
Loss of DNMT1o Disrupts Imprinted X Chromosome Inactivation and Accentuates Placental Defects in Females
The maintenance of key germline derived DNA methylation patterns during preimplantation development depends on stores of DNA cytosine methyltransferase-1o (DNMT1o) provided by the oocyte. Dnmt1omat−/− mouse embryos born to Dnmt1Δ1o/Δ1o female mice lack DNMT1o protein and have disrupted genomic imprinting and associated phenotypic abnormalities. Here, we describe additional female-specific morphological abnormalities and DNA hypomethylation defects outside imprinted loci, restricted to extraembryonic tissue. Compared to male offspring, the placentae of female offspring of Dnmt1Δ1o/Δ1o mothers displayed a higher incidence of genic and intergenic hypomethylation and more frequent and extreme placental dysmorphology. The majority of the affected loci were concentrated on the X chromosome and associated with aberrant biallelic expression, indicating that imprinted X-inactivation was perturbed. Hypomethylation of a key regulatory region of Xite within the X-inactivation center was present in female blastocysts shortly after the absence of methylation maintenance by DNMT1o at the 8-cell stage. The female preponderance of placental DNA hypomethylation associated with maternal DNMT1o deficiency provides evidence of additional roles beyond the maintenance of genomic imprints for DNA methylation events in the preimplantation embryo, including a role in imprinted X chromosome inactivation.
Published in the journal:
. PLoS Genet 9(11): e32767. doi:10.1371/journal.pgen.1003873
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003873
Summary
The maintenance of key germline derived DNA methylation patterns during preimplantation development depends on stores of DNA cytosine methyltransferase-1o (DNMT1o) provided by the oocyte. Dnmt1omat−/− mouse embryos born to Dnmt1Δ1o/Δ1o female mice lack DNMT1o protein and have disrupted genomic imprinting and associated phenotypic abnormalities. Here, we describe additional female-specific morphological abnormalities and DNA hypomethylation defects outside imprinted loci, restricted to extraembryonic tissue. Compared to male offspring, the placentae of female offspring of Dnmt1Δ1o/Δ1o mothers displayed a higher incidence of genic and intergenic hypomethylation and more frequent and extreme placental dysmorphology. The majority of the affected loci were concentrated on the X chromosome and associated with aberrant biallelic expression, indicating that imprinted X-inactivation was perturbed. Hypomethylation of a key regulatory region of Xite within the X-inactivation center was present in female blastocysts shortly after the absence of methylation maintenance by DNMT1o at the 8-cell stage. The female preponderance of placental DNA hypomethylation associated with maternal DNMT1o deficiency provides evidence of additional roles beyond the maintenance of genomic imprints for DNA methylation events in the preimplantation embryo, including a role in imprinted X chromosome inactivation.
Introduction
Genomic methylation patterns are initially differentially acquired during male and female gametogenesis by the action of the DNMT3a cytosine methyltransferase and its accessory protein DNMT3L [1]–[3]. Germline derived DNA methylation patterns are subsequently extensively reprogrammed during preimplantation development, and while methylation is lost at many sites across the genome, it is maintained on imprinted genes [4]. Two forms of DNMT1, DNMT1s and DNMT1o, maintain DNA methylation until the blastocyst stage. DNMT1s, the full length Mr 190,000 form of DNMT1, is expressed at all cleavage stages of preimplantation development, and appears to maintain DNA methylation at all stages except the 8-cell stage [5]–[8]. DNMT1o is missing the amino-terminal-most 118 amino acids of DNMT1s and is the Mr 175,000 oocyte-derived version of DNMT1 that is only expressed in oocytes and preimplantation embryos. DNMT1o protein is present only in the cytoplasm throughout early embryo development with the exception of the 8-cell stage where it is found also in all eight nuclei [8]–[13]. Although oocytes of Dnmt1Δ1o/Δ1o female mice are devoid of DNMT1o protein, they establish normal maternal genomic imprints during oogenesis through the action of DNMT3a enzymes. Once fertilized however, the resulting preimplantation embryos fail to maintain methylation patterns at the differentially methylated domains (DMDs) of imprinted loci [12]. Mouse embryos born to Dnmt1Δ1o/Δ1o female mice lose methylation on imprinted genes between the 8-cell and 16-cell stages and most embryos subsequently die before birth [5], [12], [14]. Embryonic methylation at loci other than imprinted genes, including repeat sequences, was unaffected by the DNMT1o deficiency.
The embryos of Dnmt1Δ1o/Δ1o female mice have a variety of structural abnormalities [14]. The basis of the variation has been attributed to the failure to maintain complete DMD methylation of imprinted genes at a single stage of preimplantation development [12], [14]. Loss of methylation on half of the normally methylated alleles of imprinted genes at the 8-cell stage is followed by cell divisions whereby methylation is maintained presumably by embryo-derived DNMT1s. The presence of DMDs for imprinted genes on many autosomes coupled with random assortment of sister chromatids was predicted to result in over 4000 epigenotypes, and provide an explanation for the highly variable phenotypes [5], [14]. Molecular studies showing variations in the methylation of imprinted genes in the embryos and placentae of Dnmt1Δ1o/Δ1o female mice provided further evidence of diverse epigenotypes underlying the phenotypic abnormalities [5], [14].
The timing of preimplantation maintenance methylation and DNMT1o activity in particular coincides with key X chromosome inactivation (XCI) events. In the female zygote, both X chromosomes appear active, but soon thereafter a series of X-inactivation events ensues that results in inactivation of the paternal X chromosome. Inactivation of the paternal X chromosome is first evident at the 2 - to 4-cell stage and this parent-specific or imprinted X-inactivation is associated with Xist expression only from the paternal X (Xp) [15]–[17]. Xist in turn recruits various chromatin modifying complexes that eventually render the X chromosome transcriptionally inactive [18]. It has also been suggested that imprinted XCI occurs in a two-step manner, with Xp repeat elements first silenced at the 2-cell stage followed by Xp genic silencing emerging only at the 8 - to 16-cell stage embryo [19]. Imprinted X chromosome inactivation or silencing of genes on the paternal X chromosome in females is complete by the blastocyst stage. Interestingly, inactivation of Xp is maintained in the extraembryonic tissues, yet reversed in the inner cell mass of blastocyst, evident as biallelic X-linked gene expression that later evolves into mosaic monoalleic expression via random XCI [16], [17].
In extraembryonic tissues, imprinted XCI is specifically observed in cells of the trophoblast lineage and in primitive endoderm-derived cells of the visceral and parietal yolk sac [20]. At the molecular level, Xist expression during the initiation and maintenance of random XCI is known to be controlled by an array of both cis - and trans-acting factors. Key cis-acting negative regulators of Xist are Tsix and Xite, a cluster harboring multiple regulatory elements and a specific enhancer of Tsix [21]–[24]. In contrast to random XCI, the precise nature and timing of the events involved in the initiation and maintenance of imprinted XCI have been difficult to define, primarily because of technical difficulties of working with different stages of preimplantation embryo development. In a recent study Ohhata, et al. overcame some of the technical obstacles by devising an inducible Tsix expression system to over-express Tsix during preimplantation development [25]. The induction of Tsix led to the repression of Xist associated with an increased methylation of the Xist promoter and reactivation of Xp in the extraembryonic lineages. The latter findings indicate that imprinted XCI can be perturbed by altering the control of Tsix/Xist expression during preimplantation development. In addition, the findings indicate that at least some components of random XCI are shared by the random and imprinted X-inactivation processes.
Recently, on examination of DNMT1o-deficient placentae [26], we detected a higher incidence of and more severe placental defects in female versus male embryos of Dnmt1Δ1o/Δ1o mothers. These observations suggested that molecular defects extend beyond autosomally imprinted genes and that X chromosome-linked methylation may also be affected following absence of methylation maintenance by DNMT1o at the 8-cell stage. Therefore, we extended our molecular analysis to both the embryo and placenta of male and female Dnmt1omat−/− conceptuses, examined methylation at a large number of autosomal and X-linked loci, and traced a DNA hypomethylation defect within the X-inactivation center (Xic) back to preimplantation embryos, in blastocysts or shortly after DNMT1o's action in 8-cell embryos. The results uncover a new role for DNMT1o in maintaining methylation on the X chromosome of extraembryonic tissues and in upholding imprinted Xp inactivation.
Results
DNMT1o deficiency is associated with sex-specific abnormalities in extraembryonic tissues
To confirm whether female placentae, versus male placentae, were more severely affected as a result of maternal DNMT1o deficiency, we examined the morphology of extraembryonic tissues of 9.5dpc conceptuses conceived by crossing wild-type males to wild-type Dnmt1+/+, heterozygous Dnmt1Δ1o/+ and homozygous Dnmt1Δ1o/Δ1o females. The mutant Dnmt1Δ1o allele was generated by targeted deletion of exon 1o of the mouse Dnmt1 gene [12]. Conceptuses from these three crosses are designated Dnmt1omat+/+, Dnmt1omat+/− and Dnmt1omat−/−, respectively. Enlargement or hyperplasia of the extraembryonic tissue layers was a frequent finding, especially amongst female Dnmt1omat−/− conceptuses (Figure 1A, Figure S1). Dnmt1omat+/+ extraembryonic tissues lacked this hyperplasia, whereas Dnmt1omat+/− extraembryonic tissues displayed low levels of the abnormality (female: 5%; males: 12%). No case of severe hyperplasia was observed in Dnmt1omat+/− males, while severe hyperplasia was found in 3% of Dnmt1omat+/− females (Figure 1B). The vast majority of Dnmt1omat+/− extraembryonic tissues (92%) were similar in size in both males and females (Table S1). In striking contrast, Dnmt1omat−/− extraembryonic tissues displayed a wide range of anatomical abnormalities in both males and females compared to those of Dnmt1omat+/+ and Dnmt1omat+/− embryos (Figure 1B, Table S1, S2, S3). Female Dnmt1omat−/− conceptuses showed a major increase in extraembryonic hyperplasia (mild and severe) compared to female Dnmt1omat+/− conceptuses (47% vs. 5%, p<0.001). Furthermore, mild and severe hyperplastic abnormalities had a tendency to be more widespread in female extraembryonic tissues compared to their male counterparts (47% vs. 28%, p = 0.1). A detailed histological examination showed that cellular and structural morphology in the Dnmt1omat−/− extraembryonic tissue of females was defective at various levels (Figure S2, Supporting Text S1, Table S4, S5). In particular, increases in the giant cell population and abnormal branching to create villi were observed in the extraembryonic tissues of the Dnmt1omat−/− females. Thus, a shortage of the maternal DNMT1o enzyme during preimplantation development has more adverse effects on female, as compared to male, placental and extraembryonic tissues.
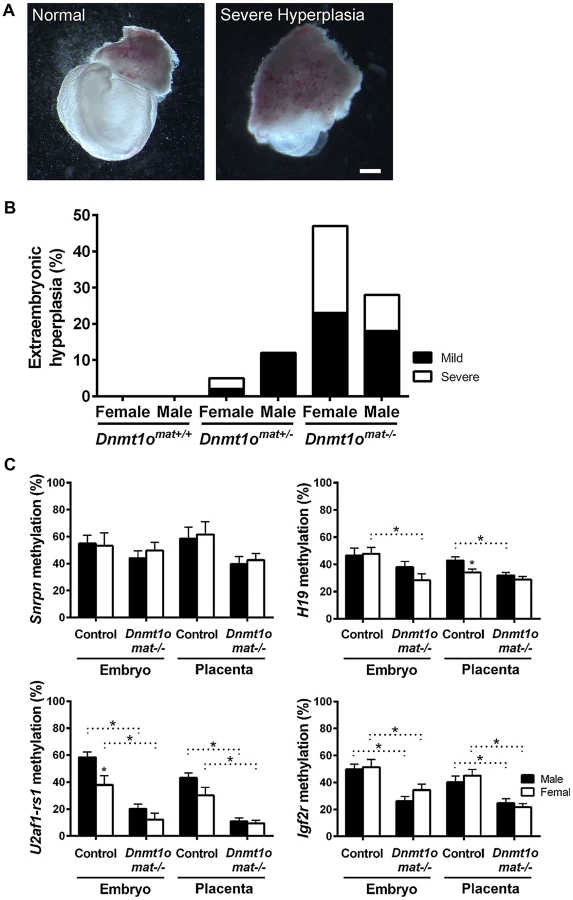
Female-specific abnormalities in Dnmt1omat−/− extraembryonic tissues are not caused by improper imprinting
Normal placental growth, development and phenotype are largely dependent on proper establishment and maintenance of genomic imprinting (reviewed in [27]). In order to ascertain that the morphological sex bias found in Dnmt1omat−/− extraembryonic tissues is independent of imprinting defects, levels of DMD methylation at imprinted loci were quantified. Previously, we provided molecular evidence that oocyte DNMT1o deficiency causes a failure in the maintenance of imprinted DMD methylation in embryos [12], which was more recently extended to include defects in the maintenance of placental DMD methylation [5]. However, sex-specific effects were not investigated in the earlier studies. Here we examined DMD methylation of H19, Snrpn, Igf2r and U2af1-rs1 in control Dnmt1omat+/+ and Dnmt1omat−/− 9.5dpc embryos and placentae of both males and females (Figure 1C). Dnmt1omat−/− animals showed a decrease in placental DMD methylation at all four imprinted loci (p<0.05); however, none of DMDs demonstrated a sex-specific effect in the Dnmt1omat−/− group. We conclude from these analyses that autosomal DMD methylation is not affected in a sex-specific manner and that differences in autosomal imprinted gene functions are not responsible for the significant female-specific increase in abnormal placental morphology.
Abnormal methylation of X-linked genes in female Dnmt1omat−/− extraembryonic tissues
Because sex-specific differences in autosomal imprinting could not account for the morphological discrepancy observed between male and female Dnmt1omat−/− extraembryonic tissues, we next investigated the possibility that X chromosome DNA methylation is altered. As previously mentioned, the major hallmark of the X chromosome is the unique form of dosage compensation that arises in females to silence one of the two X chromosomes. In the extraembryonic tissues, XCI is imprinted and is thought to be evolutionarily linked to autosomal imprinting [28]–[31]. We carried out an in-depth DNA methylation analysis of multiple CpG dinucleotides at a number of X-linked CpG islands (X-CGIs) distributed throughout the X chromosome and regions of the Xic using Sequenom MALDI-TOF mass spectrometry (MassARRAY system). We were particularly interested in CG-rich sequences associated with: 1) Xite, an enhancer that positively regulates Tsix sustained expression on the active X, 2) Tsix, which represses Xist expression, and 3) Xist, the major effector of the X-inactivation process (Figure 2). Xite, Tsix and Xist are sequences known to display differential methylation between active (Xa) and inactive (Xi) X chromosomes [21]. Data are displayed as the average methylation of all informative CpGs within each amplicon; amplicons were then averaged when more than one was assayed per region (Figure 3A). We compared 17 female 9.5dpc Dnmt1omat−/− placentae of various morphologies versus 5 female controls (Dnmt1omat+/+). The average methylation for all X-CGIs was significantly decreased in Dnmt1omat−/− versus control placentae (p<0.0001) and significant hypomethylation was found in 8/17 individual X-CGIs. X-CGIs that exhibit hypomethylation do not show any positional bias in relation to the Xic. For DNA methylation associated with the Xic, interestingly, two loci, Xite-DHS6 (in Dnmt1omat−/− placentae with either normal or abnormal morphology) and Tsix-CTCFc (in Dnmt1omat−/− placentae with abnormal morphology) revealed significant hypomethylation versus controls (p<0.05) (Figure 3A, B). These two regions are known for their intrinsic enhancer activity on Tsix [23], [24]. The 385-bp Xite-DHS6 amplicon correlates with important elements for Tsix regulation: the Tsix 5′ region, which contains the Tsix minor promoter and a DNase I hypersensitivity site (DHS), as well as Tsix transcriptional start site (TSS) and a substantial part of exon-1 (126 out of 186 bp). The Tsix-CTFCc 436-bp amplicon spans exon-3 of Tsix and a portion of the DXPas34 repeat, an element crucial in XCI and known to regulate imprinted XCI [32].


Of all regions tested, only Xist sequences displayed a trend toward hypermethylation. Because Dnmt1omat−/− mice produced conceptuses with morphologically normal and abnormal extraembryonic tissues (Figure 1), we compared the methylation of ten abnormal (mild or severe hyperplasia) Dnmt1omat−/− placentae versus controls. In this comparison, along with several X-CGIs that showed significant hypomethylation, Xist demonstrated significant hypermethylation (p<0.05) (Figure 3A, B). We next performed a cluster analysis based on the pattern of X-CGI methylation revealed by MassARRAY analysis (Figure 3A). Hierarchal clustering separates the placentae into two primary groups, one containing all 5 of the control placentae and 5 Dnmt1omat−/− placentae, in the other all 12 are Dnmt1omat−/−. Interestingly, in the group containing the control placentae, only one of the ten samples (a Dnmt1omat−/− placenta) is classified as having an abnormal morphology, whereas in the second group 9/12 samples are morphologically abnormal. This distribution is not expected by chance (χ2, p<0.01), and suggests a causative relationship between X chromosome hypomethylation and the severity of morphological abnormalities in female Dnmt1omat−/− placentae. The DNMT1o-associated X chromosome hypomethylation was restricted to extraembryonic tissue because the same MassARRAY analysis performed on female Dnmt1omat−/− embryos versus female control embryos (Dnmt1omat+/+) revealed no significant difference in X-linked gene methylation (Figure S3). From these findings, we show that sites across the X chromosome are extensively hypomethylated in female Dnmt1omat−/− extraembryonic tissues. The hypomethylation observed in the enhancer regions of Tsix and the hypermethylation detected for Xist suggest that the imprinted XCI process could be compromised in female Dnmt1omat−/− extraembryonic tissues.
Female-specific preponderance for hypomethylation of single copy loci in Dnmt1omat−/− extraembryonic tissues
In parallel, we extended our molecular DNA methylation analysis outside the X chromosome to non-DMD single-copy loci to determine whether DNMT1o deficiency was perturbing other autosomal regions. Restriction Landmark Genomic Scanning (RLGS) 2D-profiles were generated to compare the methylation levels of >2000 single-copy loci between Dnmt1omat+/− (control) and Dnmt1omat−/− 9.5dpc placentae. This method allows quantification of methylation intensities at genomic NotI restriction sites in CpG islands (CGIs) as well as in non-coding unique and repetitive sequences outside CGIs [33]. Globally, the majority of loci showed a similar methylation level in the placentae of Dnmt1omat−/− mice compared to controls, however we uncovered hypo - and hypermethylated sites in both male and female Dnmt1omat−/− placentae relative to control placentae (Figure 4A). On average, female Dnmt1omat−/− placentae had approximately twice the number of hypomethylated loci (23–26) compared with male Dnmt1omat−/− placentae (5–13). Hypermethylation was similar in all Dnmt1omat−/− placental samples (2–3 loci), except for placenta XY-P1 where a higher number of hypermethylated loci (10) was observed. Using established RLGS spot cloning methods [34], [35], 18/34 differentially methylated spots in the Dnmt1omat−/− placentae were identified (Table 1, Table S6). Loci demonstrating aberrant methylation were distributed among several chromosomes and associated mostly with CGIs and surrounding expressed sequences, either in the 5′region, body or 3′end of genes. A semi-quantitative assessment of changes in spot density shows that methylation varied in the range of 25–50% of normal methylation. Only a single genomic locus, U2af1-rs1, was found to be hypomethylated in every female and male sample. U2af1-rs1 is the only imprinted DMR identified in mouse RLGS profiles using this specific combination of restriction enzymes [36]. Its hypomethylation is consistent with findings on the same gene using the MassARRAY system (Figure 1C). Although none of the hypomethylated loci were identical among the male Dnmt1omat−/− placentae, 11 of the 23–26 hypomethylated loci among the female Dnmt1omat−/− placentae were identical. We were able to identify 6 of these 11 loci. Intriguingly, all six are positioned on various regions of the X chromosome (Table 1). Spots corresponded to genes Klhl13, Lonrf3, Pak3, Mum1l1, Map3k15 and EST CJ168414 (or BY033964). The hypomethylation observed for Klhl13, Lonrf3 and Pak3 was validated by MassARRAY over an extended segment (data included in Figure 3A). We conclude from this survey of genomic methylation that although control and Dnmt1omat−/− placentae have very similar patterns of genomic methylation, there is concentration of hypomethylated defects on X-linked loci in DNMT1o-deficient placentae.


Impairment of paternal imprinted XCI in female Dnmt1omat−/− extraembryonic tissues
Because regions within Xite, Xist and other X-linked genes in Dnmt1omat−/− placentae exhibited abnormal methylation, we next determined if paternally imprinted XCI is compromised by the loss of DNMT1o. Firstly, we sought to validate that both the maternal and paternal alleles of X chromosomes in female conceptuses were active in Dnmt1omat−/− tissues. To distinguish the parental origin of the active and inactive X chromosome alleles in female Dnmt1omat−/− extraembryonic tissues, we generated 9.5dpc conceptuses from inbred 129/Sv mothers (Dnmt1omat−/− and control Dnmt1omat+/+) and Mus musculus castaneus fathers to generate single nucleotide polymorphisms (SNPs) along the X chromosome. This analysis was carried out on visceral endoderm cells where normally the paternal X chromosome is preferentially silenced (imprinted XCI), and gene expression is solely under the control of the active maternal allele [37], [38] (reviewed in [20]). We dissected and isolated the visceral endoderm in order to analyze the expression of six X-linked genes that are situated centromerically (Mecp2 and Ogt) and telomerically (Rlim, Abc7 and Jarid1c) to the Xic (Figure 4B). As expected, for all six X-linked genes, female Dnmt1omat+/+ controls exhibited expected expression from only the maternal allele. Male Dnmt1omat−/− samples yielded the same banding pattern as they only possess one maternally derived X chromosome. However, female Dnmt1omat−/− samples demonstrated biallelic expression for all the X-linked genes (Mecp2, Ogt, Tsix, Rlim, Abc7 and Jarid1c), thus showing that the lack of maternal Dnmt1o during preimplantation triggers a relaxation event in the imprinted inactivation of the paternal X chromosome.
Identification of an early hypomethylation event at Xite in female Dnmt1omat−/− blastocysts
To identify possible early initiating events that led to the abnormal expression of paternal X-linked genes in female Dnmt1omat−/− extraembryonic tissues at 9.5dpc, we examined DNA methylation in preimplantation embryos. Since evidence of Xp genic silencing was reported to occur at the 8 - to 16-cell stage [19], in the same time window as DNMT1o action, we postulated that lack of DNMT1o prompted a cascade of events that prevented complete establishment of imprinted XCI in trophectoderm cells of blastocysts (Figure 5A). Given that the interplay among Xite, Tsix and Xist is pivotal in the control of XCI, we investigated if hypomethylation of Xite could be the initiating upstream event. In order to reliably sex and perform DNA methylation analysis on X-linked genes from single embryos we examined blastocysts, a stage shortly after the time of action of DNMT1o. A region within Xite, Xite-DHS6, which exhibited hypomethylation in female Dnmt1omat−/− extraembyronic tissues (normal and abnormal morphology, 9.5 dpc) (Figure 3A, B) was selected for classical bisulfite sequencing studies. We postulated that the initial hypomethylation event on the X chromosome would occur within the Xic. In the same blastocysts in which we examined Xite methylation, we also examined the methylation of Chic1, a neighbouring gene that displayed hypomethylation in female Dnmt1omat−/− extraembryonic tissues but was located outside the Xic (Figure 3A). For Xite-DHS6, both control and Dnmt1omat−/− male blastocysts revealed very low levels of methylation (Figure 5B, C). In female control blastocysts, the Xite-DHS6 loci averaged 40.3% of DNA methylation. In contrast, female DNMT1o-deficient blastocysts had significant DNA hypomethylation (4.6%). For Chic1, male and female control (XY-3.6% and XX-3.5%), and Dnmt1omat−/− blastocysts (XY-2.3% and XX-3.6%) displayed similar DNA methylation levels (Figure 5B, C).

Next, we used RNA FISH to determine whether the hypomethylation observed for Xite-DHS6, a strong Tsix enhancer, was associated with altered Xist expression in female Dnmt1omat−/− blastocysts. First, we carried out Xist RNA FISH and subsequently DNA FISH with an X-specific centromeric probe on the same embryos to distinguish male and female embryos. As expected, control female blastocysts showed the characteristic pattern of one Xist RNA domain in all cells. Similarly, female Dnmt1omat−/− blastocysts also exhibited the same pattern in all cells (Figure 5D). In male control and Dnmt1omat−/− blastocysts no Xist RNA signals were seen (data not shown). The results provide evidence that although an important regulatory region of Tsix is hypomethylated in Dnmt1omat−/− embryos, imprinted expression of Xist was initiated and persisted until at least the blastocyst stage.
Hypomethylation of repetitive sequences in Dnmt1omat−/− extraembryonic tissues
To determine if additional genomic regions have altered DNA methylation levels in Dnmt1omat−/− female placentae, we measured the level of DNA methylation in different retrotransposon sequences. Retrotransposon elements are CpG-rich and characteristically highly methylated, which prevents their transcriptional expression [39]. Roughly half of all mouse genomic sequences are derived from historic transposition events, and the methylation of specific categories of repeated retrotransposon sequences, such as long interspersed nuclear elements (LINE1) and Alu, are often used to gauge the extent of global DNA methylation [40], [41]. Repeat elements are also known to be enriched on X chromosomal DNA compared to the autosomal DNA and are paternally imprinted at the 2-cell stage [19]. One repeat, LINE1, shows a ∼2-fold enrichment (X-linked: 26.5% vs. non-X linked: 13.4%) on the X chromosome, with the uppermost increase measured in the region including the Xic [42]. LINE1 has also been implicated in various functions during XCI [19], [42]–[45], although its methylation has not been studied in the context of XCI.
Because the Dnmt1omat−/− female placentae were hypomethylated at various X-linked loci (Figure 3A, Table 1), we tested whether Dnmt1omat−/− female placentae have a reduction in global DNA methylation. We first quantified the methylation status of LINE1, intracisternal A particle (IAP), the major (gamma) satellite repeat (GSAT), the minor satellite repeat (MSAT) and the short-interspersed element (B1-SINE) in placental and embryonic DNA using MassARRAY and Southern blotting (Figure 6A, B, Figure S4). The Dnmt1omat−/− genotype did not influence the methylation levels of repeat elements in male or female embryos, which agrees with results from our previous study [12]. However, Dnmt1omat−/− female placentae displayed a significant reduction of methylation in 4/5 repeat sequence types versus control placentae. Interestingly, the methylation of all repeats was further reduced in female versus male Dnmt1omat−/− placentae. Although repetitive element methylation was not assayed in a chromosome-specific manner using MassARRAY or Southern blotting, the total decrease observed in females versus males is greater than could be accounted for by hypomethylation on the X chromosome alone. Furthermore, RLGS studies revealed in Dnmt1omat−/− female placentae, as compared to control females, genomic sites with slight hypomethylation (<25% differences that did not meet the threshold to be counted in Figure 4A and Table 1) that map to various types of interspersed repetitive elements located on various autosomes (Figure S5). These findings reveal a genome-wide repeat methylation defect in Dnmt1omat−/− extraembryonic tissue that is accentuated in females.

Analogous to our findings in Dnmt1omat−/− extraembryonic tissue, previous work has shown that female (XX) embryonic stem cells (ESCs) derived from mouse blastocysts display global hypomethylation of repetitive sequence elements [46]. This was thought to be the result of the unique metastable condition in which both X chromosomes remained active, as XX ESCs that spontaneously become XO in culture acquired higher methylation levels similar to male (XY) ESCs. In support of this finding, we derived ESC lines from XX wild-type and Dnmt1omat−/− blastocysts and found that those ESCs retaining both X chromosomes were hypomethylated at all repeat elements tested (Figure 6C). The combination of ESC and placenta data support a model in which two active X chromosomes are associated with a low level of repeat sequence methylation.
The accentuation of global repeat methylation defect in females derived from oocytes with a maternal shortage of DNMT1o suggests an association to the X chromosome. Intriguingly, we find that repetitive element methylation in Dnmt1omat−/− placentae is negatively correlated to Xist methylation, which is most strongly demonstrated with IAP repeats (r2 = 0.42, p<0.01) (Figure 6D). A positive relationship between Xite-DHS6 methylation and B1-SINE methylation (r2 = 0.71, p<0.001) was also observed. These findings suggest a positive correlation between the degree of disruption of methylation on the X chromosome as a result of DNMT1o deficiency and the severity of subsequent defects in repeat sequence methylation.
Discussion
DNMT1o deficiency generates a unique spectrum of methylation defects
Three main defects in DNA methylation were apparent in Dnmt1omat−/− conceptuses derived from Dnmt1Δ1o/Δ1o mutant oocytes. First, we noted significant reductions in methylation of autosomal DMD sequences in imprinted genes in DNMT1o-deficient conceptuses [5], [12], [14]. Both the embryo and placenta were roughly equally affected. The marked reduction in methylation across all DMD sequences examined is due to a combination of reduced embryonic maintenance methyltransferase activity during preimplantation development and an inability to re-establish methylation de novo on DMD sequences in both embryonic and extraembryonic somatic cells. Second, DNMT1o deficiency resulted in a surprising increase in Xist methylation in female placentae. The third methylation defect was hypomethylation of genomic sequences. This hypomethylation was found in extraembryonic tissues, most notably on repeated DNA sequences throughout the genome and on single-copy X chromosome sequences of female placentae. Hypomethylation of Xite within the Xic of female embryos was traced back to preimplantation blastocyst-stage embryos.
Our previous studies have provided us with an excellent understanding for the cause of reduction in DMD methylation in Dnmt1mat−/− conceptuses. DNMT1o functions at just a single time in early embryonic development, the 8-cell preimplantation stage, and its only known function is maintenance of CpG methylation on genomic sequences. We previously showed that the prominent effect of DNMT1o deficiency was the permanent loss of methylation from one-half of the normally methylated parental alleles of imprinted DMDs. This permanent loss was apparent in both the embryo and placenta. Whether non-imprinted sequences lose methylation in the absence of 8-cell DNMT1o has not been examined. However, if so, the loss in the embryo is not permanent because non-imprinted sequences were methylated to a normal level in mid-gestation Dnmt1mat−/− embryos ([12] and this report). A return to normal levels of non-imprinted methylation is consistent with the observation that global levels of genomic methylation readily return to normal upon reintroduction of DNMT1 activity in DNMT1s-deficient ES cells [47], [48].
Source of hyper - and hypomethylated X chromosomal sequences
We can reasonably surmise from the observed biallelic expression of extraembryonic X-linked genes in DNMT1o-deficient conceptuses, including the biallelic expression of Tsix, that the paternal X chromosome in 9.5dpc placentae is the source of X-linked hypomethylation and Xist hypermethylation. Methylation of Xist sequences is a key epigenetic feature that unambiguously distinguishes the active (methylated Xist) from the inactive (unmethylated Xist) X chromosome in the mouse [49], [50]. For random XCI DNA methylation is not required for the initial repression of Xist [51]. Rather, recruitment of DNA methylation to Xist sequences is secondary to Tsix-mediated recruitment of chromatin modifying enzymes to the Xist promoter [17], [52], [53]. It has been shown that Tsix-mediated Xist repression and promoter methylation operate in extraembryonic cells of the mouse, where XCI is imprinted, or paternal-specific [25], [54]. Based on the strong inverse correlation between Xist expression and its methylation, it seems paradoxical that the absence of 8-cell DNMT1o leads to Xist methylation on the paternal X chromosome in extraembryonic cells. DNMT1o must therefore act in an indirect way to maintain marks that reinforce imprinted XCI. A possible mode of action is via effects on Tsix expression, which is essential for preventing inactivation of the maternal X chromosome in extraembryonic cells with imprinted XCI [25], [54]. The current understanding of XCI is that Tsix is controlled by Xite, a region that specifically enhances Tsix expression [21]–[24]. The Xite locus contains three CGIs showing low levels of methylation in oocytes, associated with a single active X, and hypermethylation in sperm, consistent with an inactive paternal X [21]. One of the three CGIs overlaps a DNAse hypersensitive site (Xite-DHS6) and the minor Tsix promoters. In female ES cells, this region is found undermethylated on both parental alleles, prior to the onset of XCI, however upon cellular differentiation and initiation of XCI, Xite-DHS6 becomes hypermethylated on both the maternal and paternal alleles [21]. Considering these and other data, it was suggested that Xite might be a candidate for a gametic XCI imprint [21]. Furthermore, Xite enhancing action seems influential predominantly at the onset of XCI, since its deletion results in a deficiency of Tsix expression at, but not prior to, the onset of random XCI [23].
Based on the aforementioned published models of Xite function, we can now understand how DNMT1o and Xite converge to regulate imprinted XCI. In our DNMT1o deficiency model, we showed that Xite-DHS6 was hypomethylated in XX blastocysts (∼40% vs ∼5%); these results indicate that regions of Xite are targeted by DNMT1o for methylation maintenance at the 8-cell stage, the same stage at which Xp genic silencing is initiated. It is expected that loss of Xite methylation will be associated with an increase of Xite activity, which would lead to Tsix expression on the paternal X chromosome; these events would then instigate a process leading to Xist repression on the paternal X chromosome (Figure 7 for model). This process does not appear to be immediate, as shown by our RNA FISH results and the persistence of Xist RNA expression in Dnmt1omat−/− trophectoderm cells of blastocysts. The delay in this proposed Tsix-induced Xist repression may be due to a developmental program of epigenetic changes thought to occur on Xist sequences, culminating in methylation and silencing of its promoter. These last two events need not be coincidental. In this regard, the rapid repression of Xist by forced Tsix expression on the inactive paternal X chromosome in an inducible transgenic model of Tsix expression may be due to supranormal levels of Tsix [25]. Nevertheless, even this rapidly induced Xist repression is readily reversed, as evidenced by reappearance of Xist transcripts when paternal X chromosome Tsix expression is turned off. Our results are reminiscent of and may be analogous to those we reported previously for imprinted genes in the Dnmt1omat−/− model [5]. While imprinted sequences were hypomethylated in blastocysts, expression remained monoallelic (imprinted) with biallelic expression only seen later in development in 7.5dpc embryos.

A specific effect of DNMT1o methylation on extraembryonic methylation in females might also explain the opposite dependence of DNA methylation on imprinted XCI observed by Sado et al. (2000). They observed that random XCI was abnormal but imprinted XCI was normal in the absence of zygotically produced DNMT1 protein [55]. Our results suggest that such preservation of imprinted XCI in the absence of zygotic DNMT1 could be due to activity of maternally derived stocks of DNMT1 and DNMT1o protein [6] in homozygous Dnmt1−/− mutant embryos derived from heterozygous Dnmt1+/− parents.
We must also consider another connection between genome-wide hypomethylation of repeated sequences and loss of extraembryonic imprinted (paternal-specific) XCI in Dnmt1omat−/− embryos. It has been proposed that repeated sequences are premarked in male germ cells, which would then be recognized after fertilization to construct a more definitive, permanent paternal-specific mark in extraembryonic cell types [19]. Loss of DNMT1o may directly (if the premark is DNA methylation) or indirectly influence the stability or inheritance of this premark in mid-preimplantation. In one specific model we can propose based on our data, reduction in repeat methylation due to the absence of DNMT1o occurs across the genome (X chromosome and autosomes) and remains reduced upon further development in both male and female extraembryonic cell types, while recovering to normal levels in both male and female embryonic cells. Hypomethylated female XX ES cells, particularly those from DNMT1o-deficient embryos, may model early female Dnmt1omat−/− embryonic cells, prior to recovery of genomic methylation and the presumed onset of random XCI. Extraembryonic tissues from Dnmt1omat−/− conceptuses and XX ESCs represent two situations where two active X chromosomes are maintained for an unnaturally prolonged period. Both of these conditions result in a global reduction of repeat methylation, and our data also suggests the possibility of a functional relationship between the Xic and regulation of global repeat methylation (further discussed in Supporting Text S2).
Morphological and functional integrity of female and male placentae compromised by DNMT1o deficiency
How can we account for the histomorphological abnormalities in Dnmt1omat−/− placentae, including the striking differences between female and male E9.5 Dnmt1omat−/− placentae? We can assume that these structural abnormalities are due primarily to methylation abnormalities and their collateral effects, which are most likely transcriptional defects occurring in cells of the developing placentae. Two epigenetic abnormalities were described in Dnmt1omat−/− placentae, defects in imprinted DMD methylation, and hypomethylation of repeated sequences and X-linked sequences. DMD methylation abnormalities occurred to the same extent in male and female Dnmt1omat−/− E9.5 placentae, and we propose that the histomorphological abnormalities in male Dnmt1omat−/− placentae were due entirely or nearly entirely (because there was hypomethylation of repeated sequence, presumably autosomal, in male Dnmt1omat−/− placentae) to such DMD methylation abnormalities. It is known that disruptions in the integrity of the strict parent-specific methylation of imprinted DMD sequences can lead to dysregulation of imprinted gene expression, including dysregulation of placentally expressed imprinted genes. Disruption in the monoallelic imprinted expression of individual placentally expressed genes can result in profound histomorphological abnormalities [20] and therefore it is likely that dysregulation of imprinted gene expression alone in Dnmt1omat−/− placentae could account for all of the male Dnmt1omat−/− morphological defects.
We further propose that hypomethylation of X chromosome sequences in female Dnmt1omat−/− placentae accounts for the histomorphological abnormalites unique to the female Dnmt1omat−/− placentae. How might this X chromosome hypomethylation lead to gross and microscopic morphological abnormalities? A likely scenario is that this hypomethylation initially occurred at the time of, or soon after, the loss of DNMT1o in preimplantation development, and that it persisted in extraembryonic cell types (see above). The hypomethylation, via an unknown mechanism, would lead to dysregulation in the expression of X-linked genes and associated female-specific extraembryonic morphological abnormalities. Failure in imprinted XCI via the deregulation of Xist is known to lead to morphological aberrations. Female conceptuses that contain an Xist deletion by paternal transmission have severe growth retardation and die in early embryogenesis [56]. Some Xist−/− conceptuses that fail to undergo gastrulation show elongated ectoplacental cones and expanded yolk sacs. Authors stipulate that the instability caused by the two active X chromosomes is responsible for the imprinted lethal phenotype observed in the mutant Xist females.
Since in the Dnmt1omat−/− females, only 50% of the placental cells are affected, one might ask why loss of imprinted XCI in 50% of the cells is not compensated by the cells presumably retaining imprinted XCI. Evidence from a recent study suggests that compensation does occur. Thus while there were clear female-specific placental abnormalities at 9.5dpc (this study), by 17.5 dpc there were no differences in the embryo-to-placenta weight ratios or placental phenotypes between Dnmt1omat−/− males and females [26]. The exact nature of this compensation is unknown, but we can speculate that it is a combination of viability of 50% of cells retaining normal imprinted X-inactivation, death of many cells not expressing Xist and compensatory random X-inactivation that rescues a fraction of the Xist non-expressors.
In conclusion, the work presented here furthers our understanding on the maintenance of methylation patterns during early embryo development. This study is the first to provide evidence of additional roles for DNMT1o beyond the maintenance of genomic imprinting in preimplantation embryos, including an unexpected role in imprinted XCI. Although further investigation is needed to comprehend how DNA methylation regulates Xite and the mechanisms behind the relaxation of imprinted XCI, these findings provide novel mechanistic concepts into the counting mechanism.
Materials and Methods
Details of experimental procedures are provided in the Supporting Information section (Supporting Text S3).
Ethics statement
All animal procedures were carried out in accordance with the Canadian Council of Animal Care.
Assessment of extraembryonic hyperplasia and histological analysis
Embryos and placentae of different genotypes (Dnmt1omat+/+, Dnmt1omat+/−, Dnmt1omat−/−) were collected from the uteri at 9.5dpc and extraembryonic hyperplasia was assessed by microscopy. For the histological analysis, fixed 9.5dpc implantation sites were sectioned and stained with hematoxylin/eosin (H&E).
DNA methylation analyses
Quantitative DNA methylation analyses were accomplished using isolated and bisulfite treated DNA from 9.5dpc Dnmt1omat+/+ and Dnmt1omat−/− embryos and placentae. Candidate regions of imprinted DMDs, X chromosome-CGI and repeat elements were amplified (primers see Table S7) and the quantitative DNA methylation state was analyzed using matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry [57].
Sanger bisulfite sequencing was used to analyzed the methylation status in single male and female control (Dnmt1omat+/+) and mutant (Dnmt1omat−/−) 3.5dpc blastocysts. Isolated DNA was modified by sodium bisulfite, and Xite-DHS6 and Chic1 were amplified by PCR (primers see Table S7), subcloned and sequenced.
Restriction landmark genomic scanning (RLGS) was used to assess genome-wide methylation in 9.5dpc control (Dnmt1omat+/−) and Dnmt1omat−/− placentae [33]. Sufficient DNA was obtained from single control and Dnmt1omat−/− 9.5dpc placentae, however in the case of smaller samples, pools of similar placentae (same sex and similar morphology) were used to obtain sufficient tissue for DNA extraction and RLGS studies (samples XY P4 and XX P4).
Southern blots on 9.5dpc Dnmt1omat+/+ and Dnmt1omat−/− placentae were performed as described [58] and visualized by autoradiography. Minor satellite probes were constructed by PCR amplification of mouse genomic DNA [59] as previously described [60], [61]. DNA was digested with either MspI or its methylation-sensitive isoschizomer HpaII. Membranes were stripped and reprobed according to the manufacturer's recommended conditions (Hybond; GE Healthcare/Amersham).
RNA and DNA FISH
RNA and DNA FISH were performed, as previously described [62], on Dnmt1omat+/+ and Dnmt1omat−/− preimplantation embryos using an Xist probe (pEFB, [63]) and a X chromosome probe (DXwas70, [64]).
Allele specific expression assay by RT-PCR
RNA was extracted from the visceral endoderm layers of Dnmt1omat+/+ and Dnmt1omat−/− yolk sacs from 9.5dpc conceptuses. Following reverse transcription, X chromosome linked genes containing polymorphisms were amplified with the use of flanking primers (Table S7), and the PCR products were digested and separated on acrylamide gels.
Statistical analysis
Comparisons in the extraembryonic hyperplasia and histological studies were first subjected to Levene's test for homogeneity of variance. One-way ANOVA was used for equal variance groups, while the non-parametric test Kruskal-Wallis was applied to unequal variance groups. Comparisons in methylation of imprinted DMDs and repeat elements were made using the Holm-Sidak method for multiple pair-wise comparisons, whereas analysis of X-CGI methylation was accomplished using an unprotected t test of the difference between means.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Bourc'hisD, XuGL, LinCS, BollmanB, BestorTH (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294 : 2536–2539.
2. HataK, OkanoM, LeiH, LiE (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129 : 1983–1993.
3. KanedaM, OkanoM, HataK, SadoT, TsujimotoN, et al. (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429 : 900–903.
4. ReikW, DeanW, WalterJ (2001) Epigenetic reprogramming in mammalian development. Science 293 : 1089–1093.
5. CirioMC, MartelJ, MannM, ToppingsM, BartolomeiM, et al. (2008) DNA methyltransferase 1o functions during preimplantation development to preclude a profound level of epigenetic variation. Dev Biol 324 : 139–150.
6. CirioMC, RatnamS, DingF, ReinhartB, NavaraC, et al. (2008) Preimplantation expression of the somatic form of Dnmt1 suggests a role in the inheritance of genomic imprints. BMC Dev Biol 8 : 9.
7. KuriharaY, KawamuraY, UchijimaY, AmamoT, KobayashiH, et al. (2008) Maintenance of genomic methylation patterns during preimplantation development requires the somatic form of DNA methyltransferase 1. Dev Biol 313 : 335–346.
8. RatnamS, MertineitC, DingF, HowellCY, ClarkeHJ, et al. (2002) Dynamics of Dnmt1 methyltransferase expression and intracellular localization during oogenesis and preimplantation development. Dev Biol 245 : 304–314.
9. CardosoMC, LeonhardtH (1999) DNA methyltransferase is actively retained in the cytoplasm during early development. J Cell Biol 147 : 25–32.
10. CarlsonLL, PageAW, BestorTH (1992) Properties and localization of DNA methyltransferase in preimplantation mouse embryos: implications for genomic imprinting. Genes Dev 6 : 2536–2541.
11. DohertyAS, BartolomeiMS, SchultzRM (2002) Regulation of stage-specific nuclear translocation of Dnmt1o during preimplantation mouse development. Dev Biol 242 : 255–266.
12. HowellCY, BestorTH, DingF, LathamKE, MertineitC, et al. (2001) Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104 : 829–838.
13. MertineitC, YoderJA, TaketoT, LairdDW, TraslerJM, et al. (1998) Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development 125 : 889–897.
14. ToppingsM, CastroC, MillsPH, ReinhartB, SchattenG, et al. (2008) Profound phenotypic variation among mice deficient in the maintenance of genomic imprints. Hum Reprod 23 : 807–818.
15. HuynhKD, LeeJT (2003) Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 426 : 857–862.
16. MakW, NesterovaTB, de NapolesM, AppanahR, YamanakaS, et al. (2004) Reactivation of the paternal X chromosome in early mouse embryos. Science 303 : 666–669.
17. OkamotoI, OtteAP, AllisCD, ReinbergD, HeardE (2004) Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303 : 644–649.
18. SilvaJ, MakW, ZvetkovaI, AppanahR, NesterovaTB, et al. (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4 : 481–495.
19. NamekawaSH, PayerB, HuynhKD, JaenischR, LeeJT (2010) Two-step imprinted X inactivation: repeat versus genic silencing in the mouse. Mol Cell Biol 30 : 3187–3205.
20. HembergerM (2002) The role of the X chromosome in mammalian extra embryonic development. Cytogenet Genome Res 99 : 210–217.
21. BoumilRM, OgawaY, SunBK, HuynhKD, LeeJT (2006) Differential methylation of Xite and CTCF sites in Tsix mirrors the pattern of X-inactivation choice in mice. Mol Cell Biol 26 : 2109–2117.
22. LeeJT (2005) Regulation of X-chromosome counting by Tsix and Xite sequences. Science 309 : 768–771.
23. OgawaY, LeeJT (2003) Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell 11 : 731–743.
24. StavropoulosN, RowntreeRK, LeeJT (2005) Identification of developmentally specific enhancers for Tsix in the regulation of X chromosome inactivation. Mol Cell Biol 25 : 2757–2769.
25. OhhataT, SennerCE, HembergerM, WutzA (2011) Lineage-specific function of the noncoding Tsix RNA for Xist repression and Xi reactivation in mice. Genes Dev 25 : 1702–1715.
26. HimesKP, KoppesE, ChailletJR (2013) Generalized disruption of inherited genomic imprints leads to wide-ranging placental defects and dysregulated fetal growth. Dev Biol 373 : 72–82.
27. FowdenAL, CoanPM, AngioliniE, BurtonGJ, ConstanciaM (2011) Imprinted genes and the epigenetic regulation of placental phenotype. Prog Biophys Mol Biol 106 : 281–288.
28. HuynhKD, LeeJT (2005) X-chromosome inactivation: a hypothesis linking ontogeny and phylogeny. Nat Rev Genet 6 : 410–418.
29. LeeJT (2003) Molecular links between X-inactivation and autosomal imprinting: X-inactivation as a driving force for the evolution of imprinting? Curr Biol 13: R242–254.
30. ReikW, LewisA (2005) Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet 6 : 403–410.
31. WagschalA, FeilR (2006) Genomic imprinting in the placenta. Cytogenet Genome Res 113 : 90–98.
32. CohenDE, DavidowLS, ErwinJA, XuN, WarshawskyD, et al. (2007) The DXPas34 repeat regulates random and imprinted X inactivation. Dev Cell 12 : 57–71.
33. OkazakiY, OkuizumiH, SasakiN, OhsumiT, KuromitsuJ, et al. (1995) An expanded system of restriction landmark genomic scanning (RLGS Ver. 1.8). Electrophoresis 16 : 197–202.
34. SmiragliaDJ, Kazhiyur-MannarR, OakesCC, WuYZ, LiangP, et al. (2007) Restriction landmark genomic scanning (RLGS) spot identification by second generation virtual RLGS in multiple genomes with multiple enzyme combinations. BMC Genomics 8 : 446.
35. YuL, LiuC, BennettK, WuYZ, DaiZ, et al. (2004) A NotI-EcoRV promoter library for studies of genetic and epigenetic alterations in mouse models of human malignancies. Genomics 84 : 647–660.
36. HatadaI, SugamaT, MukaiT (1993) A new imprinted gene cloned by a methylation-sensitive genome scanning method. Nucleic Acids Res 21 : 5577–5582.
37. TakagiN, SasakiM (1975) Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 256 : 640–642.
38. WestJD, PapaioannouVE, FrelsWI, ChapmanVM (1978) Preferential expression of the maternally derived X chromosome in extraembryonic tissues of the mouse. Basic Life Sci 12 : 361–377.
39. SchulzWA, SteinhoffC, FlorlAR (2006) Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol 310 : 211–250.
40. BestorTH, TyckoB (1996) Creation of genomic methylation patterns. Nat Genet 12 : 363–367.
41. KloseRJ, BirdAP (2006) Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31 : 89–97.
42. BaileyJA, CarrelL, ChakravartiA, EichlerEE (2000) Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci U S A 97 : 6634–6639.
43. ChaumeilJ, Le BacconP, WutzA, HeardE (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20 : 2223–2237.
44. ChowJC, CiaudoC, FazzariMJ, MiseN, ServantN, et al. (2010) LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141 : 956–969.
45. LyonMF (1998) X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet 80 : 133–137.
46. ZvetkovaI, ApedaileA, RamsahoyeB, MermoudJE, CromptonLA, et al. (2005) Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet 37 : 1274–1279.
47. BorowczykE, MohanKN, D'AiutoL, CirioMC, ChailletJR (2009) Identification of a region of the DNMT1 methyltransferase that regulates the maintenance of genomic imprints. Proc Natl Acad Sci U S A 106 : 20806–20811.
48. TuckerKL, TalbotD, LeeMA, LeonhardtH, JaenischR (1996) Complementation of methylation deficiency in embryonic stem cells by a DNA methyltransferase minigene. Proc Natl Acad Sci U S A 93 : 12920–12925.
49. BarrH, HermannA, BergerJ, TsaiHH, AdieK, et al. (2007) Mbd2 contributes to DNA methylation-directed repression of the Xist gene. Mol Cell Biol 27 : 3750–3757.
50. BeardC, LiE, JaenischR (1995) Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev 9 : 2325–2334.
51. SadoT, OkanoM, LiE, SasakiH (2004) De novo DNA methylation is dispensable for the initiation and propagation of X chromosome inactivation. Development 131 : 975–982.
52. OhhataT, HokiY, SasakiH, SadoT (2008) Crucial role of antisense transcription across the Xist promoter in Tsix-mediated Xist chromatin modification. Development 135 : 227–235.
53. SunBK, DeatonAM, LeeJT (2006) A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell 21 : 617–628.
54. SadoT, WangZ, SasakiH, LiE (2001) Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 128 : 1275–1286.
55. SadoT, FennerMH, TanSS, TamP, ShiodaT, et al. (2000) X inactivation in the mouse embryo deficient for Dnmt1: distinct effect of hypomethylation on imprinted and random X inactivation. Dev Biol 225 : 294–303.
56. MarahrensY, PanningB, DausmanJ, StraussW, JaenischR (1997) Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 11 : 156–166.
57. EhrichM, NelsonMR, StanssensP, ZabeauM, LiloglouT, et al. (2005) Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A 102 : 15785–15790.
58. TraslerJM, HakeLE, JohnsonPA, AlcivarAA, MilletteCF, et al. (1990) DNA methylation and demethylation events during meiotic prophase in the mouse testis. Mol Cell Biol 10 : 1828–1834.
59. LehnertzB, UedaY, DerijckAA, BraunschweigU, Perez-BurgosL, et al. (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13 : 1192–1200.
60. MichaudEJ, van VugtMJ, BultmanSJ, SweetHO, DavissonMT, et al. (1994) Differential expression of a new dominant agouti allele (Aiapy) is correlated with methylation state and is influenced by parental lineage. Genes Dev 8 : 1463–1472.
61. WalshCP, ChailletJR, BestorTH (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20 : 116–117.
62. HeardE, MongelardF, ArnaudD, AvnerP (1999) Xist yeast artificial chromosome transgenes function as X-inactivation centers only in multicopy arrays and not as single copies. Mol Cell Biol 19 : 3156–3166.
63. MakW, BaxterJ, SilvaJ, NewallAE, OtteAP, et al. (2002) Mitotically stable association of polycomb group proteins eed and enx1 with the inactive x chromosome in trophoblast stem cells. Curr Biol 12 : 1016–1020.
64. DistecheCM, TantravahiU, GandyS, EisenhardM, AdlerD, et al. (1985) Isolation and characterization of two repetitive DNA fragments located near the centromere of the mouse X chromosome. Cytogenet Cell Genet 39 : 262–268.
65. SadoT, Ferguson-SmithAC (2005) Imprinted X inactivation and reprogramming in the preimplantation mouse embryo. Hum Mol Genet 14 (Spec No 1) R59–64.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 11
Nejčtenější v tomto čísle
- and Are Required for Growth under Iron-Limiting Conditions
- Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland cAMP/PKA Signaling Defects in the Neurofibromatosis-1 Growth Deficiency
- The Light Skin Allele of in South Asians and Europeans Shares Identity by Descent
- RNA∶DNA Hybrids Initiate Quasi-Palindrome-Associated Mutations in Highly Transcribed Yeast DNA