biol Chem 276 28058 28067
7. SarkarM
LeventisPA
SilvescuCI
ReinholdVN
SchachterH
2006 Null mutations in Drosophila N-acetylglucosaminyltransferase I produce defects in locomotion and a reduced life span. J Biol Chem 281 12776 12785
8. OkajimaT
ReddyB
MatsudaT
IrvineKD
2008 Contributions of chaperone and glycosyltransferase activities of O-fucosyltransferase 1 to Notch signaling. BMC Biol 6 1
9. LuhnK
LaskowskaA
PielageJ
KlambtC
IpeU
2004 Identification and molecular cloning of a functional GDP-fucose transporter in Drosophila melanogaster. Exp Cell Res 301 242 250
10. IshikawaHO
HigashiS
AyukawaT
SasamuraT
KitagawaM
2005 Notch deficiency implicated in the pathogenesis of congenital disorder of glycosylation IIc. Proc Natl Acad Sci U S A 102 18532 18537
11. SelleckSB
2001 Genetic dissection of proteoglycan function in Drosophila and C. elegans. Semin Cell Dev Biol 12 127 134
12. NybakkenK
PerrimonN
2002 Heparan sulfate proteoglycan modulation of developmental signaling in Drosophila. Biochim Biophys Acta 1573 280 291
13. HainesN
IrvineKD
2003 Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol 4 786 797
14. DongX
ZavitzKH
ThomasBJ
LinM
CampbellS
1997 Control of G1 in the developing Drosophila eye: rca1 regulates Cyclin A. Genes Dev 11 94 105
15. Van VactorDJr
KrantzDE
ReinkeR
ZipurskySL
1988 Analysis of mutants in chaoptin, a photoreceptor cell-specific glycoprotein in Drosophila, reveals its role in cellular morphogenesis. Cell 52 281 290
16. KanieY
Yamamoto-HinoM
KarinoY
YokozawaH
NishiharaS
2009 Insight into the regulation of glycan synthesis in Drosophila chaoptin based on mass spectrometry. PLoS ONE 4 e5434 doi:10.1371/journal.pone.0005434
17. KrantzDE
ZipurskySL
1990 Drosophila chaoptin, a member of the leucine-rich repeat family, is a photoreceptor cell-specific adhesion molecule. EMBO J 9 1969 1977
18. Hirai-FujitaY
Yamamoto-HinoM
KanieO
GotoS
2008 N-Glycosylation of the Drosophila neural protein Chaoptin is essential for its stability, cell surface transport and adhesive activity. FEBS Lett 582 2572 2576
19. FreezeHH
1995 Lectin Affinity Chromatograpy, Current Protocol in Protein Science;
ColiganJE
DunnBM
PloeghHL
SpeicherDW
WingfieldPT
Hoboken, NJ John Wiley & Sons, Inc
20. AokiK
PerlmanM
LimJM
CantuR
WellsL
2007 Dynamic developmental elaboration of N-linked glycan complexity in the Drosophila melanogaster embryo. J Biol Chem 282 9127 9142
21. EcheverriCJ
PerrimonN
2006 High-throughput RNAi screening in cultured cells: a user's guide. Nat Rev Genet 7 373 384
22. NaitoY
YamadaT
MatsumiyaT
Ui-TeiK
SaigoK
2005 dsCheck: highly sensitive off-target search software for double-stranded RNA-mediated RNA interference. Nucleic Acids Res 33 W589 591
23. DietzlG
ChenD
SchnorrerF
SuKC
BarinovaY
2007 A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 151 156
24. KuhnU
WahleE
2004 Structure and function of poly(A) binding proteins. Biochim Biophys Acta 1678 67 84
25. CarmodySR
WenteSR
2009 mRNA nuclear export at a glance. J Cell Sci 122 1933 1937
26. CassoDJ
LiuS
IwakiDD
OgdenSK
KornbergTB
2008 A screen for modifiers of hedgehog signaling in Drosophila melanogaster identifies swm and mts. Genetics 178 1399 1413
27. StowersRS
SchwarzTL
1999 A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics 152 1631 1639
28. UiK
NishiharaS
SakumaM
TogashiS
UedaR
1994 Newly established cell lines from Drosophila larval CNS express neural specific characteristics. In Vitro Cell Dev Biol Anim 30A 209 216
29. HurtJA
ObarRA
ZhaiB
FarnyNG
GygiSP
2009 A conserved CCCH-type zinc finger protein regulates mRNA nuclear adenylation and export. J Cell Biol 185 265 277
30. BrownNH
1994 Null mutations in the alpha PS2 and beta PS integrin subunit genes have distinct phenotypes. Development 120 1221 1231
31. OlofssonB
PageDT
2005 Condensation of the central nervous system in embryonic Drosophila is inhibited by blocking hemocyte migration or neural activity. Dev Biol 279 233 243
32. RuaudAF
LamG
ThummelCS
2010 The Drosophila nuclear receptors DHR3 and betaFTZ-F1 control overlapping developmental responses in late embryos. Development 137 123 131
33. AshrafSI
GangulyA
RooteJ
IpYT
2004 Worniu, a Snail family zinc-finger protein, is required for brain development in Drosophila. Dev Dyn 231 379 386
34. LinYR
ReddyBV
IrvineKD
2008 Requirement for a core 1 galactosyltransferase in the Drosophila nervous system. Dev Dyn 237 3703 3714
35. RendicD
SharrowM
KatohT
OvercarshB
NguyenK
2010 Neural-specific {alpha}3-fucosylation of N-linked glycans in the Drosophila embryo requires Fucosyltransferase A and influences developmental signaling associated with O-glycosylation. Glycobiology
36. SunB
SalvaterraPM
1995 Characterization of nervana, a Drosophila melanogaster neuron-specific glycoprotein antigen recognized by anti-horseradish peroxidase antibodies. J Neurochem 65 434 443
37. SmithRD
LupashinVV
2008 Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr Res 343 2024 2031
38. LeonardR
RendicD
RabouilleC
WilsonIB
PreatT
2006 The Drosophila fused lobes gene encodes an N-acetylglucosaminidase involved in N-glycan processing. J Biol Chem 281 4867 4875
39. StrasserK
MasudaS
MasonP
PfannstielJ
OppizziM
2002 TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417 304 308
40. Pascual-GarciaP
Rodriguez-NavarroS
2009 A tale of coupling, Sus1 function in transcription and mRNA export. RNA Biol 6 141 144
41. GatfieldD
Le HirH
SchmittC
BraunIC
KocherT
2001 The DExH/D box protein HEL/UAP56 is essential for mRNA nuclear export in Drosophila. Curr Biol 11 1716 1721
42. ForlerD
RabutG
CiccarelliFD
HeroldA
KocherT
2004 RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol Cell Biol 24 1155 1167
43. FarnyNG
HurtJA
SilverPA
2008 Definition of global and transcript-specific mRNA export pathways in metazoans. Genes Dev 22 66 78
44. FreezeHH
2007 Congenital Disorders of Glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med 7 389 396
45. GongWJ
GolicKG
2003 Ends-out, or replacement, gene targeting in Drosophila. Proc Natl Acad Sci U S A 100 2556 2561
46. YanoH
Yamamoto-HinoM
AbeM
KuwaharaR
HaraguchiS
2005 Distinct functional units of the Golgi complex in Drosophila cells. Proc Natl Acad Sci U S A 102 13467 13472
47. HunterS
ApweilerR
AttwoodTK
BairochA
BatemanA
2009 InterPro: the integrative protein signature database. Nucleic Acids Res 37 D211 215
48. ThomasPD
CampbellMJ
KejariwalA
MiH
KarlakB
2003 PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13 2129 2141
49. YanoH
Yamamoto-HinoM
GotoS
2009 Spatial and temporal regulation of glycosylation during Drosophila eye development. Cell Tissue Res
50. NiranjanakumariS
LasdaE
BrazasR
Garcia-BlancoMA
2002 Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26 182 190
51. SallesFJ
StricklandS
1999 Analysis of poly(A) tail lengths by PCR: the PAT assay. Methods Mol Biol 118 441 448
52. JangBC
Munoz-NajarU
PaikJH
ClaffeyK
YoshidaM
2003 Leptomycin B, an inhibitor of the nuclear export receptor CRM1, inhibits COX-2 expression. J Biol Chem 278 2773 2776
53. BerglundAC
SjolundE
OstlundG
SonnhammerEL
2008 InParanoid 6: eukaryotic ortholog clusters with inparalogs. Nucleic Acids Res 36 D263 266
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding