
Investigation and Functional Characterization of Rare Genetic Variants in the Adipose Triglyceride Lipase in a Large Healthy Working Population
Recent studies demonstrated a strong influence of rare genetic variants on several lipid-related traits. However, their impact on free fatty acid (FFA) plasma concentrations, as well as the role of rare variants in a general population, has not yet been thoroughly addressed. The adipose triglyceride lipase (ATGL) is encoded by the PNPLA2 gene and catalyzes the rate-limiting step of lipolysis. It represents a prominent candidate gene affecting FFA concentrations. We therefore screened the full genomic region of ATGL for mutations in 1,473 randomly selected individuals from the SAPHIR (Salzburg Atherosclerosis Prevention program in subjects at High Individual Risk) Study using a combined Ecotilling and sequencing approach and functionally investigated all detected protein variants by in-vitro studies. We observed 55 novel mostly rare genetic variants in this general population sample. Biochemical evaluation of all non-synonymous variants demonstrated the presence of several mutated but mostly still functional ATGL alleles with largely varying residual lipolytic activity. About one-quarter (3 out of 13) of the investigated variants presented a marked decrease or total loss of catalytic function. Genetic association studies using both continuous and dichotomous approaches showed a shift towards lower plasma FFA concentrations for rare variant carriers and an accumulation of variants in the lower 10%-quantile of the FFA distribution. However, the generally rather small effects suggest either only a secondary role of rare ATGL variants on the FFA levels in the SAPHIR population or a recessive action of ATGL variants. In contrast to these rather small effects, we describe here also the first patient with “neutral lipid storage disease with myopathy” (NLSDM) with a point mutation in the catalytic dyad, but otherwise intact protein.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001239
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001239
Summary
Recent studies demonstrated a strong influence of rare genetic variants on several lipid-related traits. However, their impact on free fatty acid (FFA) plasma concentrations, as well as the role of rare variants in a general population, has not yet been thoroughly addressed. The adipose triglyceride lipase (ATGL) is encoded by the PNPLA2 gene and catalyzes the rate-limiting step of lipolysis. It represents a prominent candidate gene affecting FFA concentrations. We therefore screened the full genomic region of ATGL for mutations in 1,473 randomly selected individuals from the SAPHIR (Salzburg Atherosclerosis Prevention program in subjects at High Individual Risk) Study using a combined Ecotilling and sequencing approach and functionally investigated all detected protein variants by in-vitro studies. We observed 55 novel mostly rare genetic variants in this general population sample. Biochemical evaluation of all non-synonymous variants demonstrated the presence of several mutated but mostly still functional ATGL alleles with largely varying residual lipolytic activity. About one-quarter (3 out of 13) of the investigated variants presented a marked decrease or total loss of catalytic function. Genetic association studies using both continuous and dichotomous approaches showed a shift towards lower plasma FFA concentrations for rare variant carriers and an accumulation of variants in the lower 10%-quantile of the FFA distribution. However, the generally rather small effects suggest either only a secondary role of rare ATGL variants on the FFA levels in the SAPHIR population or a recessive action of ATGL variants. In contrast to these rather small effects, we describe here also the first patient with “neutral lipid storage disease with myopathy” (NLSDM) with a point mutation in the catalytic dyad, but otherwise intact protein.
Introduction
Despite immense efforts, the genetic contribution to many complex phenotypes and diseases is still incompletely understood [1]. Two opposite hypotheses about the nature of genetic variation underlying common diseases have been proposed. The “common disease – common variant” hypothesis argues that the genetic basis of complex diseases is formed by the cumulative action of frequent polymorphisms (here defined by a minor allele frequency (MAF) >5%) with small impact [2], while the “common disease – rare variant” hypothesis rather advocates a strong impact of rare variants with large effects and high penetrance [3], [4]. Genome-wide association studies (GWAS) based on the former rationale led to the identification of an impressive wealth of new loci and pathways [5], [6], but left a large fraction of the estimated heritability still unexplained [7]. Since effects of rare variants are not captured by conventional GWAS, they were discussed to be a major source to explain this “missing heritability” [1], [7]–[9].
Studies resequencing the coding regions of candidate genes for example for lipoprotein metabolism [10]–[13], obesity [14], renal function [15], type 1 diabetes [16] and cancer predisposition [17]–[19] follow the rationale that mutations with large effects are likely to be enriched in the extremes of the phenotype distribution [8], [10]. These studies detected several rare mutations and underscored the power of the “extreme phenotype-approach” to identify new functional variants and provide substantial insights into the molecular function of genes [20]–[22]. However, the influence of rare variants outside of phenotypic extremes and their relevance in the general population has not yet been addressed in depth. Some expectations about the effect size of rare variants may currently be too optimistic and rare variants with modest but still detectable effects are likely to exist [23]. In addition, resequencing studies so far focused on coding regions, neglecting promoter regions and introns. Finally, recently resequenced genomes revealed that current single nucleotide polymorphism (SNP) databases are still far from being complete [24]–[29].
Rare genetic variants at a population level have not been investigated for PNPLA2 (more commonly known as Adipose Triglyceride Lipase, ATGL [MIM *609059]) which is a highly interesting candidate gene in the lipolytic cascade. The protein encoded by the ATGL/PNPLA2 gene catalyzes the rate-limiting step of lipolysis, i.e. the breakdown of triacylglycerol to diacylglycerol [30]–[32], and represents the main lipase for basal and hormone stimulated lipolysis [33]. The human ATGL protein consists of 504 amino acids divided into an N-terminal part containing the catalytic patatin domain (amino acids 10 to 178) and a more loosely defined regulatory C-terminus [34] (Figure 1). The predicted catalytic dyad is formed by a serine residue at position 47 and an aspartate residue at position 166 [33]. Moreover, a putative hydrophobic lipid binding domain at positions 315–360 and two phosphorylation sites with unknown function at Ser404 and Ser428 were described [30], [35].

Two proteins are known to regulate ATGL activity in an opposite manner. Interaction with ABHD5 (also known as CGI-58, [MIM *604780]) enhances the catalytic activity up to 20-fold in mice and still 4-fold in humans [36], while the G0/G1 switch 2 protein (G0S2; [no MIM number assigned]) inhibits ATGL activity by interacting with the patatin domain, as described very recently by Yang and colleagues [37]. Additionally, regulation by nutritional status, insulin, PPARA (MIM +170998) and PPARG (MIM *601487) was reported [38], [39].
Mutations in ATGL and ABHD5 have provided the molecular basis underlying neutral lipid storage disease (NLSD) in humans. NLSD is a group of rare, autosomal recessive disorders characterized by excessive accumulation of trigylceride-containing cytoplasmic droplets in peripheral blood smears (referred to as Jordans' anomaly) and other tissues of the body including skin, bone marrow, muscles and cultured fibroblasts. Excessive lipid storage leads to variable forms of clinical presentations including ichthyosis, skeletal and cardiac myopathy and hepatic steatosis. Additionally, some cases have been reported with ataxia, hearing loss, or mental retardation. A recently proposed classification subdivides NLSD into two distinct groups [40]. Depending on whether or not the patients suffer from skin involvement, they are diagnosed with either neutral lipid storage disease with ichthyosis (NLSDI, also known as Chanarin Dorfman Syndrome [MIM #275630] or neutral lipid storage disease with myopathy (NLSDM, [MIM #610717]), respectively. Importantly, this classification finds its molecular basis in the affected genes. Mutations in ATGL cause NLSDM, and mutations in ABHD5 cause NLSDI [41].
On the other hand, polymorphisms and haplotypes in ATGL were associated with reduced plasma levels of free fatty acids (FFA) and triglycerides, increased plasma levels of glucose and an elevated risk for type 2 diabetes in the Utah Obesity Case-Control Study [42]. Common haplotypes have also been associated with increased triglycerides in Greenland Inuits [43]. However, no association of rare variants with obesity was observed in a recent case-control study [14].
The aim of this study was to thoroughly investigate the influence of rare variants in the ATGL gene on FFA levels in a large healthy working population both by association and biochemical studies. This was achieved by (1) screening the genomic region of the ATGL gene for novel variations, (2) in-vitro evaluation of residual enzymatic activity and intracellular localization of all detected protein variants and (3) genetic association studies on FFA levels. All investigations were done in the SAPHIR population, which is an observational study in healthy unrelated Caucasian subjects. Study participants were recruited in the years 1999–2002 by health-screening programs in large companies in and around the Austrian city of Salzburg. For further details see the Methods section below. Our study in a healthy population revealed a pronounced allelic heterogeneity due to mostly rare and even private mutations with, however, modest effects of FFA plasma concentrations.
Besides this screening approach and in order to oppose the mutations found in heterozygote state in a healthy population to an example of a homozygous state, we describe here also the first patient with NLSDM caused by a point mutation within the catalytic dyad (p.D166G).
Results
Mutation screening
We screened a total of 11.6 megabases for mutations corresponding to the complete ATGL gene region in 1,473 individuals randomly selected from the SAPHIR Study. We detected 20 rare and 34 private variants, but also one common polymorphism, which were not yet reported in dbSNP build 131 (Table 1 and Figure 2). An exhaustive list is given in Table S1. Taken together, rare variants in ATGL were present in 7.7% of the screened individuals (n = 113).


Only about half of the SNPs stored in dbSNP for ATGL were detected in our population. However, confirmation of 14 out of 17 independently validated SNPs corroborates the sensitivity of our approach. The inspection of the database entries of the three undetected validated SNPs revealed that two of them were originally detected in samples of different ethnicity than SAPHIR (rs12270408 in Blacks and rs28535188 in Asians) and the third one (rs10544581) was located in a low complexity region.
Although representing a general healthy working population, 39 (2.6%) of the individuals presented rare missense and even nonsense mutations in ATGL (Table 2). Most notably, we even identified a heterozygous frameshift variation at aspartate 244, which introduces a premature translation stop at amino acid 254 and thus produces a dysfunctional protein totally lacking lipid droplet binding and catalytic activity (see below). Moreover, two frequent non-synonymous SNPs were present in the population.
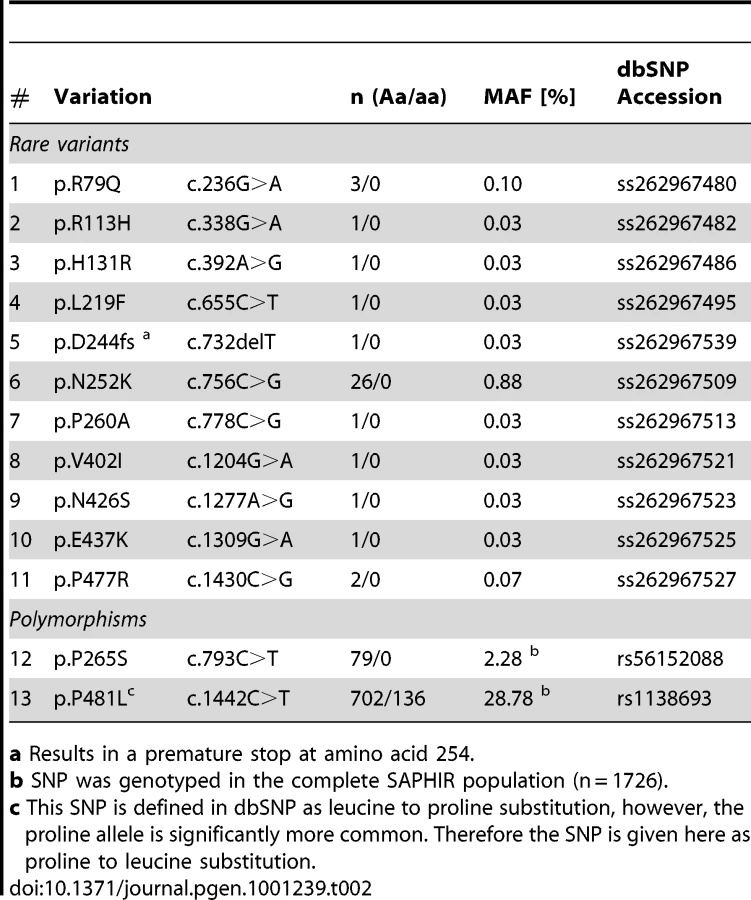
The mutation density was highest in the upstream region of ATGL (1 every 71 bases) and similar in introns and coding regions (1 every 132 and 115 bases, respectively) (Figure 2 and Table 1). Strikingly, about three quarters of all detected variants in ATGL in the present study were until recently unknown.
Functional studies of ATGL variants
We investigated the intracellular localization and in-vitro triglyceride hydrolase activity of all detected protein variants including the two frequent SNPs rs56152088 and rs1138693 (p.P265S and p.P481L, respectively). All mutants were readily expressed by Cos-7 cells and, except for the frame shift mutation p.D244fs, still correctly localized to the lipid droplets (Figures S1 and S2). In line with previous reports [34], the loss of the C-terminus effectively abolished the binding of the truncated p.D244fs protein to the lipid droplets (Figure 3).

Both basal and ABHD5-stimulated catalytic activities showed large variability ranging from total inactivity (p.D244fs) over strong impairment (p.R113H, p.V402I) to nearly wild type activity (Figure 4). All mutants with residual activity were still stimulated by ABHD5.

Identification of the first NLSDM patient with a point mutation in the catalytic dyad
During our screening in the SAPHIR Study, a 20 year old male was referred to our laboratory with the suspicion of NLSDM. Clinical investigations had revealed a dilation of the right cardiac ventricle with severely impaired function, diffuse cardiac fibrosis and vacuolated neutrophiles. However, no ichthyosis was observed. Cultured fibroblasts showed a 1.54-fold accumulation of [9,10-3H]-oleate, when compared to 5 healthy controls (51±6.6×10−3 vs. 33±5.5×10−3 counts per mg protein/24 hours; measurements done in quadruplicates).
In order to corroborate the NLSDM diagnosis, we included the exons and splice sites of ATGL of the patient and both parents in our mutation screening, revealing a homozygous c.497A>G mutation (p.D166G) disrupting the catalytic dyad (Figure 1). Parents were consanguineous and heterozygous for the mutation. Biochemical investigations showed intact intracellular localization, but total loss of catalytic activity (Figure 3 and Figure 4). This is the first proof of the crucial importance of the aspartate 166 in humans, which was so far only demonstrated by in vitro experiments in mice [35], [41].
Conservation and bioinformatic prediction
Except for valine 402, all mutated amino acids were well conserved in mammals, vertebrates and, partly, even in other bilateria (Figure 4). The catalytic aspartate at position 166 was conserved in all species from man to fruit fly and both asparagine 252 and arginine 113 were conserved in all but one species. However, also several C-terminal variations affected positions were rather conserved in mammals and vertebrates.
The performance of the bioinformatic prediction tools Polyphen-2, SIFT and PMUT was quite ambiguous (Figure 4, bottom). They concordantly predicted a deleterious effect of the N-terminal mutations p.R79Q, p.R113H and p.D166G, but all failed to predict the strong activity decrease of p.V402I. The predictions for the C-terminal variations were generally partially inconclusive, probably due to the lower degree of conservation of the C-terminus, its less well defined function and the rather small impact of the variants on the protein function.
Genetic association studies
Finally, we aimed to evaluate the genetic association of rare variants with FFA levels in the SAPHIR population using both a continuous and an “extreme phenotype” approach. FFA levels were available for a subgroup of 1253 individuals out of the 1473 individuals screened for variants. Figure S3 shows the distribution and corresponding percentiles of the FFA levels. Effect estimates from a linear model on log(FFA) for each of the rare variants showed an accumulation of increased plasma levels for variants in the promoter region (Figure S4). For the other classes of rare variants the effect direction was rather inconclusive, suggesting a rather heterogeneous effect of rare ATGL variants on the population if present in heterozygous state. Overall, FFA levels showed a slight shift towards lower FFA values for rare variant carriers, which seemed to be primarily driven by the missense variant p.N252K (p = 0.023) (Figure 5). No clear association of any other missense variant was observed.

Next we assessed the impact of rare ATGL variants in the phenotypic extremes, using the lower and upper 10%-quantiles of FFA values. The conditional density plot (Figure 6) showed an excess of rare variants in the lower tail of the FFA distribution. It indicates that using the bottom and top 10% of the distribution provides a good discrimination between individuals with at least one or no rare variants without losing too many data and therefore information.

Fifteen rare variants (11.8%) were found in the group of low FFA values compared to 7 (5.6%) in the group of high FFA values. This difference in proportions was statistically significant (p = 0.036; Beta-test [44]). Especially p.N252K and, surprisingly, variants in the introns were contributing to this excess of rare variants in the lower FFA quantile (Figure 7).
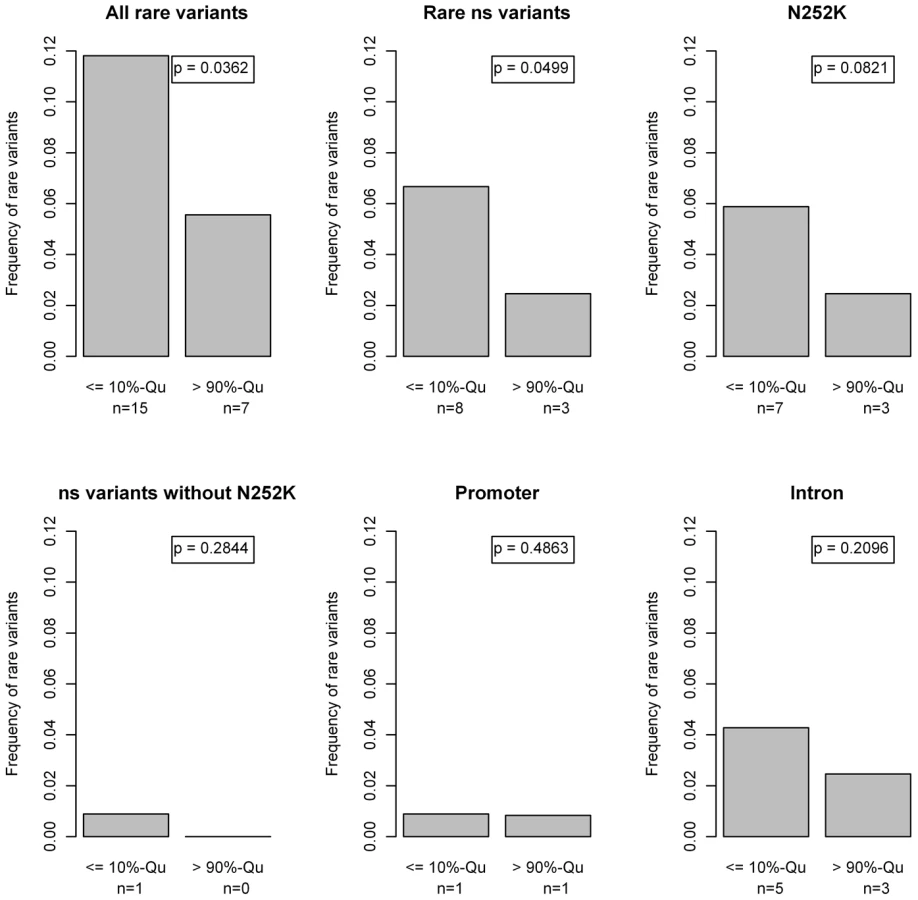
Table 3 gives a comparison of potential confounders between the two tail groups. However, none of these parameters were associated with both FFA levels and rare variants (data not shown), therefore excluding confounder effects.

Discussion
Mutation screening in SAPHIR and functional evaluation of missense variants
We investigated the influence of rare genetic variants in ATGL on FFA levels by an unbiased approach in a healthy working population by genetic association studies followed by functional investigations. The biochemical characterization of the detected missense variants demonstrated the presence of several mutated but still operative alleles with largely varying residual activity even in a healthy population. However, one variant showed a total loss of function (p.D244fs) and three variants presented markedly impaired catalytic activities (p.R79Q, p.R113H and p.V402I).
The effects of p.D244fs and p.V402I were against our expectations. The p.D244fs variant represents the shortest human nonsense mutation, which has been functionally investigated so far. Unlike most previously described forms of truncated ATGL [34], [35], p.D244fs totally lacked catalytic activity, probably due to critical changes in the overall protein structure produced by a frameshift in the α/β hydrolase fold. A similar finding for a synthetic 178X construct was recently described by Kobayashi et al. [45].
The distinct activity decrease of p.V402I was puzzling, since V402 is a C-terminal, non-conserved amino acid. However, most neighboring amino acids appear to be highly conserved (Figure S5), thus suggesting a still unknown regulatory role of this amino acid stretch. Intriguingly, Duncan et al. recently reported the binding of still unidentified proteins to the C-terminus of murine ATGL [35].
Genetic association studies
Our population-wide association studies on FFA levels in the SAPHIR Study were rather inconclusive and showed mostly non-significant trends towards lower FFA levels for rare variant carriers. These trends were primarily driven by the p.N252K variant (Figure 5). Heterozygous carriers of this variant showed a decrease in mean FFA levels of 15% compared to the wild type (6.70 mg/dl vs. 7.86 mg/dl). This is in line with previous results in the Utah Obesity Case Control study, where we observed a decrease of about 12% [42]. Interestingly, p.N252K did not show a pronounced decrease in enzyme activity, although it affects one of the best conserved amino acids among all evaluated variants (Figure 4). This suggests that the association of p.N252K with FFA levels may be based on mechanisms other than the mere modification of the catalytic activity. On the other hand, given the nearly wild type-like residual activity and the lower conservation grade of the affected amino acids, the lack of association of the more frequent variations p.P265S and p.P481L seems well plausible.
Since no clear effect of rare variants on the whole population was observed we next investigated the impact of rare variants in the outer quantiles of the FFA distribution. This uncovered an excess of rare variants in the lower 10%-quantile, which was to a large extend driven by p.N252K. However, the accumulation of rare variants was even more pronounced when considering all rare variant carriers, due to a contribution of rare intronic variants (Figure 7). On the other hand, no accumulation of other missense variants was seen.
Since enzyme variants were frequently shown to follow a recessive model, the observed lack of significant associations of ATGL variants with FFA levels may indicate possible recessive effects of missense mutations in ATGL. This was also observed in NLSDM carriers [40]. Accordingly, even the heterozygote carrier of p.D244fs did not present any abnormalities in FFA levels (Figure S4).
Patient with NLSDM due to a point mutation in the catalytic dyad
NLSDM-causing mutations were frequently reported to be located in the C-terminus, thus keeping intact the patatin domain and the catalytic dyad [33]. Only two mutations in the patatin domain have been described so far: a retrotransposon insertion into exon 3 [46] and a frameshift mutation in exon 4 (p.Q160fs) [47], [48]. The latter also causes a mutation of the active site D166, but possible effects on enzyme inactivation were not investigated [33]. We describe the first patient with a point mutation of the catalytic aspartate (p.D166G), which is sufficient to effectively abolish the catalytic activity (Figure 4), despite of a still intact binding to the lipid droplets (Figure 3). The seemingly well pronounced binding of the D166G reminds to the recently reported p.P195L loss of function mutation [40], which showed an slightly increased binding to lipid droplets too [34], which might be caused by a decreased protein turn-over rate. In line with previous reports [40], [45], [48], the patient aged 19 presented a severe cardiomyopathy and is currently awaiting a cardiac transplant. This confirms the potentially highly deleterious effects of functionally impaired ATGL alleles.
Although both the muscles and the heart are not classical lipid storage tissues, the observation of myopathy and cardiomyopathy is in line with observations in ATGL-ko mice, which die from cardiac insufficiency at about week 12 from birth and show reduced exercise tolerance [33], [49]. Recent studies have provided some insights in the underlying mechanisms [33], [49], [50]. By using mice lacking ATGL in all tissues except the cardiac muscle, Schoiswohl et al have demonstrated a crucial role of ATGL for the supply of the working muscle with fatty acids [50]. Conversely, the cardiomyopathy is thought to be produced by a defect in the utilization of FFAs by the cardiomyocytes [33]. The cardiac muscle is not able to autonomously synthesize fatty acids, but rather relies on the exogenous supply from plasma. The uptaken FFAs are however not directly utilized for β-oxidation, but first re-esterified to triglycerides, which, in case of an ATGL deficiency, then accumulate in the cardiomyocytes. This issue is further exacerbated by the fact that the FFA uptake machinery still remains fully induced, despite the impaired triglyceride breakdown [33]. This may then produce the observed cardiac hypertrophy and subsequent cardiac failure.
Strengths and limitations of our study
The resequencing of individuals with extreme phenotypes has been very successful to identify new functional variants and shed light on the molecular function of involved proteins [20]–[22], [51]. However the total impact of rare variants on traits in healthy populations is still unclear. The rare variant hypothesis has been repeatedly invoked to explain at least partially the missing heritability issue [1], [7]–[9], and postulates a high allelic heterogeneity consisting of individually rare but collectively common rare variants [3]. However, the well established impact of rare variants on the tails of the phenotypic distribution alone may not sufficiently explain a considerable portion of heritability and current expectation might be too optimistic [23]. Therefore, besides the highly deleterious rare variants in the outmost tails, also a number of rare variants with modest but still detectable effects should exist. These effects may, however, not be strong enough to drive their carriers into the outmost tails of the distribution, but rather contribute to the overall genetic distribution.
The main strengths of our study are (1) the unbiased approach based on a large healthy working population, (2) the functional follow-up of all detected missense variants, and (3) the statistical evaluation using both on a continuous and dichotomous approach. Only a few similar studies have been carried out before [13], [15], [20], and none has so far evaluated also intronic and promoter variants.
However, since a healthy population rather reflects the general variability in the population and will likely not be enriched for functional variants, the size of our screening population could be considered a limitation of our study, as it bears the risk of underestimating the role of rare variants due to insufficient statistical power. Due to lack of power, statistical tests were not corrected for multiple testing. Thus, the association results presented can merely be seen as a trend, which has to be followed in further, even larger studies.
Finally, since coding mutations are most prone to exert a severe impact on the protein function, we restricted the functional evaluation only to coding variants, providing the so far largest catalog of functionally investigated ATGL variants. However, the additional exhaustive investigation of all non-coding variants detected was beyond the scope of this study. Nevertheless, we are well aware that also non-coding variants can exhibit strong influence the phenotype [52]. Therefore, we cannot definitely rule out the presence of unidentified regulatory variants.
Conclusions
Although ATGL represents a crucial player in the lipolysis cascade, the role of rare genetic variants in ATGL remains elusive. We found several rare genetic variants and observed a trend towards lower FFA levels in carriers, as wells as an accumulation of rare variants in the lower 10%-quantile of FFA levels. However, the effects were rather small and suggested a secondary role of rare ATGL variants on the FFA levels in a healthy population.
Our study demonstrates that even in a healthy population, rare variants are collectively common and provide a large amount of allelic heterogeneity. However, at least in our study, their role on a quantitative trait in a healthy population outside of phenotypic extremes was rather small. Whether this implies that the impact of rare variants is, more generally, limited to the outer percentiles of the phenotypic distribution of a quantitative trait, remains to be investigated in further studies.
Methods
SAPHIR population
SAPHIR (Salzburg Atherosclerosis Prevention program in subjects at High Individual Risk) is an observational study involving 643 females aged 50 to 70 years and 1083 males aged 40–60 recruited between 1999 and 2002 in large companies in and around the Austrian city of Salzburg [53]. The differential age range between the sexes was chosen to match the cardiovascular risk, which is lower for women but matches the risk of men aged 10 years plus. All subjects were unrelated and of Caucasian origin. Exclusion criteria were severe obesity (BMI >40 kg/m2), established coronary artery, cerebrovascular or peripheral arterial disease, congestive heart failure, valvular heart disease, chronic alcohol (more than three drinks/day) or drug abuse and pregnancy. At baseline, a detailed personal and family history of all study participants was assessed via standardized questionnaires and anthropometric parameters were measured in the course of a physical examination. Venous blood was collected after an overnight fast and plasma samples were either used immediately for analysis or were stored frozen at −80°C. Plasma FFA concentrations were measured by using a colorimetric assay (Wako, USA) and DNA was extracted from total blood using the Invisorb Blood Universal Kit (Invitek, Germany). Informed consent was obtained from each participant and the study was carried out in accordance with the local ethics committee.
Patient with NLSDM due to a point mutation in the catalytic dyad
A 19-year-old male of consanguineous South Asian parents, who was previously well and has no significant family history apart from an older sister who died of an unknown cardiac problem at the age of 9 (no post-mortem was carried out) presented to his local Accident and Emergency Department with acute episode of shortness of breath and generally unwell. He was subsequently admitted under the care of cardiology team for further investigations. He had normal developmental milestones, was not on any medications and was due to enroll first year third level education. On admission, he had normal heart rate and blood pressure, his body mass index was below 25 kg/m2. Routine blood tests including full blood count, renal, bone profiles and creatinine kinase (CK) were all normal. His liver function tests were also essentially normal apart from mildly raised γ-glutamyl transferase (GGT) suggestive of mild fatty liver which was confirmed on a liver ultrasound. No ischaemic changes were noted on 12-lead electrocardiogram (ECG). An echocardiogram showed severely dilated both ventricles with left ventricle end-diastolic diameter of 72 mm (normal <49±4 mm) and ejection fraction of 14% (normal >58±7%). He denied any musculo-skeletal symptoms and no history of ichthyosis was reported in the patient or the family.
He subsequently developed weakness of his left arm associated with impaired co-ordination, and brain magnetic resonance imaging showed a small cardio-embolic infarct in the right parietal and cerebellum areas. 24 hr electrocardiogram showed episodes of non-sustained broad complex tachycardia and multiple ventricular ectopics. He was subsequently warfarinised and has had implantable cardioverter defibrillator (ICD) and cardiac resynchronization therapy (CRT) inserted, and being placed on cardiac transplant waiting-list. General metabolic and autoimmune screenings for cardiomyopathy were negative apart from Jordan's anomaly observed on peripheral blood smear histology, suggestive of a neutral lipid storage disorder.
Detection of genetic variants
Mutation screening was performed using the Ecotilling technology. Ecotilling is a mutation detection technology based on mismatch cleavage and enables sensitive high throughput detection of rare mutations in 1.5 kb large PCR fragments and up to 768 pooled samples per run [54]. It therefore dramatically lowers the costs for rare variant screening in large populations by introducing a cost-effective and robust prescreening step capable of both detecting and exactly localizing several mutations per amplicon and therefore significantly easing the subsequent follow-up steps. Moreover, the intrinsic double detection of each mutation ensures high specificity [54]. A flowchart detailing the Ecotilling process is provided in Figure S6. Briefly, Ecotilling screening was performed as described before with minor modifications [54] and using fourfold pooled DNA. The genomic region of ATGL (1506 bp before exon 1 to 106 bp after end of exon 10) was amplified in 8 overlapping PCRs using LI-COR IRDye labeled primers (Metabion GmbH, Germany). After heteroduplex formation (99°C for 10 min, 70 cycles: 70°C for 20 s, decreasing 0.3°C per cycle), 4 µl PCR product were incubated at 45°C with 1.5 µl reaction buffer (1 M MgSO4, 0.1 M HEPES pH 7.0, 0.1 M KCl, 0.02% Triton X-100, 2 mg/ml BSA), 0.1 µl purified Cel-I (diluted 1∶10,000) and 9.4 µl PCR grade water (Lonza, Belgium). The digestion was stopped with 5 µl 75 mM EDTA, purified by Sephadex G50 (GE Healthcare, Sweden), mixed with 5 µl of formamide loading buffer and reduced to approximately 1.5 µl at 85°C. 0.8 µl were then loaded on the LI-COR 4300 DNA Analyzer (LI-COR Biosciences, USA). The assay design and evaluation approach was described in detail earlier [55]. For primer sequences and PCR conditions see Tables S2, S3, S4, S5. The Ecotilling approach was applied in 1473 randomly selected individuals from the SAPHIR Study.
Each rare mutation was then confirmed in each affected sample by sequencing. Common SNPs present in multiple samples were confirmed in at least one sample to ensure the identity of the respective Ecotilling signal. Sequencing was performed using on an Applied Biosystems (ABI) 3130xl Genetic Analyzer the BigDye Terminator v1.1 cycle sequencing kit (ABI Applied Biosystems, USA) using a modified protocol. For details see Tables S4 and S5. Genotyping of rs56152088 (p.P265S) and rs1138693 (p.P481L) in the entire population was performed with an ABI 7900HT Fast Real-Time PCR System using ABI Taqman assays.
Site-directed mutagenesis
Point mutations were inserted into the eukaryotic expression vector pEYFP-C1 (BD Biosciences Clontech, USA) encoding the sequence of human ATGL and a yellow-green fluorescent protein (YFP) [34] using the Invitrogen GeneTailor Site-Directed Mutagenesis System. Fidelity of picked clones was verified by restriction enzyme digestion and bidirectional sequencing. PCR conditions and primer sequences are given in Tables S6, S7, S8.
Expression of human ATGL mutant proteins
Expression of recombinant YFP-ATGL proteins and controls (wild type ATGL and β-galactosidase (LacZ)) was performed in Cos-7 cells (ATCC CRL-1651; ATCC, USA). Cos-7 cells were cultured at 37°C, 7.5% CO2 and 95% humidity in standard growth medium consisting of Dulbecco's Eagle modified medium (GIBCO, USA) supplemented with 10% fetal calf serum, (Sigma, USA), 100 µg/ml Streptomycin and 100 U/ml Penicillin (GIBCO). For transfection, cells were seeded at a density of 9*105 cells/10 cm2 dish, cultured in standard growth medium for 24 hours and transfected in serum-free medium with 6 µg plasmid DNA using Metafectene (Biontex, Germany).
Isolation of cytoplasmatic extracts for triglyceride hydrolase assay
After 48 hours in regular growth medium, cells were collected in PBS buffer, pelleted at 290 g for 3 minutes and resuspended in assay buffer (0.25 M sucrose, 1 mM EDTA, 1 mM DTT, 20 µg/ml leupetine, 2 µg/ml antipain, 1 µg/ml pepstatin, pH 7.0). All further steps were performed on ice. To generate cytoplasmatic extracts, cells were disrupted by sonication (Sonicator 4000 with Microtip Probe, Qsonica LLC, USA) and centrifugated at 1000 g (5 min, 4°C). Total protein concentration was determined by a colorimetric assay (Protein Assay, Bio-Rad Laboratories, USA) with BSA as standard.
Western blot analysis
The expression levels were determined by Western blotting using 20 µg cytoplasmatic extract, a rabbit anti-GFP antibody (1∶5,000) (Abcam, USA) and a goat anti-rabbit IgG antibody conjugated with horseradish peroxidase (1∶10,000) (Vector Laboratories, USA). Chemoluminescence was induced using the GE Amersham ECL Plus Kit and detected with a Typhoon 9400 Imager (GE Healthcare). Relative expression rates were determined with the ImageQuant Software 5.2 (GE Healthcare) using the local median method for background correction (Figure S7). To exclude the saturation of the gel bands, which might affect correct quantification, amounts from 9 to 50 µg protein extract of the lysate with the highest expression levels (p.V402I) were blotted without reaching signal saturation (data not shown).
Triglyceride hydrolase assay
Triglyceride hydrolase activity was determined as the amount of FFA released by 15 µg cytoplasmatic extract from a radiolabeled substrate containing [9,10-3H]-triolein in 1 hour [34], [56]. The wild type protein and each mutant were tested for basal and ABHD5 stimulated activity. Determination of ABHD5 stimulated activity was measured by adding purified murine GST-ABHD5 [36]. The concentration of ABHD5 leading to maximal stimulation of ATGL was determined before. All measurements were done in triplicates and corrected for Cos-7 background activity and varying expression levels (see Figure S7).
Intracellular localization
Cos-7 cells were seeded on microscope slides in six well plates at a density of 1.5*105 cells/well and transfected with 1 µg DNA per well. 24 hours after transfection, cells were incubated for 20 hours with regular growth medium containing 400 µM fat free BSA-complexed oleic acid to induce lipid droplet generation. Lipid droplets were finally stained by incubating the cells with 1 µg/ml Bodipy 558/568 C12 (Invitrogen, USA) for 1 hour. The localization of the YFP-tagged ATGL mutants was determined by laser scanning microscopy using a Leica SP5 confocal microscope (Leica Microsystems, Germany). YFP fluorescence was excited at 514 nm and emission was detected between 520 and 540 nm. BODIPY 558/568 C12 was excited at 561 nm and detected between 565–600 nm (Figures S1 and S2. Selected mutants are shown in Figure 3).
Lipid accumulation in p.D166G fibroblasts of the patient with NLSDM
Cultured fibroblasts were incubated as monolayers in multi-well plates in Kreb's ringer pH 7.0, 220 µmol/L [9,10-3H]oleic acid complexed to α-cyclodextrin (25mg/ml) for 5 hours at 37°C. The supernatant was removed, cells were washed in phosphate buffered saline and digested with 1N sodium hydroxide. Total accumulated tritium counts in the incubated cell digests were measured by liquid scintillation counting in patient and simultaneous normal controls and expressed in relation to fibroblast protein.
Statistical and bioinformatic analysis
For each of the variants, linear regression was performed on the logarithmized FFA values. To strengthen the power of the analysis including rare variants, they were grouped into carriers of rare variants (“all rare variants”) and non-carriers. This group was refined to several subgroups, including carriers of non-synonymous variants, variants in the promoter region, intronic variants and carriers of p.N252K. To follow the hypothesis that rare variants contribute to the “extremes” of a phenotype, FFA values were a priori categorized into their lower and upper 10% quantiles. The choice of this cutpoint was evaluated by a conditional density plot showing the probability of rare variants conditional on FFA values (Figure 6).
Proportion differences of rare variants between these extreme groups of FFA were tested using the Beta-Test proposed by Li et al [44].
All amino acid mutations were submitted to Polyphen-2 using the HumDiv dataset [57], SIFT [58] and PMUT [59]. Evaluation of non-coding SNPs was carried out as described before [60]. Promoter prediction was done with the GenomatixSuite PE (Genomatix GmbH, Germany). Conservation was evaluated using ClustalW [61] on the Uniprot Homepage (http://www.uniprot.org). Accession numbers of the sequences used are given in Table S9.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ManolioTA
CollinsFS
CoxNJ
GoldsteinDB
HindorffLA
2009 Finding the missing heritability of complex diseases. Nature 461 747 753
2. ReichDE
LanderES
2001 On the allelic spectrum of human disease. Trends Genet 17 502 510
3. PritchardJK
2001 Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69 124 137
4. BodmerW
BonillaC
2008 Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40 695 701
5. KronenbergF
2008 Genome-wide association studies in aging-related processes such as diabetes mellitus, atherosclerosis and cancer. Exp Gerontol 43 39 43
6. FrazerKA
MurraySS
SchorkNJ
TopolEJ
2009 Human genetic variation and its contribution to complex traits. Nat Rev Genet 10 241 251
7. MaherB
2008 Personal genomes: The case of the missing heritability. Nature 456 18 21
8. GoldsteinDB
2009 Common genetic variation and human traits. N Engl J Med 360 1696 1698
9. SchorkNJ
MurraySS
FrazerKA
TopolEJ
2009 Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev 19 212 219
10. CohenJC
KissRS
PertsemlidisA
MarcelYL
McPhersonR
2004 Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305 869 872
11. CohenJC
PertsemlidisA
FahmiS
EsmailS
VegaGL
2006 Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A 103 1810 1815
12. KotowskiIK
PertsemlidisA
LukeA
CooperRS
VegaGL
2006 A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 78 410 422
13. RomeoS
PennacchioLA
FuY
BoerwinkleE
Tybjaerg-HansenA
2007 Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet 39 513 516
14. AhituvN
KavaslarN
SchackwitzW
UstaszewskaA
MartinJ
2007 Medical sequencing at the extremes of human body mass. Am J Hum Genet 80 779 791
15. JiW
FooJN
O'RoakBJ
ZhaoH
LarsonMG
2008 Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40 592 599
16. NejentsevS
WalkerN
RichesD
EgholmM
ToddJA
2009 Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324 387 389
17. AzzopardiD
DallossoAR
EliasonK
HendricksonBC
JonesN
2008 Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res 68 358 363
18. FearnheadNS
WinneyB
BodmerWF
2005 Rare variant hypothesis for multifactorial inheritance: susceptibility to colorectal adenomas as a model. Cell Cycle 4 521 525
19. FearnheadNS
WildingJL
WinneyB
TonksS
BartlettS
2004 Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc Natl Acad Sci U S A 101 15992 15997
20. RomeoS
YinW
KozlitinaJ
PennacchioLA
BoerwinkleE
2009 Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest 119 70 79
21. ZhaoZ
Tuakli-WosornuY
LagaceTA
KinchL
GrishinNV
2006 Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 79 514 523
22. FahmiS
YangC
EsmailS
HobbsHH
CohenJC
2008 Functional characterization of genetic variants in NPC1L1 supports the sequencing extremes strategy to identify complex trait genes. Hum Mol Genet 17 2101 2107
23. Carvajal-CarmonaLG
2010 Challenges in the identification and use of rare disease-associated predisposition variants. Curr Opin Genet Dev 20 277 281
24. AhnSM
KimTH
LeeS
KimD
GhangH
2009 The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res 19 1622 1629
25. BentleyDR
BalasubramanianS
SwerdlowHP
SmithGP
MiltonJ
2008 Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 53 59
26. KimJI
JuYS
ParkH
KimS
LeeS
2009 A highly annotated whole-genome sequence of a Korean individual. Nature 460 1011 1015
27. LevyS
SuttonG
NgPC
FeukL
HalpernAL
2007 The diploid genome sequence of an individual human. PLoS Biol 5 e254 doi:10.1371/journal.pbio.0050254
28. WangJ
WangW
LiR
LiY
TianG
2008 The diploid genome sequence of an Asian individual. Nature 456 60 65
29. WheelerDA
SrinivasanM
EgholmM
ShenY
ChenL
2008 The complete genome of an individual by massively parallel DNA sequencing. Nature 452 872 876
30. ZimmermannR
StraussJG
HaemmerleG
SchoiswohlG
Birner-GruenbergerR
2004 Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306 1383 1386
31. JenkinsCM
MancusoDJ
YanW
SimsHF
GibsonB
2004 Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279 48968 48975
32. VillenaJA
RoyS
Sarkadi-NagyE
KimKH
SulHS
2004 Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279 47066 47075
33. ZechnerR
KienesbergerP
HaemmerleG
ZimmermannR
LassA
2009 Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res 50 3 21
34. SchweigerM
SchoiswohlG
LassA
RadnerFP
HaemmerleG
2008 The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J Biol Chem 283 17211 17220
35. DuncanRE
WangY
AhmadianM
LuJ
Sarkadi-NagyE
2009 Characterization of desnutrin functional domains:Critical residues for triacylglycerol hydrolysis in cultured cells. J Lipid Res 51 309 317
36. LassA
ZimmermannR
HaemmerleG
RiedererM
SchoiswohlG
2006 Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 3 309 319
37. YangX
LuX
LombesM
RhaGB
ChiYI
2010 The G(0)/G(1) Switch Gene 2 Regulates Adipose Lipolysis through Association with Adipose Triglyceride Lipase. Cell Metab 11 194 205
38. KershawEE
HammJK
VerhagenLA
PeroniO
KaticM
2006 Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 55 148 157
39. KershawEE
SchuppM
GuanHP
GardnerNP
LazarMA
2007 PPARgamma regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am J Physiol Endocrinol Metab 293 E1736 E1745
40. FischerJ
LefevreC
MoravaE
MussiniJM
LaforetP
2007 The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet 39 28 30
41. SchweigerM
LassA
ZimmermannR
EichmannTO
ZechnerR
2009 Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab 297 E289 E296
42. SchoenbornV
HeidIM
VollmertC
LingenhelA
AdamsTD
2006 The ATGL gene is associated with free fatty acids, triglycerides, and type 2 diabetes. Diabetes 55 1270 1275
43. JohansenCT
GallingerZR
WangJ
BanMR
YoungTK
2010 Rare ATGL haplotypes are associated with increased plasma triglyceride concentrations in the Greenland Inuit. Int J Circumpolar Health 69 3 12
44. LiQ
ZhangH
YuK
2010 Approaches for Evaluating Rare Polymorphisms in Genetic Association Studies. Hum Hered 69 219 228
45. KobayashiK
InoguchiT
MaedaY
NakashimaN
KuwanoA
2008 The Lack of the C-terminal Domain of Adipose Triglyceride Lipase Causes Neutral Lipid Storage Disease through Impaired Interactions with Lipid Droplets. J Clin Endocrinol Metab 93 2877 2884
46. AkmanHO
DavidzonG
TanjiK
MacdermottEJ
LarsenL
2010 Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul Disord 20 397 402
47. AkiyamaM
SakaiK
OgawaM
McMillanJR
SawamuraD
2007 Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 36 856 859
48. OhkumaA
NonakaI
MalicdanMC
NoguchiS
OhjiS
2008 Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul Disord 18 671 674
49. HuijsmanE
van de ParC
EconomouC
van der PoelC
LynchGS
2009 Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am J Physiol Endocrinol Metab 297 E505 E513
50. SchoiswohlG
SchweigerM
SchreiberR
GorkiewiczG
Preiss-LandlK
2010 Adipose triglyceride lipase plays a key role in the supply of the working muscle with fatty acids. J Lipid Res 51 490 499
51. YinW
RomeoS
ChangS
GrishinNV
HobbsHH
2009 Genetic Variation in ANGPTL4 Provides Insights into Protein Processing and Function. J Biol Chem 284 13213 13222
52. EpsteinDJ
2009 Cis-regulatory mutations in human disease. Brief Funct Genomic Proteomic 8 310 316
53. HeidIM
WagnerSA
GohlkeH
IglsederB
MuellerJC
2006 Genetic architecture of the APM1 gene and its influence on adiponectin plasma levels and parameters of the metabolic syndrome in 1,727 healthy Caucasians. Diabetes 55 375 384
54. TillBJ
ZerrT
BowersE
GreeneEA
ComaiL
2006 High-throughput discovery of rare human nucleotide polymorphisms by Ecotilling. Nucleic Acids Res 34: e99; Erratum in: Nucleic Acids Res 2006; 34 5352
55. CoassinS
BrandstätterA
KronenbergF
2008 An optimized procedure for the design and evaluation of Ecotilling assays. BMC Genomics 9 510 520
56. HolmC
OlivecronaG
OttossonM
2001 Assays of lipolytic enzymes. Methods Mol Biol 155 97 119
57. AdzhubeiIA
SchmidtS
PeshkinL
RamenskyVE
GerasimovaA
2010 A method and server for predicting damaging missense mutations. Nat Methods 7 248 249
58. NgPC
HenikoffS
2003 SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31 3812 3814
59. Ferrer-CostaC
GelpiJL
ZamakolaL
ParragaI
de la CruzX
2005 PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21 3176 3178
60. CoassinS
BrandstätterA
KronenbergF
2010 Lost in the space of bioinformatic tools: A constantly updated survival guide for genetic epidemiology. The GenEpi Toolbox. Atherosclerosis 209 321 335
61. LarkinMA
BlackshieldsG
BrownNP
ChennaR
McGettiganPA
2007 Clustal W and Clustal X version 2.0. Bioinformatics 23 2947 2948
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding