
Ribosome Synthesis and MAPK Activity Modulate Ionizing Radiation-Induced Germ Cell Apoptosis in
Synthesis of ribosomal RNA by RNA polymerase I (RNA pol I) is an elemental biological process and is key for cellular homeostasis. In a forward genetic screen in C. elegans designed to identify DNA damage-response factors, we isolated a point mutation of RNA pol I, rpoa-2(op259), that leads to altered rRNA synthesis and a concomitant resistance to ionizing radiation (IR)-induced germ cell apoptosis. This weak apoptotic IR response could be phenocopied when interfering with other factors of ribosome synthesis. Surprisingly, despite their resistance to DNA damage, rpoa-2(op259) mutants present a normal CEP-1/p53 response to IR and increased basal CEP-1 activity under normal growth conditions. In parallel, rpoa-2(op259) leads to reduced Ras/MAPK pathway activity, which is required for germ cell progression and physiological germ cell death. Ras/MAPK gain-of-function conditions could rescue the IR response defect in rpoa-2(op259), pointing to a function for Ras/MAPK in modulating DNA damage-induced apoptosis downstream of CEP-1. Our data demonstrate that a single point mutation in an RNA pol I subunit can interfere with multiple key signalling pathways. Ribosome synthesis and growth-factor signalling are perturbed in many cancer cells; such an interplay between basic cellular processes and signalling might be critical for how tumours evolve or respond to treatment.
Published in the journal:
. PLoS Genet 9(11): e32767. doi:10.1371/journal.pgen.1003943
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003943
Summary
Synthesis of ribosomal RNA by RNA polymerase I (RNA pol I) is an elemental biological process and is key for cellular homeostasis. In a forward genetic screen in C. elegans designed to identify DNA damage-response factors, we isolated a point mutation of RNA pol I, rpoa-2(op259), that leads to altered rRNA synthesis and a concomitant resistance to ionizing radiation (IR)-induced germ cell apoptosis. This weak apoptotic IR response could be phenocopied when interfering with other factors of ribosome synthesis. Surprisingly, despite their resistance to DNA damage, rpoa-2(op259) mutants present a normal CEP-1/p53 response to IR and increased basal CEP-1 activity under normal growth conditions. In parallel, rpoa-2(op259) leads to reduced Ras/MAPK pathway activity, which is required for germ cell progression and physiological germ cell death. Ras/MAPK gain-of-function conditions could rescue the IR response defect in rpoa-2(op259), pointing to a function for Ras/MAPK in modulating DNA damage-induced apoptosis downstream of CEP-1. Our data demonstrate that a single point mutation in an RNA pol I subunit can interfere with multiple key signalling pathways. Ribosome synthesis and growth-factor signalling are perturbed in many cancer cells; such an interplay between basic cellular processes and signalling might be critical for how tumours evolve or respond to treatment.
Introduction
Multicellular organisms use genetically determined cell death mechanisms – most prominently apoptosis – to ensure the timely and innocuous removal of superfluous, damaged, or potentially harmful cells. Apoptosis is tightly regulated and an integral part of the delicate balance between cell proliferation and cell loss, which is essential for the formation and maintenance of tissues and organs. Disturbances leading to excessive or diminished apoptosis contribute to devastating human diseases with increasing prevalence worldwide, such as neurodegeneration, immunological disorders, and cancer.
The integrity of our genome is continuously challenged by endogenous processes (replication errors, reactive oxygen species, by-products of cellular metabolism) and by exogenous genotoxic agents – naturally occurring or iatrogenic [e.g., 1]. Depending on the cell type, cell cycle stage, metabolic state and probably also tissue context, excessive DNA damage can be a strong stimulus for cells to undergo apoptosis. Detailed knowledge of the DNA damage response network and its failures is required for our understanding of tumor formation and progression; and also for effective and safe tumor treatment, as many of the current treatments produce DNA damage to induce replicative arrest or death of rapidly proliferating tumor cells.
Nucleoli have been increasingly acknowledged as pivot systems for homeostatic regulation and stress responses [e.g.], [ 2]–[4]. Hypertrophic and irregularly shaped nucleoli were reported to be characteristic of malignant cells already at the end of the 19th century, and have gained a high prognostic value for several human neoplasias [5]. However, large nucleoli and increased ribosome biogenesis are common signatures of proliferating cells given the increased demand for protein synthesis. It has therefore been challenging to explore whether nucleolar hypertrophy in cancer cells is a mere expression of rapid proliferation, or whether the enlarged nucleoli could play a causative role in tumor development [6]. Synthesis of ribosomal RNA accounts for up to 75% of total transcriptional activity in yeast [7] and at least 50% of the synthetic effort of rapidly proliferating eukaryotic cells are expended on ribosome production [8]. Transcription of rRNA by RNA polymerase I (RNA pol I) is the rate-limiting step for ribosome synthesis [e.g., 9] and a major target of many signalling pathways of cellular growth and proliferation, e.g., Ras/ERK [10], Myc [e.g., 11], mTOR [12], pRb [13], and p53 [14]. In turn, physiological or pathological changes in rRNA transcription and processing, or in nucleolar integrity affect cellular fate decisions through altered translation, but also through more direct regulatory signalling [6], [15], [16]. Various disturbances in early steps of rRNA synthesis have strong pro-apoptotic effects [17]–[19]. Somewhat contrasting, partial depletion of ribosomal proteins in zebrafish proved to be cancerogenic [20]. Also in humans, tumor predisposing diseases could be linked to mutations in rRNA processing and ribosome assembly factors, such as Dyskeratosis congenita (to pseudouridine synthase DKC1) or Diamond-Blackfan anaemia (to various ribosomal proteins) [e.g.], [ 16,21].
The germ line of Caenorhabditis elegans has proven a versatile model to dissect the classical DNA damage responses [22] in the context of an optically transparent, living organism [23]. Proliferation arrest of mitotic germ stem cells and apoptosis of meiotic germ cells occur in two spatially separate areas and can be morphologically followed by Nomarski differential interference contrast (DIC) microscopy. Under standard laboratory conditions, germ cells at the exit from late meiotic pachytene stage alternatively progress into large oocytes or undergo apoptosis [24]. This developmental, ‘physiological’ apoptosis affects an estimated half of all oocyte precursors in an apparently stochastic manner, possibly to supply the rapidly growing oocytes with cytoplasmic constituents [reviewed in [25]]. Two regulatory pathways of global importance for cellular growth and proliferation have been associated with physiological cell death: the Rb complex [26], [27] and Ras/MAP kinase signalling [28], [29]. In contrast to physiological apoptosis, DNA damage-induced apoptosis depends on CEP-1 (a functional C. elegans homolog of mammalian p53 [30], [31]), which transcriptionally upregulates the two pro-apoptotic BH3-only proteins EGL-1 and CED-13 [32]. In the classical model (schematic in Fig. S13A), EGL-1 activates the core apoptotic machinery by binding to CED-9/Bcl-2, therewith releasing CED-4/Apaf-1 [33], which in turn oligomerises and serves as a platform for activation of the effector caspase CED-3 [reviewed in [34]].
Many of the factors involved in the maintenance of genome integrity are evolutionarily conserved in C. elegans. In this work, we characterise the mutation rpoa-2(op259) – isolated in an unbiased forward genetic screen – which affects the second largest subunit of RNA polymerase I (RNA pol I) and thus an essential and highly conserved gene. Taking advantage of this viable allele, we identified causal links between ribosome synthesis and cellular checkpoints in the context of a whole organism. We show that rpoa-2(op259) leads to stimulation of stress signalling and notably to chronic activation of the pro-apoptotic CEP-1/p53 pathway. It simultaneously interferes with Ras/MAPK signalling, possibly by increasing LIP-1 phosphatase activity, and thereby modulates apoptosis downstream of CEP-1, likely at the level of CED-9. Only the combination of these two effects explains the apoptotic phenotype of the RNA pol I mutant under normal conditions and in response to irradiation.
Results
Genetic screen for DNA damage response genes yields an RNA polymerase I subunit
We searched for additional regulators of cellular DNA damage responses with a forward genetic (EMS mutagenesis) screen (see Methods). Of 2'000 haploid genomes tested, one candidate mutation, op259, led to reduced apoptosis of meiotic germ cells following ionising radiation (IR) (Fig. 1A). In op259, the basal levels of germ cell corpses were lower than wild type, and IR-induced apoptosis was strongly reduced (Fig. 1B). op259 mutants also showed a reduced apoptotic response to ultraviolet light (UV-C) (Fig. S1A), speaking for a more global impairment of damage-induced apoptosis.

Genetic mapping and molecular characterisation of op259 mutants (see Methods) led to the identification of a C→T transition in the second exon of F14B4.3 (Fig. S2A), resulting in a Proline to Serine change (P70S). F14B4.3 codes for the second-largest subunit of eukaryotic RNA polymerase I; we hence named the gene rpoa-2.
Several lines of evidence suggest that this missense mutation is the cause of the apoptotic defect in op259 mutants. First, we tried to phenocopy rpoa-2(op259) by knockdown of F14B4.3 using RNAi by feeding [35], [36]. Whereas worms grown on control RNAi conditions showed a strong increase of the cell corpse number upon irradiation, F14B4.3(RNAi)-treated animals had only few germ cell corpses (Fig. S1C). Second, transgenic expression of wild-type RPOA-2 could mostly rescue the apoptotic phenotype of rpoa-2(op259) (Fig. S1B). Third, rpoa-2(op259) failed to complement the independent deletion allele rpoa-2(ok1970) available from a public source [37]. Because this allele is homozygous lethal, we generated transheterozygous rpoa-2(ok1970/op259) animals by crossing rpoa-2(op259) males with rpoa-2(ok1970)/hT2 hermaphrodites. rpoa-2(ok1970/op259) were viable and shared the apoptotic phenotype of rpoa-2(op259) homozygotes (Text S1 and Fig. S1E). Taken together, our observations confirm that rpoa-2(op259) is the cause of the observed apoptotic defects.
The core subunits of the DNA-dependent RNA polymerases (Table S1) are evolutionarily highly conserved [38], particularly the catalytic (largest and second-largest) subunits [39]. Alignment of the RPOA-2 protein sequence with its homologs reveals that the P70S mutation affects a highly conserved residue at the beginning of a well-conserved stretch of amino acids both when comparing diverse orthologous eukaryotic RNA pol I β-subunits (Fig. S2B) as well as the three paralogous C. elegans β-subunits of RNA pol I, II, and III (Fig. S2C). In the crystal structure of the RNA pol II core complex from yeast ([40], PDB entry 1I50), the proline corresponding to the residue mutated in RPOA-2 maps to a region that lies distant to the catalytic centre of the polymerase, towards an outer surface of the core complex (not shown), suggesting that the synthesis of RNA is preserved and possibly explaining why the op259 mutants, unlike the ok1970 null animals, are viable.
Consistent with the expected function of RPOA-2, we found that a YFP::RPOA-2 transgene under the control of its endogenous promoter is ubiquitously expressed, and localises mainly to the nucleolus (Fig. S3 and Fig. S8B). To test for potential effects of the op259 mutation on RPOA-2 expression and localisation, we compared YFP-tagged mutant and wild-type protein. In spite of variable expression levels between different transgenic strains, YFP::RPOA-2(P70S) consistently showed stronger relative cytoplasmic fluorescence than YFP::RPOA-2(wt) (Fig. S3).
In conclusion, we found that op259 is a viable hypomorphic mutation in the essential rpoa-2 gene and thus offers a valuable means to investigate disturbances in the process of ribosome synthesis in a living system.
Cell cycle arrest and DNA repair are normal in rpoa-2(op259) mutants
Many C. elegans DNA damage response mutants show not only defective apoptosis but also defects in DNA repair and cell cycle arrest of proliferating germ cells [23], [41]. In wild-type worms, DNA damage induces cell cycle arrest of mitotic germ cells until the damage has been repaired [42]. Nuclear growth and cytoplasmic expansion meanwhile persist, resulting in a visible enlargement of the germ cell nuclei [43]. Chromatin staining of isolated gonads from control animals showed a uniform pool of nuclei in the mitotic compartment, which was interspersed with larger nuclei shortly after irradiation; at 6 hours, almost all nuclei were considerably enlarged (Fig. 1C). The nuclei in rpoa-2(op259) gonads showed a similar response (while the nucleoli grew only little), confirming that germ cells in the mutant do arrest following IR.
To assess DNA repair in rpoa-2(op259) germ lines, we used the fluorescent reporter RAD-54::YFP expressed from the transgene opIs257 [44]. RAD-54 is involved in homologous recombination at sites of DNA double strand breaks (DSB). In addition to its role in meiotic recombination, RAD-54 is also recruited to repair foci when DNA has been damaged exogenously [44], [45]. We found that meiotic cells in rpoa-2(op259) animals had similar numbers of RAD-54::YFP foci both under control conditions (0.3±0.1 vs. 0.4±0.1 (95% CI) foci per nucleus), and following IR (3.7±0.3 vs. 3.4±0.4 foci at 3 hours). In the mitotic region, rpoa-2(op259) and wild-type worms alike had very few foci under control conditions; and upon irradiation, the number strongly increased in both strains (Fig. 1D). Twenty-four hours after IR, we noticed a strong association of RAD-54::YFP foci with enlarged cells (Fig. 1D), i.e., cells arrested in the cell cycle, likely due to unrepaired DNA damage. The numbers of RAD-54::YFP foci per large mitotic cell were 6.4±1.8 (95% CI) for wild-type and 6.7±1.0 for rpoa-2(op259) gonads. Taken together, our observations indicate that rpoa-2(op259) mutants have no gross defect in cell cycle arrest response or DSB-repair.
Abnormal germ cell proliferation and differentiation at sensitive conditions
rpoa-2(op259) mutants develop and reproduce less rapidly than wild-type worms. A full generation cycle at 20°C took at least 24 hours longer in rpoa-2(op259) than in the wild type (Fig. S4A). Most significantly delayed was the progression from the L4 stage to the appearance of the first eggs on the plate. Further, the egg-laying rate of rpoa-2(op259) animals in young adulthood was reduced in comparison to wild type (Fig. S4B); however, the total number of progeny laid per animal was less divergent (179±32 vs. 200±38, n = 6). rpoa-2(op259) mutants had no clear signs of oocyte retention or stacking of embryos that would explain a lower output; we thus considered that germ cell proliferation or maturation might be slowed down and assessed germ line dynamics in early adulthood (Fig. S5A and Text S1). The total number of germ cells indeed increased at a slower rate in rpoa-2(op259) mutants (Fig. S5B). However, in contrast to many mutants of germ line proliferation characterised so far [e.g.], [ 46], [47, chapters in 48], the numerical proportions of the distinct germ cell stages – mitotic, transition or mid-late pachytene cells – did not significantly deviate from the wild-type pattern; at 24 hours after onset of egg laying, wild type and rpoa-2(op259) gonads showed very similar germ cell populations (174 vs. 201 mitotic, 516 vs. 511 meiotic pachytene cells) (Fig. S5C). Thus, rpoa-2(op259) mutants have a reduced germ cell production rate, not so much due to an altered mitotic cell pool but most likely due to slower cell cycles. A lower rate of cells exiting late meiotic pachytene might thus contribute to the lower number of apoptotic cells in rpoa-2(op259) mutants. However, reduced proliferation alone is unlikely to explain the lower apoptosis rate and weak IR-response in rpoa-2(op259) gonads (see further results).
rpoa-2(op259) mutants were smaller than wild type at the L4 stage. The mutants however caught up during adulthood and exceeded the wild-type length by 10% at the third day of adulthood. Older animals also contained significantly more intestinal and interstitial lipids, indicative of metabolic alterations. Knockdown of certain translation initiation factors had been shown to considerably prolong adult lifespan in C. elegans [e.g.], [ 49,50]. In contrast, rpoa-2(op259) animals had a slightly shortened median survival (Fig. S4C).
Whereas rpoa-2(op259) worms were fertile at 15°C or 20°C, animals raised at 25°C were sterile. Most rpoa-2(op259) worms had not switched from spermatogenesis to oogenesis by 18 to 24 hours after L4 as wild-type worms would [51], and more than half of all gonads revealed the presence of small cells resembling pre-diakinetic germ cells between the most proximal oocyte and the uterus (Fig. S6A). At later stages, these cell clusters expanded to large masses of innumerable cells (Fig. S6B). DAPI staining of these tumours revealed a chromatin pattern typical for mitotic germ cells (Fig. S6B). A similar proximal proliferation (Pro) phenotype had previously been described by Hubbard and colleagues [52]. Interestingly, the conditions that provoked this Pro phenotype were mutations or RNAi knockdowns of genes involved in rRNA processing [53]. Formation of the proximal tumours in these conditions involves events in early germ line development (as many germ cell proliferation phenotypes do [e.g.], [ 54,55]) and is likely a result of spatiotemporal mismatch between early germ cells and the signalling environment [56]. Similarly, we found in temperature shift experiments that the critical developmental stage that determines tumor formation and fertility in rpoa-2(op259) mutants was the L3 larval stage (not shown).
We observed a further distinct germ line differentiation defect in a small fraction of rpoa-2(op259) mutant gonads: the ectopic presence of large, apparently mature oocytes in the distal arm, followed proximally by apparently normal early pachytene nuclei (Fig. S7A). The penetrance of this defect, which is described in further detail in the Supplementary Results section (Text S1) as Gogo phenotype, was clearly enhanced by irradiation of mutant animals at adult stage. In contrast to the Pro phenotype, which arose independently of cep-1 and ced-3 (not shown), the Gogo phenotype could be suppressed by loss of cep-1 function (Fig. S7C).
In summary, rpoa-2(op259) mutants have a delayed germ line development and a reduced germ cell proliferation rate, and show at least two distinct germ cell maturation disorders under restrictive conditions. One of these, the proximal tumor phenotype, is shared with other mutants of ribosome synthesis.
rpoa-2(op259) mutants have altered ribosome synthesis
Considering that rpoa-2 codes for the second-largest subunit of RNA pol I, we wished to determine the effect of the rpoa-2(op259) mutation on ribosomal RNA synthesis. Germ cell nucleoli in C. elegans make up a significant fraction of the nuclear volume and can be readily observed by DIC microscopy. Nucleolar volume was reduced by more than 50% in rpoa-2(op259) mutant gonads (Fig. 2B). Additionally, the nucleoli in rpoa-2(op259) mutants often exhibited an enlargement of a substructure visible by DIC (Text S1 and Fig. 2A). Similar changes could also be observed in somatic nucleoli (Fig. S8A).

Eukaryotic nucleoli form around the tandem rDNA repeat units, which are transcribed by RNA pol I as one precursor rRNA (pre-rRNA) that is subsequently processed to generate the 18S, 5.8S and 28S rRNA [e.g., 57]. The 18S rRNA together with 33 canonical ribosomal proteins (in yeast) forms the 40S subunit (SSU); the 26S (28S in mammals), 5.8S and the RNA pol III-transcribed 5S rRNAs combine with 46 ribosomal proteins to the 60S subunit (LSU).
Mature ribosomal RNA is thought to constitute about 60% of total cellular RNA in yeast [58]. To quantify such highly abundant RNA by qRT-PCR, we adopted the competimer strategy to the worm (see Methods). We designed competimer primer sets for amplicons of the three RNA pol I transcribed rRNAs (Fig. S11A and Table S4). On average, the relative levels of 18S, 5.8S or 26S in rpoa-2(op259) were only about 70% of wild type (Fig. 2C). For the short-lived precursors (pre-rRNA), we used primers that annealed to the transcribed spacers ets, its1 or its2, and would thus amplify the cDNAs of transcripts that had not yet been processed in the respective regions, but not the fully mature rRNAs. Overall, pre-rRNA levels were only slightly reduced in rpoa-2(op259) mutants (Fig. 2C). Measurements of in vivo rRNA synthesis activity using 5-fluorouridine (5-FU) incorporation also suggested that rRNA transcription is grossly normal in rpoa-2(op259) mutants (Text S1 and Fig. S9B).
Eukaryotic pre-rRNA is cleaved at specific sites in a well-defined sequence of exo - and endonucleolytic events [e.g., 59], and more than 100 nucleotides are modified in human rRNA, to yield the mature ribosomal RNAs. Numerous non-ribosomal factors and small nucleolar RNPs are required for this processing and for the assembly and nuclear export of ribosomal subunits [e.g.], [ 60,61]. Defects of certain early steps of eukaryotic rRNA processing can lead to accumulation of pre-rRNA or to processing by alternative routes, eventually resulting in a characteristic pattern of processing intermediates that can be separated by electrophoresis. For instance, pro-1 mutants in C. elegans differently process rRNA at the internal transcribed spacer its2, similar to yeast mutants of its homolog Ipi3 [53].
We examined rpoa-2(op259) mutant worms for aberrant rRNA intermediates using digoxigenin (DIG)-labelled antisense RNA probes to various regions of the polycistronic pre-rRNA (ets1, its1, its2; and 18S, 5.8S, 26S rRNAs) (Text S1 and Fig. S11A). We did not detect significant alterations in the levels of early pre-rRNA species or shorter processing intermediates that retained transcribed spacer sequences (not shown). However, we noticed a distinct band between the outstanding 26S and 18S rRNA bands already when separating total RNA in denaturing agarose gels and staining with EtBr (Fig. 2E). This product was more prominent in rpoa-2(op259) samples than in wild-type extracts. We found that this molecule corresponded to a truncated version of the 26S rRNA (26S-short), with a processed 3′ end without signs of polyadenylation (Text S1 and Fig. S11D). We assessed the quantitative difference of the 26S-short band between wild type and various mutants from northern blots with probes complementary to the 5′ terminus. The intensity relative to the 26S rRNA was on average 1.4-fold higher in rpoa-2(op259) than in wild type (Fig. 2F).
Truncated versions of ribosomal RNA had been described in other species; in mammalian cells, truncated 28S rRNA were observed in the context of apoptosis or of viral infection. We did, however, not find a dependence of the 26S-short band on apoptosis execution (tested with ced-3(lf)) or on the multi-exonuclease exosome (tested with crn-3(lf)) (Fig. 2F; for details and references, see Text S1). We further looked at the two rRNA processing mutants pro-2(na27) (Noc2 in yeast) and pro-3(ar226) (Sda1), which share the Pro phenotype with rpoa-2(op259). Similarly to rpoa-2(op259), the 26S-short to 26S rRNA ratio was about 1.4-fold that of wild type for both mutants. The accumulation might thus result from defects in certain steps of rRNA processing, and it supports an effect of rpoa-2(op259) on ribosome synthesis beyond transcription of ribosomal RNA.
Quantitative or qualitative changes in transcription and processing of ribosomal RNA likely alter the composition and the activity of ribosomes, and therefore might influence translation. Interfering with translation regulation mechanisms often leads to alterations in protein expression that are confined to a subset of factors rather than being global [e.g.], [ 62,63]. Expression of some genes or groups of genes might thus be differentially affected in rpoa-2(op259) – even with a background of largely normal translation. To determine the effect of the rpoa-2(op259) mutation on the proteome level, we performed mass spectrometric analysis, applying a label-free quantitation technique on whole worm protein extracts. We could identify a total of 342 proteins in wild-type samples and 343 in rpoa-2(op259); of these, 328 proteins were present in both samples and could be compared quantitatively. Seventy proteins were ribosomal; thus, most of the total 80 ribosomal proteins were represented. Strikingly, in the mutant, most ribosomal proteins were reduced to about 50–70% of wild type when normalised by their fraction of the total in each sample. The average of the normalised ratios between rpoa-2(op259) and wild type of individual ribosomal proteins was 0.66 (0.69 for SSU and 0.64 for LSU) (Fig. 2D). Collectively, ribosomal proteins formed 21.8% of total protein in wild type, whereas in rpoa-2(op259) they represented a mere 14.2%.
Altogether, we have found that rpoa-2(op259) mutants have no significant decrease of ribosomal RNA transcription, but a clear reduction of mature rRNA levels and a similar reduction in the abundance of ribosomal proteins. Cells in rpoa-2(op259) animals might therefore have a smaller pool of mature ribosomal subunits and thus a somewhat reduced protein synthesis capacity. In addition, we have characterised an unconventional RNA of relatively high abundance, which has the nucleotide sequence of a 3′-terminally truncated, non-polyadenylated 26S rRNA, and which has higher relative abundance in rpoa-2(op259) and in mutants of rRNA processing.
Impaired rRNA processing reduces irradiation-induced apoptosis
Based on the data above, we considered three alternatives how the rpoa-2(op259) mutation might lead to disturbed cell death. First, the apoptotic defects in rpoa-2(op259) animals might be a result of globally reduced translation; DNA damage-induced apoptosis is induced through CEP-1-dependent transcriptional upregulation of the BH3 domain protein EGL-1 [30], [64] and therefore clearly requires new protein synthesis. Second, alterations in ribosome biogenesis and the nucleolus might affect germ cell apoptosis through mechanisms that do not necessarily involve protein synthesis. Third, the role of RPOA-2 in apoptosis regulation might be an independent, specific characteristic of this second largest polymerase subunit or even of the mutated site, and might not be directly linked with the overall performance of RNA pol I as a transcription apparatus. To distinguish between these possibilities, we tested the effect of knocking down the expression of other RNA pol I core subunits or associated factors, as well as of other factors involved in ribosome biogenesis or protein synthesis (Table S2 and Table S3). We chose the L3/L4 stage for initiation of RNAi by feeding to minimize detrimental effects on germ line development. Knockdown of RNA polymerase subunits frequently resulted in fertility and growth defects, but did, with the exception of F14B4.3 (rpoa-2) and F36A4.7 (ama-1) not lead to defective DNA damage-induced apoptosis (Table S2). A possible explanation for the apoptosis defect in ama-1 (which codes for the largest RNA pol II subunit) is that the reduction of mRNA transcription might become rate-limiting for the rapid transcriptional upregulation of pro-apoptotic BH3-only factors that is required for DNA-damage induced apoptosis or critical for the expression levels of other pro-apoptotic factors.
rRNA processing - and ribosomal assembly factors are critical for the maturation of the small 40S and the large 60S ribosomal subunits. There is a high number of factors involved specifically in the synthesis of either of the two subunits as well as factors needed for both [61]. Interestingly, several worm mutants of rRNA processing factors had been isolated from one genetic screen, based on their conditional proximal proliferation (Pro) phenotype described above [52], [53]. We thus looked for apoptotic defects in the viable Pro mutants pro-2(na27) and pro-3(ar226). pro-2(na27) animals indeed failed to exhibit increased apoptosis upon IR at the permissive 20°C, similarly to rpoa-2(op259) (Fig. 1E). This strengthens the notion that rpoa-2(op259) and pro-2(na27) have very similar phenotypes and that they likely result in similar molecular defects.
We addressed the question of whether failure in properly processing rRNA might more generally lead to changes in apoptosis. RNAi knockdown of nucleolar proteins, of rRNA processing factors and of representative ribosomal proteins indeed frequently blocked IR-induced apoptosis. However, knockdown of several of these factors entailed strong germ line defects that became visible soon after apoptosis scoring, so that an interpretation is difficult in these cases (Text S1 and Table S3). Nevertheless, these observations support the notion that proper rRNA processing and ribosome synthesis are critical for IR-induced apoptosis of germ cells.
Finally, we looked at the effect of interfering with translation. eIF4E has been recognised as a central factor in translation initiation and is frequently altered in various proliferative diseases [65]. C. elegans has five homologs of eIF4E, coded for by the genes ife-1 to ife-5 [66]. The five isoforms show specificity for translation of distinct sets of mRNAs [e.g.], [ 67,68]. We tested the major isoforms for somatic translation, ife-2, and for germ line translation, ife-1. The ife-1 loss-of-function alleles ok1978 (Fig. 1F) and bn127 (data not shown) reduced apoptosis in response to IR, whereas ife-2(lf) animals were not obviously defective (Fig. 1F). We also used the bacterial toxin cycloheximide (CHX) to pharmacologically block translation (see Text S1). At 500 µg/ml, most of the IR response was abolished, whereas baseline apoptosis remained unaffected (Fig. S12A). However, this dose also led to massive impairment of animal health and growth. Lower doses that only marginally impaired development had little to no effect on apoptosis (see Text S1).
In summary, we found that interfering with different steps of ribosome synthesis can lead to inhibition of germ cell apoptosis. Chemically interfering with protein translation also inhibits germ cell apoptosis, but only under conditions that are so drastic that they result in widespread defects. By contrast, rpoa-2(op259) animals appear largely normal and healthy and with a largely normal proteome profile (excepting ribosomal proteins), suggesting that the rpoa-2(op259) mutation is able to reduce apoptotic irradiation response without drastically hampering global translation. Even though we can herewith not exclude that the apoptotic phenotype is due to rate-limiting factors of apoptosis pathways whose expression is critically sensitive to particular alterations of the translation apparatus, the disturbed IR response is likely a more direct effect of abnormal ribosome biogenesis.
rpoa-2(op259) mutants lack physiological germ cell death and show weak apoptotic response to high CEP-1/p53 activity
The data above suggest that rpoa-2(op259) might selectively interfere with DNA damage-induced apoptotic signalling. CEP-1 – a functional homolog of mammalian p53 family proteins [30], [69] – is a key player in this pathway, mediating apoptosis mainly by transcriptionally activating the two pro-apoptotic BH3-only proteins EGL-1 and CED-13 [30], [64]. We evaluated the possibility that insufficient activation of CEP-1 is responsible for the weak IR response in rpoa-2(op259) by measuring transcript levels of EGL-1 and CED-13 by qRT-PCR. To our surprise, we found that both EGL-1 and CED-13 were efficiently induced in irradiated rpoa-2(op259) mutants (Fig. 3A). Moreover, the levels in non-irradiated rpoa-2(op259) control animals were much higher than in the wild type. Increased basal levels of EGL-1 and CED-13 were CEP-1-dependent, as they were abrogated in cep-1(gk138) rpoa-2(op259) double mutants (Fig. 3A). These observations suggest that rpoa-2(op259) mutant animals suffer from a chronic activation of CEP-1/p53, while at the same time being surprisingly resistant to this activation. Consistent with the hypothesis that rpoa-2(op259) mutants are resistant to CEP-1-dependent apoptosis, we found that rpoa-2(op259) could suppress the strongly increased baseline levels of germ cell apoptosis in the dsDNA break repair mutant rad-51(lg8701) (Fig. S13C) as well as the hypersensitivity to irradiation-induced apoptosis in in the Abl kinase mutant abl-1(ok171) (Fig. S13B), both of which involve CEP-1 [70].

Intriguingly, cep-1(gk138) rpoa-2(op259) double mutants had extremely few corpses, far less than either cep-1(gk138) or rpoa-2(op259) alone. We used the engulfment defective background ced-6(n1813) for better numerical ‘resolution’ of germ cell corpses around the low baseline levels and could confirm these findings (Fig. 3B and Fig. 3C). Thus, loss of cep-1 function not only suppresses the remaining moderate levels of IR-induced death in rpoa-2(op259) gonads, but also eliminates baseline cell death. This result is surprising in so far that most of the basal ‘physiological’ apoptosis observed in adult gonads normally is CEP-1-independent [30]. A hypothesis consistent with all the above information would be that rpoa-2(op259) mutants show a reduced sensitivity not only to DNA damage-induced apoptosis, but also to physiological germ cell death cues. The basal apoptosis observed in non-irradiated rpoa-2(op259) could then be explained by the increased CEP-1 activity observed in these mutants.
Considering this broader defect in germ cell apoptosis downstream of CEP-1-induced EGL-1 transcription, the rpoa-2(op259) mutation ought to influence cell death at the level or downstream of the core apoptotic machinery. In experiments that we describe in detail in the Supplementary Results (see Text S1) we observed that rpoa-2(op259) had no influence on the transcript levels of the core factors CED-4 and CED-3 and no obvious effect on the CED-4 expression pattern in pachytene stage germ cells (Fig. S14C). By contrast, rpoa-2(op259) showed a strong genetic interaction with ced-9(RNAi) or with the hypomorphic ced-9(n1653) mutation (Fig. S15C and Fig. S15A). The combination rpoa-2(op259); ced-9(n1653) led to highly excessive apoptosis, which could be suppressed by egl-1(lf) (Fig. S15B). These findings suggest that rpoa-2(op259) impacts the apoptotic machinery at the level of CED-9.
rpoa-2(op259) mutants have low germ line levels of activated MPK-1
MAPK signalling pathways are central regulators of cell proliferation and growth. In the C. elegans gonad, Ras/MAPK is required for ‘physiological’ germ cell death and has recently also been shown to modulate DNA damage-induced apoptosis [71]. In addition to the lack of physiological apoptosis, rpoa-2(op259) mutants showed a shift of the appearance of the first large oocyte to the proximal arm of the gonad, another germ line feature reminiscent of reduced Ras/MAP kinase activity. This suggested that the loss of CEP-1-independent baseline germ cell death in rpoa-2(op259) could be due to reduced Ras/MAPK pathway activity. Hypothetically, such a reduction might also contribute to the weak irradiation response in rpoa-2(op259).
To measure the levels of activated MPK-1 in situ, we performed immunofluorescence analysis on dissected gonads. MAPK-YT, an antibody to di-phosphorylated ERK with cross-reactivity to activated MPK-1 [72] detects high ppMPK-1 levels in the most proximal oocytes and distinct accumulation in the region of late pachytene cells, whereas the distal gonad is generally devoid of signal [73]. We co-stained dissected gonads for ppMPK-1; for total MPK-1; and for dsDNA. For wild-type worms, we observed the signal pattern described above (Fig. 4A). By contrast, in rpoa-2(op259) gonads, fluorescence intensity of activated MPK-1 was clearly reduced in the late pachytene region, where germ cells start maturing into oocytes or become apoptotic (Fig. 4A and Fig. 4B). These findings support that the low levels of germ cell apoptosis in rpoa-2(op259) mutants could be due to reduced Ras/MAPK activity.

Overactivation of Ras/MAPK renders germ cells hypersensitive to irradiation-induced apoptosis and restores IR response in rpoa-2(op259) mutants
The EGFR/Ras/ERK signalling cascade has been studied in much detail in the context of vulval development in C. elegans [e.g., 74]. Briefly (Fig. 5B), binding of the LIN-3 (EGF) signal to LET-23 (EGFR) and activation of the receptor tyrosine kinase activity leads to activation of the Ras GTPase homolog LET-60, and via a kinase cascade, to phosphorylation of the ERK homolog MPK-1. The phosphatase LIP-1 antagonises MAPK activity through dephosphorylation of MPK-1. LIP-1 is also an important regulator of germ cell proliferation [75], oocyte maturation [76], and germ cell apoptosis [77].
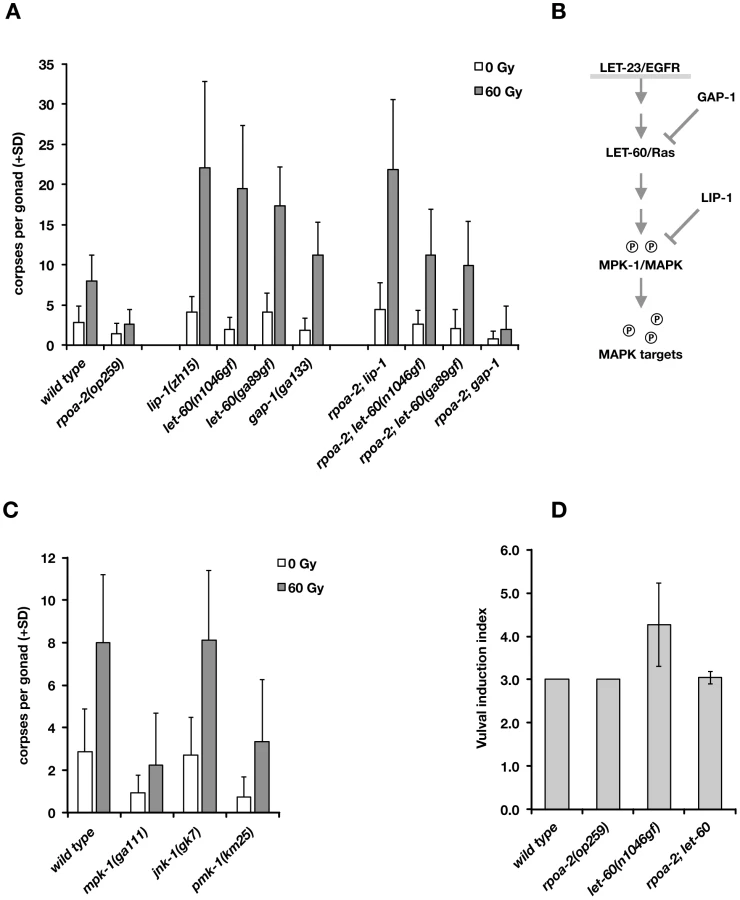
We genetically tested whether an increase in Ras/MAPK pathway activity would compensate for the abolished ‘physiological’ cell death in rpoa-2(op259) and what the effect would be on irradiation-induced germ cell apoptosis. We generated double mutants between rpoa-2(op259) and the let-60 gain-of-function alleles let-60(n1046gf) and let-60(ga89), which both lead to constitutive moderate activation of Ras GTPase activity [77]. In both strains, germ line anatomy was largely normal at 20°C; yet, the cell corpse number upon irradiation rose significantly higher than in wild type (Fig. 5A). Thus, both let-60(gf) alleles can at least partially compensate for the apoptotic defect of rpoa-2(op259).
The germ lines of lip-1(zh15) loss-of-function mutants had slightly increased baseline apoptosis and were strongly hypersensitive to IR (Fig. 5A), consistent with a recent report [71]. rpoa-2(op259); lip-1(zh15) double mutant worms also had higher than wild-type levels of germ cell death at baseline and were equally hypersensitive to IR as lip-1(zh15) animals, indicating that lip-1(zh15) is fully epistatic to rpoa-2(op259) in terms of IR-induced germ cell apoptosis. To evaluate the possibility that the rpoa-2(op259) mutation affected LIP-1, we performed anti-LIP-1 immunostaining [75] on extruded gonads of adult animals (see Text S1): the wild-type pattern of membrane-association puncta was lost in the late pachytene region of rpoa-2(op259), leaving a relatively higher cytoplasmic signal (Fig. S17B). We also looked at the somatic expression pattern of a Plip-1::GFP transcriptional reporter [78] in late larval stages, and found a strongly increased signal in hypodermal cells (Fig. S17A). The genetic and expression data support that rpoa-2(op259) might reduce MAPK activity by activating the phosphatase LIP-1. The observation of antagonistic effects of rpoa-2(op259) and let-60(gf) on germ cell apoptosis levels would fit a model where rpoa-2(op259) stimulates phosphatase activity and counteracts phosphorylation of MAPK resulting from constitutive MAPK-kinase activation by let-60(gf).
We and others previously described mutations that lead to increased baseline levels of germ cell death [79]. gla-3 mutants, which show high levels of CEP-1-independent apoptosis, have increased Ras/MAPK activity both in vulval development and in the germ line [80]. We tested whether reduced gla-3 would also render germ cells hypersensitive to IR. Due to very close linkage of gla-3, cep-1, and rpoa-2, we used gla-3(RNAi) instead of a mutation. Germ cell death was increased to about 25 corpses per gonad in gla-3(RNAi) treated worms; upon irradiation, this number rose massively, to 50 corpses on average (Fig. S16A). gla-3(RNAi) therefore does not only increase baseline apoptosis, but it potently enhances irradiation response. In rpoa-2(op259) mutants, gla-3(RNAi) increased the corpse number to approximately wild-type levels. Moreover, gla-3(RNAi) significantly restored the IR response in rpoa-2(op259) mutants (Fig. S16A), similarly to let-60(gf).
We also tested whether lip-1(zh15), let-60(n1046gf) or gla-3(RNAi) would restore CEP-1-independent – i.e. ‘physiological’ – germ cell apoptosis in rpoa-2(op259). Indeed, all these conditions re-established baseline apoptosis in cep-1(gk138) rpoa-2(op259) at levels that were similar to cep-1(gk138) (Fig. S16B).
Remarkably, the conditions leading to activation of Ras/MAPK increased the cell corpse number in rpoa-2(op259) without a significant effect on the slow (germ line) development of this mutant. Taken together, these observations suggest that rpoa-2(op259) animals suffer from reduced Ras/MAPK pathway activity in the germ line, which can account for both the reduced physiological germ cell death and the reduced sensitivity to DNA damage-induced apoptosis in this mutant.
rpoa-2(op259) antagonises let-60(gf) effects on vulval development
To determine whether the negative effect of rpoa-2(op259) on MAPK signalling extends beyond the germ line, we looked at vulval development, where MPK-1 controls the cell fates of vulval precursor cells. let-60(gf) hyperactivates MPK-1 and leads to extra inductions of vulval precursor cells and to multiple vulvae (Muv phenotype) in the adult animal [81]. The excessive vulval inductions caused by let-60(gf) were almost completely suppressed in rpoa-2(op259); let-60(n1046gf) double mutants (Fig. 5D). To exclude the possibility that rpoa-2(op259) acts downstream of mpk-1 in this model, we tested epistasis with the MPK-1 target LIN-1. In its non-phosphorylated state, LIN-1 acts as a negative regulator of genes that regulate vulva formation [82]; it is inhibited through phosphorylation by MPK-1. Loss of lin-1 function thus mimics Ras/MAPK overactivity downstream of mpk-1. We found that all rpoa-2(op259); lin-1(n304) animals had the maximal number of four extra vulvae, like lin-1(n304) single mutants (50 out of 50 animals in both strains), indicating that the effect of the rpoa-2(op259) mutation is at the level or upstream of mpk-1.
ERK and p38 MAPK activity are required for wild-type apoptosis levels upon IR
Finally, we tested mutants of MPK-1 and two other MAP kinase pathways in C. elegans, PMK-1/p38 and JNK-1/Jnk, for apoptotic IR response. The mpk-1(ga111) allele is temperature-sensitive; at 25°C, many gonads show the mpk-1 loss-of-function pachytene-exit defect and brood size is strongly reduced, while at 20°C, germ cell progression into oocytes still occurs and viable progeny are produced. Even at this permissive temperature, mpk-1(ga111) animals had lower than wild-type levels of ‘physiological’ germ cell death and showed a reduced response to IR (Fig. 5C). We also observed an apoptotic defect with RNAi knockdown of mpk-1 (not shown). These results confirm the importance of MPK-1 in DNA damage-induced germ cell death.
The MAP kinase p38 family homolog PMK-1 is involved in the response to danger signals, such as infectious agents or excess transition metals [83], [84]; lack of functional PMK-1 sensitizes C. elegans to toxic effects of pathogens [85]. We tested whether this pathway might also play a role in irradiation-induced apoptosis. Indeed, pmk-1(km25) animals had reduced numbers of apoptotic germ cell corpses at baseline or following irradiation (Fig. 5C). In contrast, animals with the jnk-1(gk7) loss-of-function mutation had normal, if not slightly increased levels of germ cell corpses following IR (Fig. 5C).
As described above, MAPK signalling has a strong impact on irradiation-induced apoptosis. mpk-1(rf) and rpoa-2(op259) mutants had only weak IR response; conversely, elevating MAPK activation had a potentiating effect on apoptosis levels, most significantly following IR. We wondered whether the role of MAPK pathway activity was solely to facilitate apoptosis, independently of the additional pro-apoptotic cues, or whether the pathway might actually be activated by irradiation responses, and examined the effect of IR three hours after treatment. The ppMPK-1 signal in the late pachytene region was indeed stronger in IR treated wild-type worms than in non-irradiated controls (Fig. 4A), as also recently reported [71]. By contrast, rpoa-2(op259) mutants did not significantly respond to irradiation, and ppMPK-1 signal intensity remained lower than in non-irradiated wild-type worms (Fig. 4B).
Taken together, our observations support the hypothesis that rpoa-2(op259) animals have low activation of Ras/MAPK, which likely is responsible for the reduced germ cell apoptosis observed in these animals. Ras/MAPK activity is a prerequisite for germ cell death and seems to sensitise cells for further pro-apoptotic signals. Possibly, the early increase in Ras/MAPK pathway activity following IR additionally boosts germ cell death in response to this genotoxic and cytotoxic treatment.
Discussion
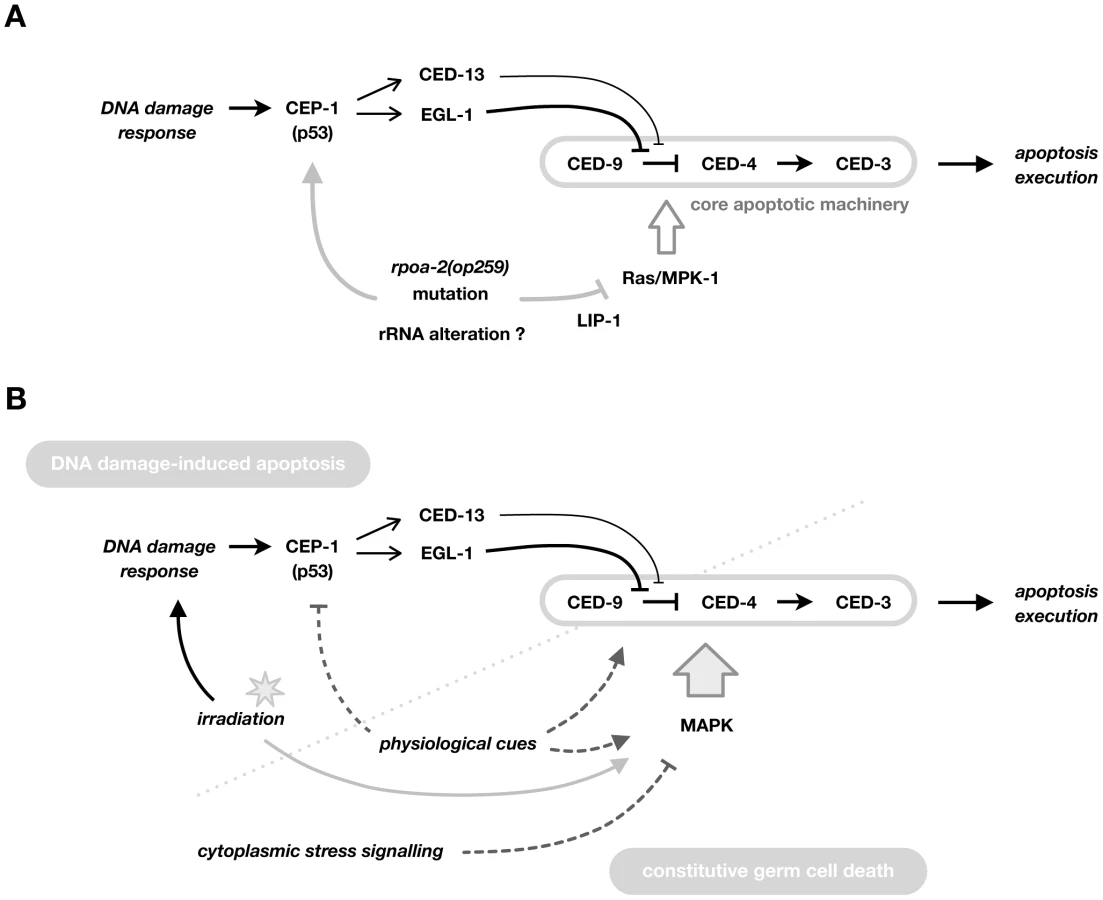
Here, we describe our characterisation of a point mutation in the second largest subunit of C. elegans RNA pol I. This mutation results in distinct, partly conditional germ line phenotypes, such as ectopic germ cell proliferation and tumor formation, precocious germ cell maturation, and reduced germ cell apoptosis. Germ cells in rpoa-2(op259) mutants fail to activate apoptosis following X-ray irradiation despite a normal CEP-1-induced upregulation of pro-apoptotic EGL-1. Importantly, CEP-1/p53 activity in non-irradiated mutants is higher than in wild-type controls, but without a concomitant increase in baseline germ cell death. We found that this block in apoptosis is most likely due to reduced levels of activated MPK-1, itself possibly a result of altered LIP-1 phosphatase activity. Hyperactivation of Ras/MAPK signalling is sufficient to restore IR-induced germ cell apoptosis levels in rpoa-2(op259). Ras/MAPK has long been recognised as a prerequisite for ‘physiological’ germ cell apoptosis. Our findings, together with two recent reports [71], [86] extend its role to setting the levels of cell death following irradiation. Whereas the first of these studies suggested a modulatory effect of MPK-1 on CEP-1, we find that it regulates apoptosis most likely downstream of CEP-1 activation, possibly at the level of CED-9/Bcl-2.
Transcription and processing of ribosomal RNA in rpoa-2(op259)
The rpoa-2(op259) mutation affects a highly conserved residue in one of the two main catalytic subunits of the RNA polymerase. With various assays, we detected no strong reduction of early pre-rRNA transcripts, but a significant drop of mature ribosomal RNAs, and an according reduction of ribosomal proteins to approximately 70% of wild type. Thus, rpoa-2(op259) mutant cells likely have a reduced pool of mature ribosomal subunits, either as a cumulative result of steadily subnormal rRNA transcription, or due to a post-transcriptional effect of rpoa-2(op259) on rRNA maturation and ribosome assembly. Supporting the latter possibility, transcription of rRNA has been shown to be interdependent and co-regulated with rRNA processing and pre-ribosome assembly, especially of the SSU [87], [88]. Intriguingly, a mutation in yeast Rpa135, the RPOA-2 homolog, also shows clear evidence for such a link [89]. Northern blot analyses of rpoa-2(op259) worm RNA extracts revealed an increase of an RNA band which we were able to characterise as a truncated, non-polyadenylated version of the 26S rRNA. Increased 26S-short levels were also present in the rRNA processing mutant pro-2(na27), which shares various phenotypic aspects with rpoa-2(op259), as well as in mutants of wdr-46 (UTP7 in yeast, involved in small subunit assembly) [90]. Even though the relevance of this molecule remains speculative (see Text S1), its relative increase in rpoa-2(op259) indicates altered rRNA processing or turnover and thus an effect of the rpoa-2(op259) mutation beyond transcription. Given the localisation of the P70S substitution on the surface of the RPOA-2 protein, it is conceivable that this mutation disrupts the interaction between the RNA pol I core complex and associated proteins required for proper assembly or disassembly of processing factors, e.g., of the SSU processosome.
Nucleolar integrity and p53 activation
rpoa-2(op259) mutants show increased CEP-1/p53 activation in the absence of exogenous DNA damage. Studies over the last two decades clearly place the nucleolus and ribosome biogenesis at the crossroads of cellular metabolism, cell cycle regulation, growth control, cellular stress responses, aging, and cell death [e.g., 91]. Nucleolar disruption has been recognised as a major hallmark of cellular stress; it can result from reduced rRNA synthesis [e.g., 18] and is possibly even recruited as a mechanistical step towards cellular fate in response to stress like DNA damage [2], [15]. A key function is attributed to the regulation of p53 by nucleolar integrity or nucleolar disintegration. Mdm2, the major ubiquitin ligase and negative regulator of p53, is controlled by the nucleolar tumor suppressor protein ARF (p19ARF). ARF is released upon nucleolar breakdown, eventually leading to p53 stabilisation [e.g., 4]. The Mdm2–p53 interaction is also target of ribosomal proteins, e.g., RPL11 or RPL26, that are released from the nucleolus following different types of cellular stress [92].
rpoa-2(op259) mutant germ cells have smaller than wild-type nucleoli and enlarged nucleolar substructures, indicating altered nucleolar physiology. Unfortunately, the proposed molecular link between nucleolar integrity and p53 stability cannot be easily transferred from mammals to the worm. Whereas p53 is functionally – and less tightly by sequence – conserved in CEP-1 [30], [31], no homolog of Mdm2 has been found in C. elegans (it is conceivable that sequence conservation is very low and therefore the homolog has not been identified, or that alternative ubiquitin ligases regulate CEP-1 stability), and there is no obvious C. elegans homolog of ARF. Nevertheless, loss of nucleolar proteins was found to increase resistance to pathogenic bacteria, via a mechanism that involves CEP-1 [93]. Thus, whether by the yet to be identified homologs of ARF and Mdm2 or by alternative mechanisms, it is well possible that the nucleolus is involved in stress responses in C. elegans and that CEP-1/p53 activation in rpoa-2(op259) is a result of disturbed nucleolar function. It remains to be determined whether the CEP-1::GFP-positive substructures that we observed in germ cell nucleoli [see Text S1; similar structures have also been reported in mammalian cells] are relevant for CEP-1 regulation.
Anti-apoptotic effects of ribosome synthesis defects
Contrasting with the expected pro-apoptotic effect of increased CEP-1 signalling, germ cell apoptosis levels were not increased in rpoa-2(op259). We show that there is an additional effect of rpoa-2(op259) in this in vivo system – reduced Ras/MAPK activity – that eventually blocks the pro-apoptotic drive from increased CEP-1/p53 activity, leading to lower than wild-type levels of germ cell death both under normal growth conditions and following DNA damage (Fig. 6A).
How might disturbances in ribosome production lead to reduced Ras/MAPK signalling? rpoa-2(op259) counteracts Ras/MAPK activation not only in the germ line, but also e.g., during vulval development. It is possible that disturbed ribosome production, beyond the specific nucleolar signalling described above, leads to a stress response, which might include a reduction in Ras/MAPK signalling. Consistent with this hypothesis, a recent study demonstrated upregulation of pathogen - and xenobiotic-associated defences in worms that had been treated with drugs or RNAi to reduce ribosome synthesis and other essential cellular processes [94]. That study and work in many species have shown that cellular stress leads to changes in the activity of MAPK superfamily members and in their cross-talks [e.g.], [ 95,96], including possibly a reduction in MPK-1/ERK activity.
In our quantitative proteome comparison of rpoa-2(op259) and wild type adults, we have found a tendency of the group of proteins annotated with ‘stress response’ towards higher relative abundance in the mutant (Fig. S10). Additionally, we have tested four reporters of stress response genes in the rpoa-2(op259) mutant background. Three of them showed increased expression in rpoa-2(op259) (Fig. S18). Remarkably, the formerly mentioned study found that upregulation of defence genes was raised globally even if only specific tissues like intestine or hypodermis were targeted with RNAi [94]. Such cross-tissue effects are likely to be in play in rpoa-2(op259) as well; altered MAPK activity and germ cell apoptosis need not be a germ line autonomous effect of rpoa-2(op259). Indeed, we found that tissue specific knockdown of rpoa-2 specifically in the soma reduced germ cell apoptosis as much as knockdown specifically in the germ line (Fig. S1C), suggesting a more global mechanism for the transmission from altered ribosome synthesis to cell death regulation.
It is also conceivable that impaired ribosome synthesis signals starvation-like conditions. Nutritional signalling – particularly the insulin/IGF pathway – is critical for the soma-germ line interaction during germ line development and also for germ cell apoptosis [86], [97], [98]. Additionally, starvation of larval stage worms was shown to counteract the effects of let-60(gf) in vulval development [99]. rpoa-2(op259) mutants have slower larval growth than wild-type worms and show increased lipid accumulation in adults, indicating significant metabolic alterations. The rpoa-2(op259) mutation leads to reduced MPK-1 activity in the gonad and antagonises the effects of LET-60/Ras overactivity on IR-induced germ cell death and on cell fate determination in somatic development. Intriguingly, a very similar phenotypic pattern has recently been reported for the insulin signalling mutants daf-2 and pdk-1 [86]. This congruence suggests that an alteration in insulin signalling by rpoa-2(op259), issued in the germ line and/or the soma, could be the link to reduced MAPK activity. Conversely, since mammalian target of rapamycin (mTOR) coordinates ribosome synthesis in response to metabolic state [12], insulin pathway mutants could lead to altered ribosome production and thereby affect MAPK activity and apoptosis.
Ras/MAPK signalling and germ cell apoptosis
Our genetic analyses of rpoa-2(op259) confirm the recently discovered functional relevance for the Ras/MAPK pathway in regulating IR-induced apoptosis [71] and additionally indicate that Ras/MAPK activity modulates germ cell death at a genetic level downstream of CEP-1/p53, likely at the level of CED-9/Bcl-2. Several other mutants beyond rpoa-2(op259) are known to show a reduced sensitivity to IR despite functional CEP-1 activation: e.g., the pRb homolog lin-35 [26], the Sirtuin homolog sir-2.1 [100], the ceramide synthesis mutant lagr-1 [101], or the ubiquitin ligase ARF-BP1 homolog eel-1 [102]. Whether these mutations also lead to reduced MPK-1 signalling remains to be determined. Supporting the model that MAP kinases play a role in stress-induced germ cell apoptosis in parallel to, or downstream of, CEP-1/p53 activation, apoptotic response to excessive arsenite [103] or copper [84] were found to depend on various MAP kinase (ERK/JNK/p38) cascades but not on CEP-1. MAPK pathways are also required for germ cell apoptosis induced by osmotic, oxidative, or heat shock stress [104]. Finally, pathogen-driven germ cell death requires the p38 MAP kinase PMK-1 but not CEP-1 [85].
Altogether, these findings support a model in which CEP-1 and MPK-1 collaboratively establish the level of IR-induced germ cell apoptosis (Fig. 6B). Ras/MAPK activity, beyond facilitating ‘physiological’ germ cell death, is critical for how sensitive cells are towards further pro-apoptotic signals. Thus, MAP kinases might act as “master modulators” of germ cell apoptosis in C. elegans.
rpoa-2(op259) – a single mutation at the basis of multiple pathways
Our analysis of rpoa-2(op259) mutants demonstrates that a single point mutation affecting a basic cellular process can readily influence multiple key signalling networks, and that the combinatorial effect of these disturbances can lead to quite complex phenotypes. The rpoa-2(op259) mutation on the one hand leads to CEP-1/p53 activation, on the other hand reduces MAPK pathway activity (Fig. 6A). Both effects influence germ cell apoptosis, but in opposite directions; reduced Ras/MAPK activity in rpoa-2(op259) mutants abolishes ‘physiological’ germ cell death and significantly impairs CEP-1-induced apoptosis; this explains the weak IR-response as well as the reduced baseline germ cell death levels in rpoa-2(op259). At the same time, we find activation of cytoplasmic stress response factors; we can guess from the literature but have no direct evidence from our studies that the activation of these specific stress response pathways leads to the observed reduced levels of activated MAPK. Nevertheless, our observations support the notion that moderate disturbance of an elemental process can lead to very specific changes in signalling rather than an amalgam of generally disrupted cellular functioning. A similar conclusion was recently reached by Melo and Ruvkun, who showed that interfering with basic cellular processes can lead to the specific activation of pathogen-associated response pathways in C. elegans [94]. The altered signalling becomes particularly evident when tissues are additionally challenged through exogenous stimuli. Conditional germ line phenotypes in rpoa-2(op259) and in other mutants of basic cellular processes are indicative of the delicate new balance in these systems regarding cell proliferation, differentiation, and death.
As the signalling pathways and cellular processes described in this work are all highly conserved through evolution, similar mechanisms might also operate in humans. This has particularly implications for the field of cancer genomics, as it suggests that mutations in genes participating in core cellular processes might, through their modulatory effects on cancer signalling pathways, significantly contribute to the development of at least some types of cancer, and influence how these respond to therapeutic treatment.
Methods
Worm strains and culture conditions
Worms were cultured at 20°C (unless indicated otherwise) on NGM agar plates seeded with OP50 strain E. coli, according to standard procedures. Synchronisation was reached by bleaching gravid adults. The moment when the first few eggs had been laid by a synchronous worm population was defined as the 0 hour reference time point for adulthood (12 hours post L4 stage in wild-type worms at standard conditions).
Genotypes used in this study (information available on www.wormbase.org):
N2 wild type, rpoa-2(op259) (this study); rpoa-2(ok1970)/hT2, pro-2(na27), pro-3(ar226), ife-1(ok1978), ife-1(bn127), ife-2(ok306), ncl-1(e1865), hus-1(op241), cep-1(gk138), ced-6(n1813), ced-3(n717), rad-51(lg8701), abl-1(ok171), let-60(n1046), let-60(ga89), lip-1(zh15), gap-1(ga133), lin-1(n304), mpk-1(ga111), pmk-1(km25), jnk-1(gk7), crn-3(ok2269), rrf-1(pk1417), mut-7(pk204), unc-119(ed3).
opIs257[Prad-54::rad-54::yfp; unc-119(+)], opIs219[Pced-4::ced-4::gfp; unc-119(+)], opIs198[Pcep-1::cep-1::gfp; unc-119(+)], zhIs4[Plip-1::gfp; unc-119(+)], dvIs19[Pgst-4::GFP::NLS], frIs7[Pnlp-29::GFP + Pcol-12::DsRed], muIs84[Psod-3::GFP + rol-6], zcIs13[Phsp-6::GFP], opIs110[Plim-7::act-5::yfp; unc-119(+)]; opIs372[Prpoa-2::yfp::rpoa-2(+); unc-119(+)], opIs375[Prpoa-2::yfp::rpoa-2(op259); unc-119(+)], opIs413[Phus-1::yfp::rpoa-2(op259); unc-119(+)], opEx1431[Phus-1::yfp::rpoa-2(+); unc-119(+)], opEx1416[Prpoa-2rpoa-2(+); unc-119(+)].
For the [rpoa-2] transgenes, the genomic sequence of rpoa-2 was fused with optionally an YFP tag and the endogenous promoter - and 3′UTR sequences in the Lazyboy cloning system (primer sequences, see Table S4). Plasmid vectors were introduced into the C. elegans germ line by ballistic transformation [105] of rpoa-2(op259); unc-119(ed3) animals. The Pro phenotype and temperature sensitive sterility at 25°C could be rescued with [rpoa-2(+)] transgenes (Fig. S6C); no rescue was attained, though, with those transgenes that did not show germ line expression.
RNAi experiments
Gene expression knockdown was performed with RNAi by feeding as described [35], [36]. Briefly, bacteria expressing double-stranded RNA (plasmids were sequenced to confirm the correct target genes) were seeded on NGM agarose plates supplemented with ampicillin and 3 mM IPTG for efficient induction. Worms were synchronised by bleaching; depending on the stage when RNAi had to be initiated, freshly hatched L1 larvae were transferred to the RNAi plates directly, or they were grown on OP50 seeded plates until reaching the appropriate stage and then washed and transferred. As controls, an empty vector RNAi clone and an unc-22 clone were always included.
DNA damage response screen
The forward genetic screen was performed as a F1 clonal screen. Because the screen was fairly laborious, we limited our search to 2000 haploid genomes, which is significantly below saturation level. Adult wild-type worms were mutagenized using the chemical alkylating agent ethyl methane sulfonate (EMS, 1.25 mM), 1000 F1 progeny were transferred singly onto new plates, and their offspring were exposed to 60 Gy of X-rays at young adulthood; multiple of these F2 animals were examined for DNA damage response defects using DIC microscopy. One candidate mutation of this screen, op259, was mapped with a combination of three-factor and two-factor mapping techniques to the right arm of chromosome I. Fine mapping and gene sequencing in the candidate region revealed a C→T transition in the second exon of F14B4.3. The mutant allele was 6× backcrossed into the wild-type genetic background prior to further characterisation.
Germ cell apoptosis
Apoptotic germ cell corpses – visible as refractile discs – were scored using Nomarski optics (DIC). Irradiation treatments were performed at the reference time point of adulthood as described above. The number of apoptotic corpses in the late meiotic pachytene region of one of the gonads (anterior or posterior) was counted in 20 worms per condition, either in a time course, or selectively at 24 hours post treatment, when the irradiation-induced corpse number was tending towards a plateau and the germ lines were still without strong signs of aging. For ionizing irradiation (IR), well-fed worms on agar plates were exposed to X-rays in an Isovolt irradiation device for 18.5 min, corresponding to a dose of 60 Gy; for UV-irradiation, worms were treated with short pulses of UV-C in a Stratalinker (254 nm) to reach the indicated energy doses.
Germ cell arrest and DNA repair foci
Gonads were extruded from adult worms and stained with Hoechst at the indicated time points after irradiation treatment, and images were acquired immediately; the fluorescent images depict slightly more than a simple axial cross-section through the gonads, as these were flattened by the preparation. The total number of mitotic cells and the fraction of enlarged cells were counted according to the chromatin-staining pattern. RAD-54::YFP foci were counted using the ImageJ [106] local intensity peak detection tool with a defined threshold.
Vulval induction
The six vulval precursor cells (VPCs) are instructed to adopt a certain fate mainly through signalling from the anchor cell (EGFR/Ras/MAPK) and by lateral crosstalk (Notch). Aberrant vulval cell inductions can result in extra vulvae (Muv) or a missing vulva (Vul) in adult animals. Cellular inductions can be accurately assessed with DIC microscopy of L4 larvae [81]. The vulval induction index represents the average number of VPCs that have adopted a 1° or 2° vulval fate (in wild type, VI index is 3.0).
qRT-PCR experiments
Adult worms were collected once the majority of the population had started laying eggs but no progeny had hatched yet (18 hours post L4 larval stage for wild type; 24 hours later for rpoa-2(op259) mutants). Gravid adults were washed from plates, cleared from bacteria and external progeny by settling in M9 buffer 3×5 min, and frozen in liquid nitrogen. Total RNA was extracted using the Nucleospin II kit (Machery and Nagel), and cDNA synthesis performed on 400 ng DNA-free RNA by Superscript III (Invitrogen). qPCR reactions were performed in triplicates, and the mean measured levels for transcripts of interest were normalised to the mean of a set of internal controls, including (in part) PGK-1, RPL-29, RPS-4 and transcripts shown to behave relatively stably between many conditions [107], namely CDC-42, PMP-3, or Y45F10D.4.
Competimers
Amplification of highly abundant RNAs can be inhibited by addition of competitive, non-extensible primers to the specific primers of the qPCR reaction [108]. Thus, only a fraction of the highly abundant template is accessible for the polymerase, and amplification efficiency decreases; signal intensity is lowered (Ct value increased) to the range of other (control) transcripts in the same, non-diluted sample. We adapted this method to quantify mature ribosomal RNA in C. elegans. Competimer primer sets were designed for amplicons of all the RNA pol I transcribed rRNAs (Table S4), and competitive primers were added to extensible primers in an excess of 4∶1.
Proteome analysis
Proteins were extracted from well-synchronised adult worms (collected as for RNA extraction, with 2 additional washes in ddH2O on ice) using glass beads and Urea/Thiourea (7 M/1 M) buffer and supplemented with 2% CHAPS and 75 mM DTT. Equal amounts of protein were digested with trypsin (100 mM Tris pH 8.5, 1 mM CaCl2), and the resulting peptides were – without further fractionation – purified on an MCX cartridge; the high sample volume after elution (5% formic acid/methanol) was reduced by evaporation, and the concentrates were desalted with C18 ZipTips. Peptide concentrations in the final samples were determined to ensure similar loading of the mass spectrometry column. Peptides were measured in triplicate LTQ-FT runs of each non-labelled sample. Estimates of individual protein abundance in the original samples were derived from spectral counting: based on the relative abundance of proteotypic peptides for the proteins identified by a ‘Mascot’ search, a quantitative value was assigned to each protein with the Scaffold 3 software [109].
MPK-1 immunofluorescence
Antibodies used for immunodetection were: MAPK-YT (Sigma M9692, 1∶100) raised against di-phosphorylated ERK and binding specifically to the homologous ppMPK-1 in C. elegans as well [72]; anti-total ERK (1∶200), cross-reacting with MPK-1; a protein-independent control antibody to dsDNA (Abcam HYB331-01; 1∶500) for normalisation. Secondary antibodies: Alexa Fluor goat anti-mouse IgG1 (MAPK-YT), goat anti-rabbit IgG (ERK), goat anti-mouse IgG2a (anti-dsDNA), all 1∶500. Blocking buffer: 10% goat serum in antibody buffer according to [110]. In our staining protocol, gonads were extruded by dissecting adult hermaphrodites 3 hours after irradiation; samples were fixed in 3% PFA for 30 min at 4°C, freeze cracked, fixed in 100% methanol for 10 min at −20°C, stained (blocking for 1 hour at RT; 1° antibodies in blocking buffer o/n at 4°C; 2° antibodies in blocking buffer for 1 hour at RT; >3 washes with PBS/Tween 0.1% between all steps) and mounted on poly-lysine coated slides. To test for specificity of the MPK-1 antibodies, we applied the immunofluorescence protocol to mpk-1(RNAi) treated worms. Loss of MPK-1 function was not very severe since most animals showed only minute phenotypes at the stage of analysis. Accordingly, ppMPK-1 and total MPK-1 signals were reduced but not completely absent. Fluorescence pictures were acquired using constant exposure settings and analysed with ImageJ.
Supporting Information
Zdroje
1. HoeijmakersJHJ (2009) DNA damage, aging, and cancer. N Engl J Med 361 : 1475–1485 doi:10.1056/NEJMra0804615
2. OlsonM (2004) Sensing Cellular Stress: Another New Function for the Nucleolus? Science's STKE 2004: pe10 doi:10.1126/stke.2242004pe10
3. MayerC, GrummtI (2005) Cellular stress and nucleolar function. Cell Cycle 4 : 1036–1038.
4. BoulonS, WestmanBJ, HuttenS, BoisvertF-M, LamondAI (2010) The nucleolus under stress. Mol Cell 40 : 216–227 doi:10.1016/j.molcel.2010.09.024
5. DerenziniM (2000) The AgNORs. Micron 31 : 117–120.
6. MontanaroL, TreréD, DerenziniM (2008) Nucleolus, ribosomes, and cancer. Am J Pathol 173 : 301–310 doi:10.2353/ajpath.2008.070752
7. RudraD, WarnerJR (2004) What better measure than ribosome synthesis? Genes Dev 18 : 2431–2436 doi:10.1101/gad.1256704
8. MossT (2004) At the crossroads of growth control; making ribosomal RNA. Current Opinion in Genetics & Development 14 : 210–217 doi:10.1016/j.gde.2004.02.005
9. LafertéA, FavryE, SentenacA, RivaM, CarlesC, et al. (2006) The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev 20 : 2030–2040 doi:10.1101/gad.386106
10. StefanovskyVY, PelletierG, HannanR, Gagnon-KuglerT, RothblumLI, et al. (2001) An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol Cell 8 : 1063–1073.
11. OskarssonT, TrumppA (2005) The Myc trilogy: lord of RNA polymerases. Nat Cell Biol 7 : 215–217.
12. MayerC, GrummtI (2006) Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 25 : 6384–6391 doi:10.1038/sj.onc.1209883
13. CavanaughAH, HempelWM, TaylorLJ, RogalskyV, TodorovG, et al. (1995) Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 374 : 177–180 doi:10.1038/374177a0
14. ZhaiW, ComaiL (2000) Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 20 : 5930–5938 doi:10913176
15. RubbiCP, MilnerJ (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22 : 6068–6077.
16. RuggeroD, PandolfiPP (2003) Does the ribosome translate cancer? Nat Rev Cancer 3 : 179–192 doi:10.1038/nrc1015
17. KalitaK, MakonchukD, GomesC, ZhengJ-J, HetmanM (2008) Inhibition of nucleolar transcription as a trigger for neuronal apoptosis. J Neurochem 105 : 2286–2299 doi:10.1111/j.1471-4159.2008.05316.x
18. YuanX, ZhouY, CasanovaE, ChaiM, KissE, et al. (2005) Genetic Inactivation of the Transcription Factor TIF-IA Leads to Nucleolar Disruption, Cell Cycle Arrest, and p53-Mediated Apoptosis. Mol Cell 19 : 77–87 doi:10.1016/j.molcel.2005.05.023
19. PontiD, TroianoM, BellenchiGC, BattagliaP, GiglianiF (2008) The HIV Tat protein affects processing of ribosomal RNA precursor. BMC Cell Biol 9 : 32 doi:1471-2121-9-32
20. AmsterdamA, SadlerKC, LaiK, FarringtonS, BronsonRT, et al. (2004) Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol 2: E139.
21. FarrarJE, VlachosA, AtsidaftosE, Carlson-DonohoeH, MarkelloTC, et al. (2011) Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood 118 : 6943–6951 doi:10.1182/blood-2011-08-375170
22. ZhouB, ElledgeS (2000) The DNA damage response: putting checkpoints in perspective. Nature 408 : 433–439.
23. StergiouL, HengartnerMO (2004) Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ 11 : 21–28.
24. GumiennyTL, LambieE, HartwiegE, HorvitzHR, HengartnerMO (1999) Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126 : 1011–1022.
25. Gartner A, Boag PR, Blackwell TK (2008) Germline survival and apoptosis. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.145.1
26. SchertelC, ConradtB (2007) C. elegans orthologs of components of the RB tumor suppressor complex have distinct pro-apoptotic functions. Development 134 : 3691–3701 doi:10.1242/dev.004606
27. ReddienPW, AndersenEC, HuangMC, HorvitzHR (2007) DPL-1 DP, LIN-35 Rb and EFL-1 E2F act with the MCD-1 zinc-finger protein to promote programmed cell death in Caenorhabditis elegans. Genetics 175 : 1719–1733 doi:10.1534/genetics.106.068148
28. ChurchDL, GuanKL, LambieEJ (1995) Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121 : 2525–2535.
29. LeeM-H, OhmachiM, ArurS, NayakS, FrancisR, et al. (2007) Multiple functions and dynamic activation of MPK-1 extracellular signal-regulated kinase signaling in Caenorhabditis elegans germline development. Genetics 177 : 2039–2062 doi:10.1534/genetics.107.081356
30. SchumacherB, HofmannK, BoultonS, GartnerA (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 11 : 1722–1727.
31. DerryW, PutzkeA, RothmanJ (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294 : 591–595 doi:10.1126/science.1065486
32. SchumacherB, SchertelC, WittenburgN, TuckS, MitaniS, et al. (2005) C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ 12 : 153–161 doi:10.1038/sj.cdd.4401539
33. Del PesoL, GonzalezVM, InoharaN, EllisRE, NúñezG (2000) Disruption of the CED-9.CED-4 complex by EGL-1 is a critical step for programmed cell death in Caenorhabditis elegans. J Biol Chem 275 : 27205–27211 doi:10.1074/jbc.M000858200
34. Conradt B, Xue D (2005) Programmed cell death. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.32.1
35. KamathR, AhringerJ (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods (San Diego, Calif) 30 : 313–321.
36. RualJ-F, CeronJ, KorethJ, HaoT, NicotA-S, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14 : 2162–2168 doi:10.1101/gr.2505604
37. CGC, Caenorhabditis Genetics Center University of Minnesota. Available: http://www.cbs.umn.edu/CGC/.
38. BushnellD, KornbergR (2003) Complete, 12-subunit RNA polymerase II at 4.1-A resolution: implications for the initiation of transcription. P Natl Acad Sci Usa 100 : 6969–6973 doi:10.1073/pnas.1130601100
39. CramerP (2002) Multisubunit RNA polymerases. Curr Opin Struct Biol 12 : 89–97.
40. CramerP (2001) Structural Basis of Transcription: RNA Polymerase II at 2.8 Angstrom Resolution. Science 292 : 1863–1876 doi:10.1126/science.1059493
41. LemmensBBLG, TijstermanM (2011) DNA double-strand break repair in Caenorhabditis elegans. Chromosoma 120 : 1–21 doi:10.1007/s00412-010-0296-3
42. HarrisJ, LowdenM, ClejanI, TzonevaM, ThomasJ, et al. (2006) Mutator Phenotype of Caenorhabditis elegans DNA Damage Checkpoint Mutants. Genetics 174 : 601–616 doi:10.1534/genetics.106.058701
43. GartnerA, MilsteinS, AhmedS, HodgkinJ, HengartnerMO (2000) A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5 : 435–443.
44. StergiouL, DoukoumetzidisK, SendoelA, HengartnerMO (2007) The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ 14 : 1129–1138.
45. StergiouL, EberhardR, DoukoumetzidisK, HengartnerMO (2011) NER and HR pathways act sequentially to promote UV-C-induced germ cell apoptosis in Caenorhabditis elegans. Cell Death Differ 18 : 897–906 doi:10.1038/cdd.2010.158
46. PepperAS-R, LoTW, KillianDJ, HallDH, HubbardEJA (2003) The establishment of Caenorhabditis elegans germline pattern is controlled by overlapping proximal and distal somatic gonad signals. Dev Biol 259 : 336–350.
47. Kimble J, Crittenden SL (2005) Germline proliferation and its control. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.13.1
48. Schedl T (2012) Germ Cell Development in C. elegans. Springer Books ISBN 9781461440154. 434 p.
49. SyntichakiP, TroulinakiK, TavernarakisN (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 445 : 922–926 doi:10.1038/nature05603
50. HansenM, TaubertS, CrawfordD, LibinaN, LeeS, et al. (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6 : 95–110 doi:10.1111/ace.2007.6.issue-1
51. Strome S (2005) Specification of the germ line. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.9.1
52. KillianD, HubbardE (2004) C-elegans pro-1 activity is required for soma/germline interactions that influence proliferation and differentiation in the germ line. Development 131 : 1267–1278 doi:10.1242/dev.01002
53. VoutevR, KillianDJ, AhnJH, HubbardEJA (2006) Alterations in ribosome biogenesis cause specific defects in C. elegans hermaphrodite gonadogenesis. Dev Biol 298 : 45–58.
54. PepperA (2003) The establishment of Caenorhabditis elegans germline pattern is controlled by overlapping proximal and distal somatic gonad signals. Dev Biol 259 : 336–350 doi:10.1016/S0012-1606(03)00203-3
55. WatersKA, ReinkeV (2011) Extrinsic and intrinsic control of germ cell proliferation in Caenorhabditis elegans. Mol Reprod Dev 78 : 151–160 doi:10.1002/mrd.21289
56. McGovernM, VoutevR, MaciejowskiJ, CorsiAK, HubbardEJA (2009) A “latent niche” mechanism for tumor initiation. Proc Natl Acad Sci USA 106 : 11617–11622 doi:10.1073/pnas.0903768106
57. Lafontaine DL, Tollervey D (2001) Ribosomal RNA. Available: http://onlinelibrary.wiley.com/doi/10.1038/npg.els.0000877/abstract.
58. WarnerJR (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24 : 437–440.
59. FaticaA, TollerveyD (2002) Making ribosomes. Curr Opin Cell Biol 14 : 313–318.
60. KresslerD, HurtE, BasslerJ (2010) Driving ribosome assembly. Biochim Biophys Acta 1803 : 673–683 doi:10.1016/j.bbamcr.2009.10.009
61. Fromont-RacineM, SengerB, SaveanuC, FasioloF (2003) Ribosome assembly in eukaryotes. Gene 313 : 17–42.
62. SonenbergN, HinnebuschA (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136 : 731–745 doi:10.1016/j.cell.2009.01.042
63. SpriggsK, BushellM, WillisA (2010) Translational regulation of gene expression during conditions of cell stress. Mol Cell 40 : 228–237 doi:10.1016/j.molcel.2010.09.028
64. DerryWB, PutzkeAP, RothmanJH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294 : 591–595 doi:10.1126/science.1065486
65. FischerPM (2009) Cap in hand: targeting eIF4E. Cell Cycle 8 : 2535–2541.
66. KeiperB, LamphearB, DeshpandeA, Jankowska-AnyszkaM, AamodtE, et al. (2000) Functional Characterization of Five eIF4E Isoforms in Caenorhabditis elegans. J Biol Chem 275 : 10590–10596.
67. HendersonM, CronlandE, DunkelbargerS, ContrerasV, StromeS, et al. (2009) A germline-specific isoform of eIF4E (IFE-1) is required for efficient translation of stored mRNAs and maturation of both oocytes and sperm. J Cell Sci 122 : 1529–1539 doi:10.1242/jcs.046771
68. DinkovaT, KeiperB, KorneevaN, AamodtE, RhoadsR (2005) Translation of a small subset of Caenorhabditis elegans mRNAs is dependent on a specific eukaryotic translation initiation factor 4E isoform. Mol Cell Biol 25 : 100–113 doi:10.1128/MCB.25.1.100-113.2005
69. GreissS, SchumacherB, GrandienK, RothblattJ, GartnerA (2008) Transcriptional profiling in C. elegans suggests DNA damage dependent apoptosis as an ancient function of the p53 family. BMC Genomics 9 : 334 doi:1471-2164-9-334
70. DengX, HofmannER, VillanuevaA, HobertO, CapodieciP, et al. (2004) Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat Genet 36 : 906–912 doi:10.1038/ng1396
71. RutkowskiR, DickinsonR, StewartG, CraigA, SchimplM, et al. (2011) Regulation of Caenorhabditis elegans p53/CEP-1-dependent germ cell apoptosis by Ras/MAPK signaling. PLoS Genet 7: e1002238 doi:10.1371/journal.pgen.1002238
72. MillerM, NguyenV, LeeM, KosinskiM, SchedlT, et al. (2001) A sperm cytoskeletal protein that signals oocyte meiotic maturation and ovulation. Science 291 : 2144–2147 doi:10.1126/science.1057586
73. PageBD, GuedesS, WaringD, PriessJR (2001) The C. elegans E2F - and DP-related proteins are required for embryonic asymmetry and negatively regulate Ras/MAPK signaling. Mol Cell 7 : 451–460.
74. Sundaram MV (2006) RTK/Ras/MAPK signaling. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.80.1
75. LeeM-H, HookB, LamontLB, WickensM, KimbleJ (2006) LIP-1 phosphatase controls the extent of germline proliferation in Caenorhabditis elegans. EMBO J 25 : 88–96 doi:10.1038/sj.emboj.7600901
76. HajnalA, BersetT (2002) The C.elegans MAPK phosphatase LIP-1 is required for the G(2)/M meiotic arrest of developing oocytes. EMBO J 21 : 4317–4326.
77. LeeM-H, HookB, PanG, KershnerAM, MerrittC, et al. (2007) Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet 3: e233 doi:10.1371/journal.pgen.0030233
78. BersetT, HoierE, BattuG, CanevasciniS, HajnalA (2001) Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 291 : 1055–1058 doi:10.1126/science.1055642
79. LettreG, KritikouE, JaeggiM, CalixtoA, FraserA, et al. (2004) Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ 11 : 1198–1203 doi:10.1038/sj.cdd.4401488
80. KritikouEA, MilsteinS, VidalainP-O, LettreG, BoganE, et al. (2006) C. elegans GLA-3 is a novel component of the MAP kinase MPK-1 signaling pathway required for germ cell survival. Genes Dev 20 : 2279–2292 doi:10.1101/gad.384506
81. SternbergPW, HorvitzHR (1986) Pattern formation during vulval development in C. elegans. Cell 44 : 761–772.
82. MileyGR, FantzD, GlossipD, LuX, SaitoRM, et al. (2004) Identification of residues of the Caenorhabditis elegans LIN-1 ETS domain that are necessary for DNA binding and regulation of vulval cell fates. Genetics 167 : 1697–1709 doi:10.1534/genetics.104.029017
83. KimDH, FeinbaumR, AlloingG, EmersonFE, GarsinDA, et al. (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297 : 623–626 doi:10.1126/science.1073759
84. WangS, WuL, WangY, LuoX, LuY (2009) Copper-induced germline apoptosis in Caenorhabditis elegans: the independent roles of DNA damage response signaling and the dependent roles of MAPK cascades. Chem Biol Interact 180 : 151–157 doi:10.1016/j.cbi.2009.03.012
85. AballayA, DrenkardE, HilbunLR, AusubelFM (2003) Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr Biol 13 : 47–52.
86. PerrinAJ, GundaM, YuB, YenK, ItoS, et al. (2013) Noncanonical control of C. elegans germline apoptosis by the insulin/IGF-1 and Ras/MAPK signaling pathways. Cell Death Differ 20 : 97–107 doi:10.1038/cdd.2012.101
87. KosM, TollerveyD (2010) Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell 37 : 809–820 doi:10.1016/j.molcel.2010.02.024
88. GallagherJ, DunbarD, GrannemanS, MitchellB, OsheimY, et al. (2004) RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev 18 : 2506–2517 doi:10.1101/gad.1226604
89. SchneiderDA, MichelA, SikesML, VuL, DoddJA, et al. (2007) Transcription elongation by RNA polymerase I is linked to efficient rRNA processing and ribosome assembly. Mol Cell 26 : 217–229 doi:10.1016/j.molcel.2007.04.007
90. LeungCK, EmpinadoH, ChoeKP (2012) Depletion of a nucleolar protein activates xenobiotic detoxification genes in Caenorhabditis elegans via Nrf/SKN-1 and p53/CEP-1. Free Radic Biol Med 52 : 937–950 doi:10.1016/j.freeradbiomed.2011.12.009
91. BoisvertF-M, van KoningsbruggenS, NavascuésJ, LamondAI (2007) The multifunctional nucleolus. Nat Rev Mol Cell Biol 8 : 574–585 doi:10.1038/nrm2184
92. DeisenrothC, ZhangY (2010) Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29 : 4253–4260 doi:10.1038/onc.2010.189
93. FuhrmanLE, GoelAK, SmithJ, ShiannaKV, AballayA (2009) Nucleolar proteins suppress Caenorhabditis elegans innate immunity by inhibiting p53/CEP-1. PLoS Genet 5: e1000657 doi:10.1371/journal.pgen.1000657
94. MeloJA, RuvkunG (2012) Inactivation of conserved C. elegans genes engages pathogen - and xenobiotic-associated defenses. Cell 149 : 452–466 doi:10.1016/j.cell.2012.02.050
95. KyriakisJM, AvruchJ (2012) Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 92 : 689–737 doi:10.1152/physrev.00028.2011
96. JunttilaMR, LiS-P, WestermarckJ (2008) Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J 22 : 954–965 doi:10.1096/fj.06-7859rev
97. MichaelsonD, KortaD, CapuaY, HubbardE (2010) Insulin signaling promotes germline proliferation in C. elegans. Development 137 : 671–680 doi:10.1242/dev.042523
98. HubbardEJA, KortaDZ, DalfóD (2013) Physiological control of germline development. Adv Exp Med Biol 757 : 101–131 doi:_10.1007/978-1-4614-4015-4_5
99. BattuG, HoierEF, HajnalA (2003) The C. elegans G-protein-coupled receptor SRA-13 inhibits RAS/MAPK signalling during olfaction and vulval development. Development 130 : 2567–2577.
100. GreissS, HallJ, AhmedS, GartnerA (2008) C. elegans SIR-2.1 translocation is linked to a proapoptotic pathway parallel to cep-1/p53 during DNA damage-induced apoptosis. Genes Dev 22 : 2831–2842 doi:PMC2569882
101. DengX, YinX, AllanR, LuDD, MaurerCW, et al. (2008) Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 322 : 110–115 doi:10.1126/science.1158111
102. RossAJ, LiM, YuB, GaoMX, DerryWB (2011) The EEL-1 ubiquitin ligase promotes DNA damage-induced germ cell apoptosis in C. elegans. Cell Death Differ 18 : 1140–1149 doi:10.1038/cdd.2010.180
103. PeiB, WangS, GuoX, WangJ, YangG, et al. (2008) Arsenite-induced germline apoptosis through a MAPK-dependent, p53-independent pathway in Caenorhabditis elegans. Chem Res Toxicol 21 : 1530–1535 doi:10.1021/tx800074e
104. SalinasLS, MaldonadoE, NavarroRE (2006) Stress-induced germ cell apoptosis by a p53 independent pathway in Caenorhabditis elegans. Cell Death Differ 13 : 2129–2139 doi:10.1038/sj.cdd.4401976
105. PraitisV, CaseyE, CollarD, AustinJ (2001) Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157 : 1217–1226.
106. Abramoff M, Magalhaes P, Ram S (2004) Image Processing with ImageJ. Biophotonics International. Laurin Publishing Co. Inc., Vol. 11. pp. 36–42.
107. HoogewijsD, HouthoofdK, MatthijssensF, VandesompeleJ, VanfleterenJ (2008) Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol Biol 9 : 9 doi:10.1186/1471-2199-9-9
108. GoidinD (2001) Ribosomal 18S RNA Prevails over Glyceraldehyde-3-Phosphate Dehydrogenase and β-Actin Genes as Internal Standard for Quantitative Comparison of mRNA Levels in Invasive and Noninvasive Human Melanoma Cell Subpopulations. Analytical Biochemistry 295 : 17–21 doi:10.1006/abio.2001.5171
109. SearleBC (2010) Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10 : 1265–1269 doi:10.1002/pmic.200900437
110. Duerr JS (2006) Immunohistochemistry. WormBook, ed. The C. elegans Research Community, http://www.wormbook.org. doi/10.1895/wormbook.1.105.1
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 11
Nejčtenější v tomto čísle
- and Are Required for Growth under Iron-Limiting Conditions
- Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland cAMP/PKA Signaling Defects in the Neurofibromatosis-1 Growth Deficiency
- The Light Skin Allele of in South Asians and Europeans Shares Identity by Descent
- RNA∶DNA Hybrids Initiate Quasi-Palindrome-Associated Mutations in Highly Transcribed Yeast DNA