
Regulation of Feto-Maternal Barrier by Matriptase- and PAR-2-Mediated Signaling Is Required for Placental Morphogenesis and Mouse Embryonic Survival
Development of mammalian embryos is dependent on an efficient exchange of nutrients, oxygen, and waste products between the mother and the embryo. The interface between the two systems is provided by the placenta in a form of a specialized epithelium that both facilitates the transport of molecules between the mother and the embryo and screens the substances that can pass between the maternal and fetal tissues. We now show that two independent signaling pathways that include the serine proteases, matriptase and prostasin, and a G protein-coupled receptor PAR-2, are critical for the establishment of a functional feto-maternal interface by specifically regulating the barrier properties of the placental epithelium. Because aberrant formation of epithelial barriers is an underlying feature of a great variety of human developmental abnormalities, the identification of the two protease-dependent signaling pathways critical for the barrier formation in embryonic tissues may help pinpoint molecular mechanisms involved in the etiology of these conditions.
Published in the journal:
. PLoS Genet 10(7): e32767. doi:10.1371/journal.pgen.1004470
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004470
Summary
Development of mammalian embryos is dependent on an efficient exchange of nutrients, oxygen, and waste products between the mother and the embryo. The interface between the two systems is provided by the placenta in a form of a specialized epithelium that both facilitates the transport of molecules between the mother and the embryo and screens the substances that can pass between the maternal and fetal tissues. We now show that two independent signaling pathways that include the serine proteases, matriptase and prostasin, and a G protein-coupled receptor PAR-2, are critical for the establishment of a functional feto-maternal interface by specifically regulating the barrier properties of the placental epithelium. Because aberrant formation of epithelial barriers is an underlying feature of a great variety of human developmental abnormalities, the identification of the two protease-dependent signaling pathways critical for the barrier formation in embryonic tissues may help pinpoint molecular mechanisms involved in the etiology of these conditions.
Introduction
The development of eutherian mammals requires an efficient exchange of nutrients, oxygen, ions, hormones, and waste products between the maternal and fetal blood. In humans and mice, a functional feto-maternal interface is established in the placenta by the formation of a complex embryonic vascular tree that is submerged in interstitial space filled with maternal blood [1], [2]. Separating the maternal and fetal circulation is a specialized embryo-derived epithelium that functions both to facilitate the transport of molecules between the mother and the embryo, and as a barrier to screen which substances can pass between the maternal and fetal tissues and which cannot. In mice, the epithelium resides in a histologically distinct region of the placenta, termed the labyrinth, and consists of two layers of differentiated polynuclear syncytiotrophoblasts surrounding the fetal vessels and an underlying layer of mononuclear cytotrophoblasts that are in direct contact with the maternal blood [2]. The thickness of the labyrinth, the degree of vascular branching, and the level of expression of transporter proteins within the labyrinth epithelium are the chief determinants of the efficiency of nutrient transfer to the embryo [3].
The ability of the epithelia to restrict free movement of water, solutes, and larger molecules through the interstitial space between the individual epithelial cells is critical for tissue compartmentalization and protection against chemical damage, infection, dehydration, or heat loss [4], [5]. Establishment of paracellular transport barriers across the epithelial layers is primarily achieved by formation of several types of specialized cell-cell junctions that include desmosomes, adherens, and tight junctions [6]. Disruption of the junctional complexes severely compromises epithelial barrier function and is frequently linked to disease in both mice and humans [5], [7]. In the developing embryo, establishment of a functional feto-maternal barrier is critical to protect the fetus from toxins and infection by blood-born pathogens present in maternal circulation during pregnancy, as well as potential ion imbalance, unchecked diffusion of maternal hormones, or an attack by maternal immune system [8]–[12].
Matriptase is a trypsin-like cell surface-associated serine protease that plays an essential role in the homeostasis of a variety of mouse and human epithelia [13]–[15]. Mice with a complete loss of matriptase function die perinatally due to a defect in epidermal barrier function, leading to a fatal dehydration [16], [17]. Likewise, tissue-specific genetic ablation of matriptase revealed a role in epithelial barrier function in a number of other organs, including oral cavity, salivary gland, small intestine, and colon, suggesting a key role of this protease in the establishment of functional epithelial barriers of both single and multi-layered epithelia [18]. Despite a widespread expression in a number of embryonic and extraembryonic epithelia, matriptase, however, is not required for term development in humans and in most mouse strains ([13], [14], [16] and unpublished data). However, the regulation of matriptase activity is essential for the successful completion of the embryonic development, as documented by severe, matriptase-dependent, defects in placental development, neural tube closure, and overall embryonic survival in mice lacking either of the two endogenous serine protease inhibitors, HAI-1 and HAI-2 [19], [20].
Protease-activated receptors (PARs) are a subfamily of the heptahelical G protein-coupled receptors (GPCRs) activated by a proteolytic cleavage within the N-terminal extracellular region, unmasking new amino-terminal residues that then serve as tethered ligands to activate the receptors [21]–[23]. Both in mice and in humans, the subfamily consists of four members, PAR-1 through PAR-4. Mice carrying loss of function mutations in PAR-1 or PAR-2 genes suffer from partial embryonic lethality although born PAR-1 - and PAR-2-deficient animals generally show a normal postnatal development and survival [24], [25]. Furthermore, the combined loss of PAR-1 and PAR-2 leads to more than 60% decrease in pre-term and 90% decrease in pre-weaning survival, and to an array of developmental defects that include neural tube defects, edema, and an enlarged pericardium, indicating the requirement of PAR-mediated signaling for the completion of normal embryonic development [26]. Whereas PAR-1, -3, and -4 are activated in vivo by the secreted serine protease thrombin and appear to primarily function as part of the blood coagulation cascade, PAR-2 is expressed by a majority of epithelial and endothelial cells and can be activated by a number of soluble and membrane-anchored proteases with trypsin-like activity, but is not efficiently activated by thrombin [23]. Matriptase, in particular, has consistently been shown to efficiently stimulate activation of PAR-2 in a variety of cell-based assays. This, together with a highly overlapping pattern of expression in majority of epithelia, suggested that at least some of the functional effects of matriptase in vivo are mediated by PAR-2 and its downstream effectors [26]–[28].
In this study, we report the unexpected observation that matriptase and PAR-2 act during embryogenesis via functionally independent and redundant proteolytic signaling pathways. We show that mouse embryonic development and survival is strongly dependent on the genetic dosage of matriptase and PAR-2 and that the combined loss of both molecules leads to a complete embryonic lethality. Our results reveal unexpected complementary and essential roles of matriptase/prostasin - and PAR-2-dependent signaling in the establishment of placental epithelial tight junctions.
Results
Combined loss of matriptase/prostasin - and PAR-2-dependent signaling leads to embryonic lethality
Matriptase has consistently been shown to efficiently stimulate activation of PAR-2 in cell-based assays, leading to a conclusion that at least some of its physiological functions may be mediated by PAR-2 [26]–[28]. However, our recent work tentatively suggested that in the context of mouse embryonic development, matriptase and its partner protease prostasin may act independently of PAR-2, based on the findings that: (i) developmental defects observed in mice lacking endogenous matriptase inhibitor, HAI-2, can be rescued by matriptase or prostasin deficiency but not by PAR-2 deficiency, and (ii) a genetic inactivation of matriptase fails to reproduce phenotypes associated with PAR-2 deficiency in mice lacking PAR-1 [29]. The HAI-2; PAR-2 double-deficient mice (Spint2−/−;F2rl1−/−) used in that study were mostly generated by interbreeding of HAI-2; PAR-2 double-heterozygous and HAI-2; PAR-2; matriptase triple-heterozygous mice (Spint2+/−;F2rl1+/−×Spint2+/−;F2rl1+/−;St14+/−). Unexpectedly, a detailed review of the genotype distribution among the offspring from these breeding pairs at weaning showed that the survival of PAR-2-deficient mice was strongly dependent on the number of active alleles of matriptase. Thus, in offspring carrying two active alleles of matriptase (St14+/+, matriptase wildtype) about 90% of the F2rl1−/− mice survived until weaning (Figure 1A and Table S2), consistent with the previously reported 10–30% pre-term lethality among the F2rl1−/− mice [25], [26]. However, in the offspring that lacked one functional allele of matriptase (St14+/−, matriptase heterozygous), the survival of the F2rl1−/− mice decreased to less than 40% compared to the expected Mendelian distribution (Figure 1A and Table S2). To further analyze this dramatic loss of survival and to test the survival of PAR-2-deficient mice in the complete absence of matriptase, we established a new cohort of breeders double-heterozygous for both matriptase and PAR-2 (F2rl1+/−;St14+/−). Inspection of the allele distribution in newborn offspring (Figure 1B) confirmed the highly significant increase in embryonic lethality (Figure 1C) among F2rl1−/− animals carrying one active allele of matriptase (F2rl1−/−;St14+/−, 40% pre-natal survival), compared to matriptase wildtype animals (F2rl1−/−;St14+/+, 90% pre-natal survival). Furthermore, analysis of the 272 newborn mice from the F2rl1+/−;St14+/− breeding pairs did not identify any F2rl1−/−;St14−/− pups, indicating a complete pre-term lethality of F2rl1−/− mice in the absence of matriptase (Figure 1B and 1C, Table S3). Similarly, embryonic survival of matriptase-deficient mice was strongly dependent on the gene dosage of the PAR-2 gene, as indicated by73%, 35%, and 0% pre-term survival of St14−/− mice carrying two, one, or no active alleles of the PAR-2 gene, respectively (Figure 1B and C, Table S3).

Activation of matriptase during embryonic development was recently shown to be dependent on the activity of the GPI-anchored serine protease prostasin (PRSS8/CAP1), indicating that the two proteases are part of the same proteolytic signaling pathway [29]. Consistent with this finding, analysis of the 247 newborn offspring from the Prss8+/−;F2rl1+/− breeding pairs also showed a diminished survival of mice with low combined dosage of Prss8 and F2rl1 genes. Specifically, the pre-term survival of PAR-2-deficient mice decreased from 93% in prostasin wildtype animals (Prss8+/+;F2rl1−/−) to 62% and 0% in mice carrying one (Prss8+/−;F2rl1−/−) or no (Prss8−/−;F2rl1−/−) active alleles of prostasin (Figure 1D, Table S4). These data show that the pre-term survival of mice is completely dependent on either PAR-2 - or matriptase/prostasin-mediated proteolytic signaling.
Matriptase; PAR-2 double-deficient mice die at midgestation despite normal development
We next investigated the offspring of interbred F2rl1+/−;St14+/−×F2rl1+/−;St14+/− and F2rl1−/−;St14+/−×F2rl1+/−;St14+/− mice at various stages of embryonic development to further characterize the lack of term survival in mice double-deficient for PAR-2 and matriptase. Analysis of embryos extracted between E10.5–E12.5 revealed normal distribution of F2rl1 and St14 alleles, and nearly all of the F2rl1−/−; St14−/− embryos extracted before E12.5 were alive and appeared normal, indicating that the pre-implantation and the early post-implantation development and survival were not affected by the combined loss of F2rl1 and St14 gene function (Figure 1E). After E12.5, however, the survival of the F2rl1−/−;St14−/− animals dramatically decreased to 59% at E13.5, 14% at E14.5, and no living F2rl1−/−;St14−/− embryos were identified at or after E15.5 (Figure 1E and Table S5). Similarly, analysis of the offspring from F2rl+/−;Prss8+/− breeding pairs found an expected number of living Prss8−/−;St14−/− embryos at or before embryonic day 12.5, followed by a dramatic decrease in survival to 47% at E13.5, 17% at E14.5, and no surviving double-deficient embryos identified at and after E15.5 (Figure 1F and Table S6).
Matriptase, prostasin, and PAR-2 all are membrane-anchored proteins and are believed to function primarily at the surface of the expressing cells. To help us identify embryonic structures potentially affected by the combined loss of matriptase/prostasin - and PAR-2-dependent activities, we looked for cells simultaneously expressing St14, Prss8, and F2rl1 in mid-gestation embryos using the publicly available Eurexpress transcriptome atlas database [30]. All three genes were detected in a number of embryonic tissues at E14.5, showing an extensive co-expression in developing epithelia of skin, oral and nasal cavities, salivary gland, lungs, kidneys, and gastrointestinal tract (Figure 2A–C). Surprisingly, despite the widespread co-expression, a detailed inspection of the living E12.5–14.5 F2rl1−/−;St14−/− or F2rl1−/−;Prss8−/− embryos failed to identify any obvious developmental abnormality. The double-deficient mice did not develop any signs of internal bleeding, edema, or enlarged pericardium typical for many mouse strains exhibiting mid-gestational lethality (Figure 2D), and did not differ in size or total body weight from their wild-type littermate controls (Figure 2D and 2E). Furthermore, histological analysis of living E11.5–14.5 F2rl1−/−;St14−/− embryos did not reveal any obvious defects in the development of any of the epithelia with detectable levels of matriptase, prostasin, and PAR-2 gene expression (see above) or any other major organs, which are typically affected in mouse strains that exhibit mid-gestational lethality, including liver [31] (Figure 2F–I′).
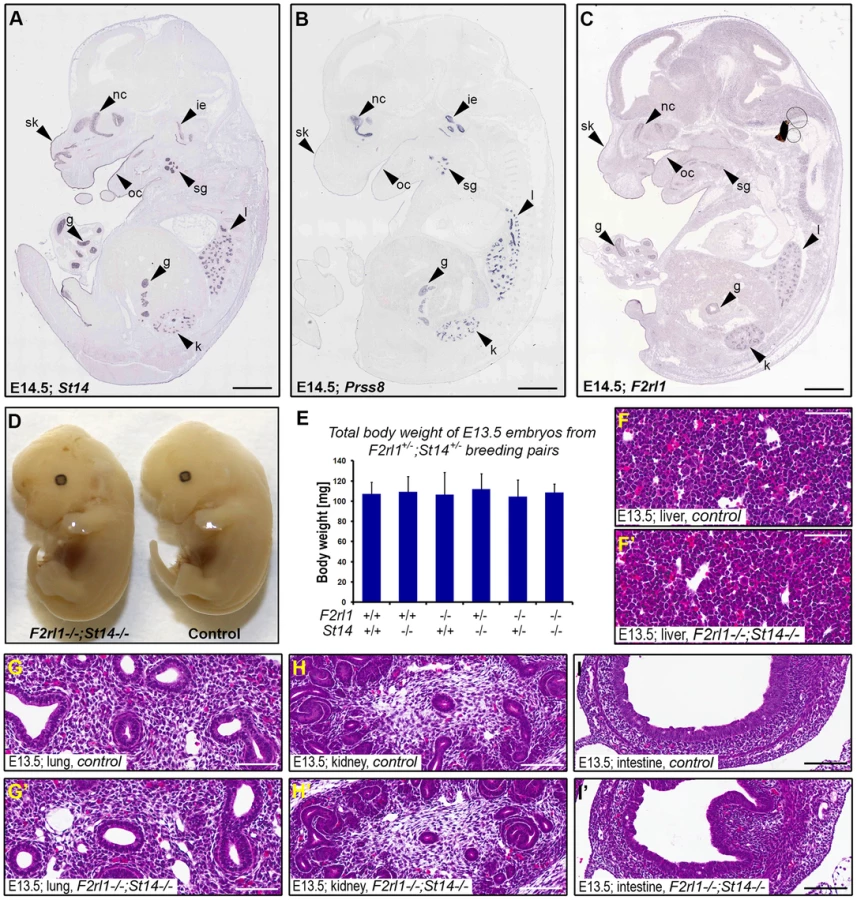
Combined loss of matriptase and PAR-2 leads to defects in development of the placental labyrinth
Failure to identify any specific defect associated with a combined loss of matriptase and PAR-2 in the embryo proper prompted us to examine development of other tissues of fetal origin. Consistent with our previous findings, immunohistochemical analysis revealed strong expression of matriptase and prostasin in the epithelial compartment of the placental labyrinth at midgestation (Figure 3A and 3B) [19], [20], [29]. To address the expression of PAR-2 in the absence of suitable antibodies, we employed a previously generated knock-in mouse strain that carries a β-galactosidase reporter construct under the control of the endogenous promoter of F2rl1 gene [32]. Analysis of the distribution of β-galactosidase activity in mouse placental tissues at E12.5 revealed that, similar to matriptase and prostasin, expression of the F2rl1 gene is found predominantly in the epithelium of the chorion and in the syncytiotrophoblast layer lining fetal endothelium within the labyrinth (Figure 3C). These data identify labyrinthine epithelium as the placental population most likely to be affected by a combined loss of matriptase/prostasin and PAR-2.
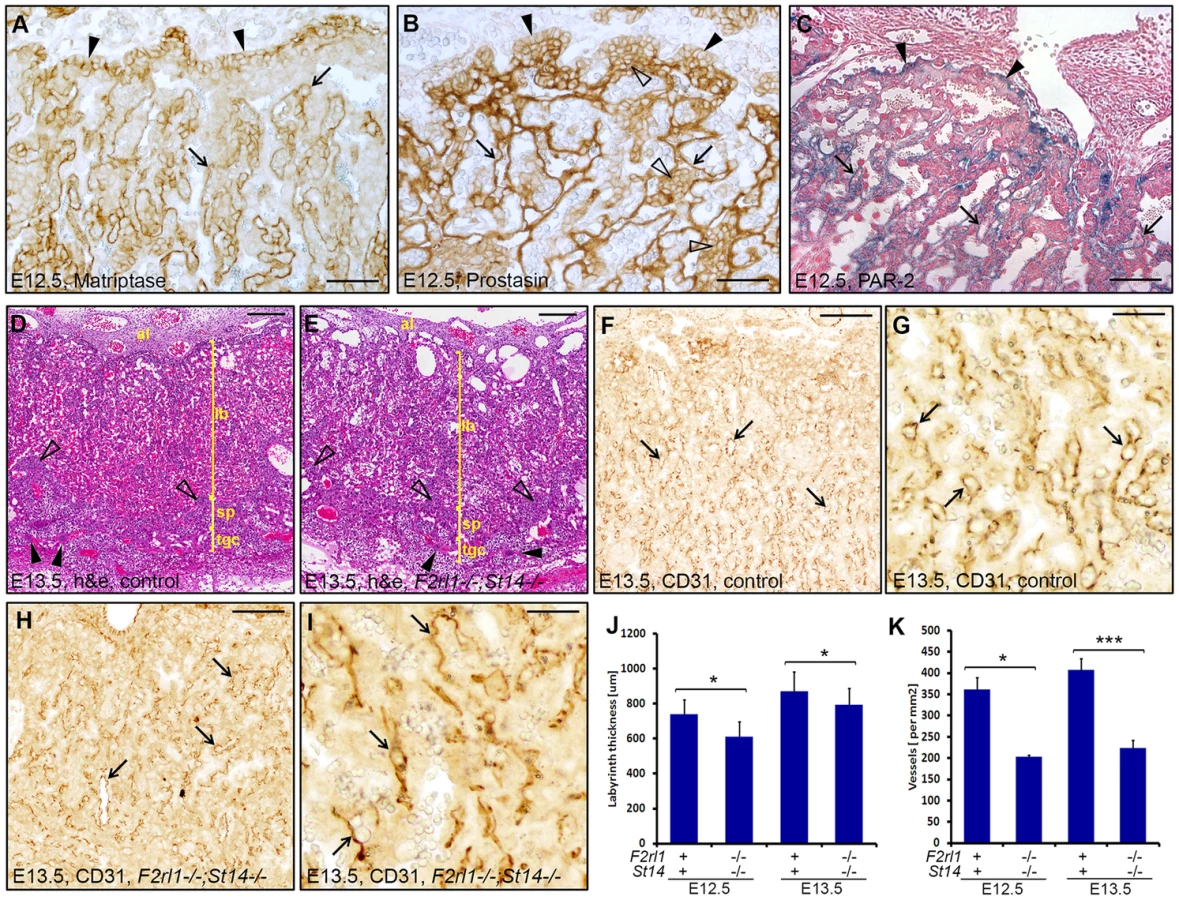
Initial histological evaluation indicated that all embryo-derived placental structures, including layers of trophoblast giant cells, spongiotrophoblasts, placental labyrinth, and allantoic mesenchyme were all formed in the placentas of F2rl1−/−;St14−/− embryos and there were no signs of placental edema (Figure 3D and 3E). Furthermore, immunohistochemical staining for the endothelial cell marker CD31/PECAM1 demonstrated the presence of a highly branched fetal vasculature in close proximity to maternal blood lacunae within the labyrinth of F2rl1−/−;St14−/− placentas (Figure 3F–3I), indicating that the combined loss of PAR-2 and matriptase does not block any of the major morphogenetic processes involved in placental development. However, a more detailed morphometric analysis revealed noticeable quantitative changes in the development of the labyrinth structure. The overall thickness of the labyrinth, defined as the maximum perpendicular distance between the undifferentiated chorionic epithelium and the labyrinth supporting spongiotrophoblast layer, was reduced by 10–15% in placentas from F2rl1−/−;St14−/− embryos at E12.5 and E13.5 (P<0.05, Figure 3J). This was also reflected in a 15% reduction of total volume of the labyrinth in F2rl1−/−;St14−/− placentas at E13.5, as determined by the Cavalieri stereological technique (Figure S1). More importantly, the complexity of the fetal-derived vasculature within the labyrinth that mediates bidirectional transport of molecules between the mother and the embryo was substantially reduced, as indicated by more than 40% decrease in the number of CD31/PECAM1-positive fetal capillary profiles found within the labyrinth area of double-deficient placentas at E12.5 and E13.5 (Figure 3F–3I and Figure 3K, P<0.01 and 0.001, respectively, Student's t-test, two-tailed). These data indicate that the combined loss of matriptase-prostasin - and PAR-2-dependent signaling interferes with the development of the feto-maternal interface.
Placental expression of matriptase rescues embryonic survival of matriptase/PAR-2 double deficient mice
To test the possibility that the loss of viability of F2rl1−/−;St14−/− mice at midgestation results from a defect in the development of placental rather than embryonic tissues, we next investigated survival of PAR-2-deficient mice with a specific inactivation of matriptase in the embryo proper. To that end, PAR-2 heterozygous breeders carrying conditional St14 allele (F2rl1+/−;St14fl/fl) were crossed to F2rl1+/−;St14+/− animals expressing Cre recombinase under the control of the endogenous Meox2 promoter. Meox2 expression is ubiquitous in mouse embryonic tissues as early as at E6.5, but is missing from any of the extraembryonic structures, including the placenta [33]. As a result, whereas both the embryos and the placentas of the Meox2-Cre+;F2rl1−/−;St14−/fl offspring would be devoid of PAR-2 expression, only the embryos will also be rendered matriptase-deficient due to the recombination of the remaining floxed St14 allele (Figure 4A). A potential survival of Meox2-Cre+;F2rl1−/−;St14−/fl embryos would therefore demonstrate a critical contribution of placental matriptase to the embryonic survival of PAR-2 and matriptase double-deficient mice, whereas lack of survival would be an indication that embryonically-expressed matriptase determines viability of these animals.

Western blot analysis of protein lysates from the E13.5 embryos and placentas showed a complete loss of expression of matriptase protein in the Meox2-Cre+;F2rl1−/−;St14−/fl mid-gestation embryos, whereas the expression of the protease in the corresponding placentas was easily detectable (Figure 4B), thus confirming the efficient, embryo-specific inactivation of the St14 conditional allele. However, despite being matriptase - and PAR-2-deficient, Meox2-Cre+;F2rl1−/−;St14−/fl embryos survived to term and were detected among the newborn offspring from the F2rl1+/−;St14fl/fl×Meox2-Cre+;F2rl1+/−;St14+/− breeding pairs in the expected ratio (Figure 4C). These mice recapitulated all of the phenotypes previously observed in matriptase knockout mice, including dry skin, and lack of whiskers, and showed no residual expression of matriptase in newborn tissues (Figure 4D–4F). These findings document that the expression of matriptase in the embryo proper is dispensable for the pre-term development and survival of PAR-2; matriptase double-deficient mice.
Matriptase and PAR-2 promote placental barrier function
The labyrinthine epithelium in mice is a principal component of the feto-maternal barrier, serving both to transport and to screen the substances passing between the maternal blood and the fetal tissues. To test whether the loss of matriptase and/or PAR-2 expression interferes with either of these functions, we next investigated the rate of molecular transport across the placental epithelium. Many nutrients, including glucose, are carried across the placenta by facilitated diffusion using an array of cell-surface transporters expressed by placental epithelial cells [34]. The efficiency of glucose uptake by the embryo can therefore be used as a quantitative measure of placental nutrient transport capacity [35]. To that end, radioactively-labeled 3-O-[methyl-14C]-D-glucose was injected into the bloodstream of pregnant females at E12.5 or E13.5, followed by embryo extraction after 2 minutes and measurement of fetal uptake of the [14C] label. Consistent with the notable underdevelopment of placental labyrinth layer in matriptase/PAR-2 double-deficient animals (see above), uptake of glucose by the F2rl1−/−;St14−/− embryos decreased by about 20% compared to wild-type littermate controls (P<0.05, Figure 5A). However, this relatively modest reduction in the transport rate together with a lack of any of the typical indications of embryo malnutrition, such as reduced size, gross underdevelopment, or enlarged pericardium, did not conclusively demonstrate an insufficient placental transport as the primary cause of embryonic lethality in the double knockouts.

In addition to the transport of gases and nutrients, placenta also fulfills a critical function of a barrier protecting the fetus against unchecked diffusion of substances, including ions, hormones, components of maternal immune system, or blood-born pathogens from maternal to fetal tissues. To test the ability of the placental epithelium to act as a barrier against passive diffusion, pregnant females were injected at E12.5 or 13.5 with the radioactively-labeled polysaccharide inulin, which can cross epithelial layers solely by a paracellular route due to lack of specific transporters [35], [36]. Interestingly, measurement of uptake of [14C] label revealed dramatic differences in inulin diffusion into the embryos of different genotypes. Thus, genetic inactivation of either matriptase or PAR-2 alone resulted in 247% and 145% increase in placental permeability (Figure 5B). This defect was further exacerbated in matriptase-deficient animals heterozygous for PAR-2 (F2rl1+/−;St14−/−, 396% increase) or PAR-2-deficient animals heterozygous for matriptase (F2rl1−/−;St14+/−, 351% increase) (Figure 5B). Finally, the highest level of inulin diffusion was observed in double-deficient embryos (F2rl1−/−;St14−/−, 589% increase, Figure 5B). These data indicate a dramatic loss of feto-maternal barrier function in mice with decreased levels of matriptase and/or PAR-2, the extent of which precisely correlates with the overall embryonic survival in these animals.
Combined loss of matriptase and PAR-2 leads to a selective decline in claudin-1 expression
Loss of placental barrier can, in principle, be a result of either: (i) an indirect effect due to an impairment of terminal differentiation and subsequent barrier acquisition within the labyrinthine epithelium, or (ii) a direct alteration of the properties of the barrier-forming epithelial cell-cell junctions in the terminally differentiated trophoblasts. To distinguish between the two possibilities we first analyzed the expression of two of the principal regulators of labyrinth differentiation, transcription factor Glial Cell Missing (GCM) a and syncytin, a fusogenic retroviral protein that mediates terminal differentiation of placental cytotrophoblasts into multinucleated syncytium [37], [38]. Analysis of placental tissues from E12.5 matriptase/PAR-2 double-deficient animals and their wild-type littermate controls did not reveal any obvious changes in the expression level of either of the two markers (Figure 5C), indicating that the process of labyrinth differentiation is not compromised by a combined loss of the two proteolytic pathways. To assess the formation of barrier-promoting epithelial cell-cell junctions, we next analyzed the levels of claudin-1 and -2, cadherins, and desmogleins 1 and 2, as the major structural components of tight junctions, adherens junctions, and desmosomes, respectively. Whereas the expression of cadherins and desmogleins was not affected by the combined loss of matriptase and PAR-2, the expression of tight junction marker claudin-1 but not claudin-2 was severely diminished (Figure 5D and 5E). Matriptase or PAR-2 single-deficient placentas did not exhibit significant changes in claudin-1 expression (Figure S2). These findings suggest that a combined activity of matriptase/prostasin and PAR-2 may regulate permeability of placental labyrinth by specifically altering properties of epithelial tight junctions.
Discussion
Inadequate placental development may lead not only to miscarriage, but also to poor health and increased risk of chronic disease in born individuals [39]. The current study for the first time reveals that the matriptase-prostasin system plays an important role in placental morphogenesis. Thus, mice with placental ablation of matriptase displayed impaired placental barrier formation, which was synergistically exacerbated by haploinsufficiency for or by complete deficiency of PAR-2. A role for the matriptase-prostasin system in placental morphogenesis would be anticipated because of the temporally and spatially coordinated expression of both membrane-anchored serine proteases and their cognate inhibitors, HAI-1 and HAI-2, in the developing placenta [19], [20], [29], [40] and the need for strict posttranslational regulation of both proteases for normal placentation to occur [19], [20], [29]. However, both matriptase-deficient humans and mice have been reported to complete embryonic development [13], [14], [16], and the role of this proteolytic cascade in development therefore has remained unclear. Because of the coordinated expression of matriptase, prostasin, HAI-1 and HAI-2 in several other developing epithelia, it now appears likely that matriptase/prostasin-initiated cellular signaling may also contribute to the development of several other organ systems in the embryo proper, acting in a redundant manner with other proteolytic systems that provide developmental backups.
Numerous studies have demonstrated a role of PAR-2 signaling, triggered by proteases of the coagulation cascade and by other trypsin-like serine proteases, in promoting both epithelial and endothelial barrier leakage by increasing paracellular permeability [41]–[45]. Our study is the first to identify a context in which PAR-2 signaling promotes epithelial barrier formation, rather than barrier leakage, thus adding another property to this multifunctional receptor. The observed specific deficiency in the expression of tight junction protein claudin-1 could suggest that the two proteolytic pathways directly promote barrier formation of terminally differentiated placental epithelium rather than doing it indirectly by providing morphogenetic signaling enabling the terminal differentiation of placental epithelium and the subsequent paracellular barrier acquisition between terminally differentiated epithelial cells. However, it should be noted that it is not yet clear whether the demise of the matriptase/PAR-2 double-deficient animals can be attributed to the observed defect in the expression of claudin-1, especially considering that claudin-1-deficient mice can complete embryonic development, at least in some mouse genetic backgrounds [46]. A previous report by Buzza and coworkers [47] suggested that matriptase regulates intestinal barrier function in a claudin-2-dependent manner. Our current analysis did not reveal any differences in claudin-2 expression in matriptase/PAR-2 double-deficient placental tissues, suggesting that different mechanisms of proteolysis-mediated regulation of tight junction function may be employed by different tissues and/or at different stages of mouse development.
Previous studies have shown that the matriptase-prostasin system is a potent activator of PAR-2 in a variety of cell-based assays [26]–[28], and PAR-2, matriptase, prostasin, HAI-1 and HAI-2 all are co-expressed in multiple developing and adult epithelia. Our genetic demonstration that the matriptase-prostasin system and PAR-2 are components of two independent and functionally redundant proteolytic pathways during placental morphogenesis therefore is unexpected and raises two unanswered questions: First, what is the identity of the placental protease(s) that activates PAR-2 in the absence of matriptase or prostasin? Second, what is the identity of the non-PAR-2 substrate(s) that is cleaved by the matriptase-prostasin cascade to promote placental morphogenesis? Regarding the former, PARs are activated by members of the S1 family of trypsin-like serine proteases, but not by other serine proteases or by other protease classes [48]–[50]. Previous transcript analysis of developing mouse embryos (in the context of neural tube closure) revealed the expression of a surprisingly large number of secreted and membrane-anchored serine proteases [20], [26]. The identification of the protease(s) that activates PAR-2 during placental morphogenesis by using a candidate epistasis analysis approach therefore may be a challenging undertaking. Regarding the latter question, the matriptase-prostasin cascade has been proposed to execute the cleavage of a wide variety of substrates, including other proteases (urokinase plasminogen activator, kallikrein-5 and -7, stromelysin-1), growth factors (hepatocyte growth factor, platelet-derived growth factor, macrophage-stimulating protein) tyrosine kinases (angiopoietin receptor, subtractive immunization M(+)HEp3 associated 135 kDa protein/CUB domain-containing protein-1, epidermal growth factor receptor) and more [15], [28], [51]–[62], reviewed in [18]. Identification of the specific proteolytic targets for the matriptase-prostasin cascade in the placenta therefore may prove difficult.
In summary, our study is the first to demonstrate a function of matriptase/prostasin - and PAR-2-dependent protease signaling in placental morphogenesis and to show that the two pathways act in an independent and functionally redundant manner to promote formation of the placental barrier.
Materials and Methods
Mouse strains
All experiments were performed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited vivarium following Standard Operating Procedures. The studies were approved by the NIDCR Institutional Animal Care and Use Committee. All studies were littermate controlled. Matriptase-deficient (St14−/−), PAR-2-deficient (F2rl1−/−), and Meox2-Cre transgenic mice have been described previously [16], [18], [33], [63]. Prostasin-deficient (Prss8−/−) mice were generated by standard blastocyst injection of C57BL/6J-derived embryonic stem cells carrying a gene trap insertion in the Prss8 gene (clone IST10122F12, Texas A&M Institute for Genomic Research, College Station, TX). Details on mouse generation will be published separately. Ear or tail clips of newborn or two week old mice were subjected to genomic DNA extraction and genotyped by PCR (see Table S1 for primer sequences).
Gene expression analysis in mouse embryos
In situ hybridization data were retrieved from the Eurexpress transcriptome atlas database [30]. Gene names St14, Prss8, and F2rl1, respectively, were individually searched to determine expression at embryonic day 14.5.
Extraction of embryonic and perinatal tissues
Breeding females were checked for vaginal plugs in the morning and the day on which the plug was found was defined as the first day of pregnancy (E0.5). Pregnant females were euthanized in the mid-day at designated time points by CO2 asphyxiation. Embryos were extracted by Caesarian section and the individual embryos and placentas were dissected and processed. Only living embryos with detectable heartbeat were used for further analysis. Tail clips or yolk sacs of individual embryos were washed twice in phosphate buffered saline, subjected to genomic DNA extraction, and genotyped by PCR (Table S1). Newborn pups were euthanized by CO2 inhalation at 0°C. For histological analysis, the embryos and newborn pups were fixed for 24 h in 4% paraformaldehyde (PFA), processed into paraffin, sectioned, and stained with hematoxylin and eosin (H&E), or used for immunohistochemistry as described below. To evaluate the development of embryonic and placental tissues, H&E-stained midline sagittal embryonic and the midline cross sections from at least three F2rl1−/−;St14−/− and three corresponding littermate controls were inspected by a certified pathologist for possible abnormalities.
Analysis of placental morphogenesis
Placental tissues from living E12.5 and E13.5 embryos were extracted and processed into paraffin as described above. To analyze the overall thickness of the placental labyrinth, a single midline cross-section at the level of the umbilical cord was stained with hematoxylin & eosin and the maximum perpendicular distance between the undifferentiated chorionic epithelium and the labyrinth-supporting spongiotrophoblast layer was measured under a light microscope by an investigator blinded to the genotypes of individual embryos. The volume of the placental labyrinth was estimated using Cavalieri's principle. Briefly, PFA-fixed placental tissue was embedded in paraffin, cross sections 200 µm apart covering the entire volume of the tissue were stained with h&e, scanned using ScanScope system (Aperio Technologies, Vista, CA), and the area of the labyrinth was measured on each section using Image J 1.46r software (National Institutes of Health, MD). The total volume of the labyrinth was then estimated as the sum of partial volumes calculated as area of the labyrinth on each section multiplied by 200 µm.
To evaluate branching of the fetal vasculature in the E12.5 and E13.5 placentas, a single midline cross section of each placenta at the level of the umbilical cord was immunostained with an anti-CD31 antibody (see below), followed by the manual counting of individual profiles of CD31-stained vessels within the placental labyrinth.
Immunohistochemistry
Antigens from 5 µm paraffin sections were retrieved by incubation for 10 min at 100°C with 1 mM EDTA, pH 8.0 for CD31 staining, or by incubation for 20 min at 100°C in 0.01 M sodium citrate buffer, pH 6.0, for all other antigens. The sections were blocked with 2% bovine serum albumin (fraction V, MP Biomedicals, Solon, OH) in phosphate-buffered saline (PBS), and incubated overnight at 4°C with 2 ug/ml rabbit anti-human CD31 (Santa Cruz Biotechnology, Santa Cruz, CA), or sheep anti-human matriptase (R&D Systems, Minneapolis, MN), primary antibodies. Bound antibodies were visualized using biotin-conjugated anti-rabbit, or -sheep secondary antibodies (Vector Laboratories, Burlingame, CA) and a Vectastain ABC kit (Vector Laboratories) using 3,3′-diaminobenzidine as the substrate (Sigma-Aldrich, St. Louis, MO). All microscopic images were acquired on an Olympus BX40 microscope using an Olympus DP70 digital camera system (Olympus, Melville, NY).
Protein extraction from mouse embryonic and newborn tissues
Placentas were extracted from embryos at E12.5 or E13.5. The embryonic portion of each placenta was manually separated from maternal decidua using a dissection microscope. The tissues were then homogenized in ice-cold 62.5 mM Tris/HCl, pH 6.8; 2% SDS; 10% glycerol buffer supplemented with 1× protease inhibitor cocktail (Sigma-Aldrich) and incubated on ice for 10 min. The lysates were centrifuged for 10 min at 20,000 g at 4°C to remove tissue debris and the supernatant was used for further analysis as described below.
Western blotting
The protein concentration in cleared lysates from embryonic, placental, and newborn tissues was determined by BCA assay (Pierce, Rockford, IL). 80 µg of total protein was loaded on 4–12% reducing SDS-PAGE and analyzed by Western blotting using a polyclonal sheep anti-human matriptase (R&D Systems), mouse anti-cow desmoglein 1 and 2 (Fitzgerald Industries International, Acton, MA), mouse anti-human Gcm1 (Abcam, Cambridge, MA), rabbit anti-human syncytinA (SantaCruz Biotechnology), rabbit anti-human pan-cadherin, rabbit anti-human GAPDH (both Cell Signaling Technology, Danvers, MA), rabbit anti-human claudin-1, and mouse anti-human claudin-2 (both Invitrogen, Carlsbad, CA) primary antibodies, and goat anti-mouse, mouse anti-rabbit (both DakoCytomation), or donkey anti-sheep (Sigma-Aldrich) secondary antibodies conjugated to alkaline phosphatase. Alkaline phosphatase activity was then visualized using nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate substrates. Where indicated, protein signal quantification was performed using ImageJ 1.46r software.
β-galactosidase staining
Pregnant female mice from breeding pairs heterozygous β-galactosidase-tagged F2rl1 knock-in mice were euthanized at E12.5 by cervical dislocation. Embryos were extracted by Caesarian section, and the individual embryos and placentas were dissected and placed in 2% PFA with 0.2% glutaraldehyde. Fixed tissues were stained overnight at room temperature in 1 mg/ml X-gal, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM magnesium chloride and 0.02% NP-40 in PBS. The tissues were post-fixed for 16 h in 4% PFA, embedded in paraffin, and sectioned. The sections were counterstained with nuclear fast red. All microscopic images were acquired on a Zeiss AxioImager Z1 light microscope (Carl Zeiss AG, Gottingen, Germany) using a Qicam FAST1394 digital camera from Qimaging.
Placental transport and feto-maternal barrier assays
100 µl of PBS containing 1 uCi of 3-O-[methyl-14C]-D-glucose or [carboxyl-14C]-inulin (both Perkin-Elmer, Hanover, MD) was injected into the tail vein of pregnant female mice from St14;F2rl1 double-heterozygous breeding pairs at 12.5 or 13.5 days of gestation. The mice were euthanized two min later by CO2 inhalation, the embryos were extracted as described above, weighted, and lysed overnight at 60°C in 500 µl of 2% potassium hydroxide solution. The lysate was then neutralized with 50 µl of 14 M hydrochloric acid, mixed with 10 ml of ScintiSafe scintillation liquid (Fisher Scientific, Pittsburg, PA), and [14C] activity was measured using Beckman LS6000IC scintillation counter (Beckman Coulter, Brea, CA). To evaluate the differences in placental function between the individual embryos, the activity was normalized to gram of embryonic tissue.
Statistical analysis
The survival of newborn and pre-weaning mice from PAR-2/matriptase double heterozygous or PAR-2/prostasin double heterozygous breeding pairs was statistically evaluated by using chi-square analysis of the observed versus the expected distribution of mice wildtype, heterozygous, and deficient for PAR-2 (F2rl1+/+; F2rl1+/−; and F2rl1−/−, respectively) among living offspring carrying two (St14+/+ or Prss8+/+), one (St14+/− or Prss8+/−), or no functional alleles (St14−/− or Prss8−/−) of the gene encoding the corresponding protease.
To evaluate the effect of matriptase or prostasin deficiency on the embryonic survival of PAR-2-deficient mice, chi-square analysis was performed on the observed versus the expected distribution of protease-expressing (St14+/+ and St14+/−, or Prss8+/+ and Prss8+/−, respectively) and protease-deficient (St14−/− or Prss8−/−) animals among the PAR-2-deficient (F2rl1−/−) embryos extracted at different embryonic stages, as indicated in the text.
Morphometric parameters of the placental development were analyzed using tissues from at least five control and five F2rl1−/−;St14−/− double-deficient animals and the observed values were statistically evaluated using a two-sample Student's t-test, two-tailed.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. RossantJ, CrossJC (2001) Placental development: lessons from mouse mutants. Nature reviews Genetics 2 : 538–548.
2. WatsonED, CrossJC (2005) Development of structures and transport functions in the mouse placenta. Physiology 20 : 180–193.
3. SandoviciI, HoelleK, AngioliniE, ConstanciaM (2012) Placental adaptations to the maternal-fetal environment: implications for fetal growth and developmental programming. Reproductive biomedicine online 25 : 68–89.
4. PreslandRB, JurevicRJ (2002) Making sense of the epithelial barrier: what molecular biology and genetics tell us about the functions of oral mucosal and epidermal tissues. Journal of dental education 66 : 564–574.
5. MarchiandoAM, GrahamWV, TurnerJR (2010) Epithelial barriers in homeostasis and disease. Annual review of pathology 5 : 119–144.
6. GuillotC, LecuitT (2013) Mechanics of epithelial tissue homeostasis and morphogenesis. Science 340 : 1185–1189.
7. LiuZ, ShiC, YangJ, ZhangP, MaY, et al. (2011) Molecular regulation of the intestinal epithelial barrier: implication in human diseases. Frontiers in bioscience 16 : 2903–2909.
8. RobbinsJR, BakardjievAI (2012) Pathogens and the placental fortress. Current opinion in microbiology 15 : 36–43.
9. BeerAE, SioJO (1982) Placenta as an immunological barrier. Biology of reproduction 26 : 15–27.
10. Delorme-AxfordE, BayerA, SadovskyY, CoyneCB (2013) Autophagy as a mechanism of antiviral defense at the maternal-fetal interface. Autophagy 9 : 2173–4.
11. RiquelmeG (2009) Placental chloride channels: a review. Placenta 30 : 659–669.
12. StulcJ (1997) Placental transfer of inorganic ions and water. Physiological reviews 77 : 805–836.
13. Basel-VanagaiteL, AttiaR, Ishida-YamamotoA, RainshteinL, Ben AmitaiD, et al. (2007) Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. American journal of human genetics 80 : 467–477.
14. AlefT, TorresS, HausserI, MetzeD, TursenU, et al. (2009) Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. The Journal of investigative dermatology 129 : 862–869.
15. SzaboR, BuggeTH (2011) Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annual review of cell and developmental biology 27 : 213–235.
16. ListK, HaudenschildCC, SzaboR, ChenW, WahlSM, et al. (2002) Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 21 : 3765–3779.
17. ListK, SzaboR, WertzPW, SegreJ, HaudenschildCC, et al. (2003) Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. The Journal of cell biology 163 : 901–910.
18. ListK, KosaP, SzaboR, BeyAL, WangCB, et al. (2009) Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. The American journal of pathology 175 : 1453–1463.
19. SzaboR, MolinoloA, ListK, BuggeTH (2007) Matriptase inhibition by hepatocyte growth factor activator inhibitor-1 is essential for placental development. Oncogene 26 : 1546–1556.
20. SzaboR, HobsonJP, ChristophK, KosaP, ListK, et al. (2009) Regulation of cell surface protease matriptase by HAI2 is essential for placental development, neural tube closure and embryonic survival in mice. Development 136 : 2653–2663.
21. VuTK, HungDT, WheatonVI, CoughlinSR (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64 : 1057–1068.
22. SohUJ, DoresMR, ChenB, TrejoJ (2010) Signal transduction by protease-activated receptors. British journal of pharmacology 160 : 191–203.
23. TraynelisSF, TrejoJ (2007) Protease-activated receptor signaling: new roles and regulatory mechanisms. Current opinion in hematology 14 : 230–235.
24. ConnollyAJ, IshiharaH, KahnML, FareseRVJr, CoughlinSR (1996) Role of the thrombin receptor in development and evidence for a second receptor. Nature 381 : 516–519.
25. DamianoBP, CheungWM, SantulliRJ, Fung-LeungWP, NgoK, et al. (1999) Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. The Journal of pharmacology and experimental therapeutics 288 : 671–678.
26. CamererE, BarkerA, DuongDN, GanesanR, KataokaH, et al. (2010) Local protease signaling contributes to neural tube closure in the mouse embryo. Developmental cell 18 : 25–38.
27. FriisS, Uzzun SalesK, GodiksenS, PetersDE, LinCY, et al. (2013) A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. J Biol Chem 288 : 19028–19039.
28. TakeuchiT, HarrisJL, HuangW, YanKW, CoughlinSR, et al. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem 275 : 26333–26342.
29. SzaboR, Uzzun SalesK, KosaP, ShyloNA, GodiksenS, et al. (2012) Reduced prostasin (CAP1/PRSS8) activity eliminates HAI-1 and HAI-2 deficiency-associated developmental defects by preventing matriptase activation. PLoS genetics 8: e1002937.
30. Diez-RouxG, BanfiS, SultanM, GeffersL, AnandS, et al. (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9: e1000582.
31. CoppAJ (1995) Death before birth: clues from gene knockouts and mutations. Trends in genetics: TIG 11 : 87–93.
32. CamererE, KataokaH, KahnM, LeaseK, CoughlinSR (2002) Genetic evidence that protease-activated receptors mediate factor Xa signaling in endothelial cells. J Biol Chem 277 : 16081–16087.
33. TallquistMD, SorianoP (2000) Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26 : 113–115.
34. DilworthMR, SibleyCP (2013) Review: Transport across the placenta of mice and women. Placenta 34 Suppl: S34–39.
35. CoanPM, AngioliniE, SandoviciI, BurtonGJ, ConstanciaM, et al. (2008) Adaptations in placental nutrient transfer capacity to meet fetal growth demands depend on placental size in mice. The Journal of physiology 586 : 4567–4576.
36. Wright C, Sibley C.P. (2011) Placental Transfer in Health and Disease; Kay H, Nelson, D.M., Wang, Y., editor: Blackwell Publishing Ltd.
37. MiS, LeeX, LiX, VeldmanGM, FinnertyH, et al. (2000) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403 : 785–789.
38. SchreiberJ, Riethmacher-SonnenbergE, RiethmacherD, TuerkEE, EnderichJ, et al. (2000) Placental failure in mice lacking the mammalian homolog of glial cells missing, GCMa. Mol Cell Biol 20 : 2466–2474.
39. GluckmanPD, HansonMA, CooperC, ThornburgKL (2008) Effect of in utero and early-life conditions on adult health and disease. The New England journal of medicine 359 : 61–73.
40. FanB, BrennanJ, GrantD, PealeF, RangellL, et al. (2007) Hepatocyte growth factor activator inhibitor-1 (HAI-1) is essential for the integrity of basement membranes in the developing placental labyrinth. Dev Biol 303 : 222–230.
41. CenacN, CoelhoAM, NguyenC, ComptonS, Andrade-GordonP, et al. (2002) Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. The American journal of pathology 161 : 1903–1915.
42. HachemJP, HoubenE, CrumrineD, ManMQ, SchurerN, et al. (2006) Serine protease signaling of epidermal permeability barrier homeostasis. The Journal of investigative dermatology 126 : 2074–2086.
43. ChinAC, LeeWY, NusratA, VergnolleN, ParkosCA (2008) Neutrophil-mediated activation of epithelial protease-activated receptors-1 and -2 regulates barrier function and transepithelial migration. Journal of immunology 181 : 5702–5710.
44. SendoT, SumimuraT, ItohY, GoromaruT, AkiK, et al. (2003) Involvement of proteinase-activated receptor-2 in mast cell tryptase-induced barrier dysfunction in bovine aortic endothelial cells. Cellular signalling 15 : 773–781.
45. GuY, GroomeLJ, AlexanderJS, WangY (2012) PAR-2 triggers placenta-derived protease-induced altered VE-cadherin reorganization at endothelial junctions in preeclampsia. Placenta 33 : 803–809.
46. FuruseM, HataM, FuruseK, YoshidaY, HaratakeA, et al. (2002) Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 156 : 1099–1111.
47. BuzzaMS, Netzel-ArnettS, Shea-DonohueT, ZhaoA, LinCY, et al. (2010) Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci U S A 107 : 4200–4205.
48. DeryO, CorveraCU, SteinhoffM, BunnettNW (1998) Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. The American journal of physiology 274: C1429–1452.
49. RamsayAJ, ReidJC, AdamsMN, SamaratungaH, DongY, et al. (2008) Prostatic trypsin-like kallikrein-related peptidases (KLKs) and other prostate-expressed tryptic proteinases as regulators of signalling via proteinase-activated receptors (PARs). Biological chemistry 389 : 653–668.
50. GrimseyN, SotoAG, TrejoJ (2011) Regulation of protease-activated receptor signaling by post-translational modifications. IUBMB life 63 : 403–411.
51. SzaboR, RasmussenAL, MoyerAB, KosaP, SchaferJM, et al. (2011) c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene 30 : 2003–2016.
52. KimC, LeeHS, LeeD, LeeSD, ChoEG, et al. (2011) Epithin/PRSS14 proteolytically regulates angiopoietin receptor Tie2 during transendothelial migration. Blood 117 : 1415–1424.
53. ChenM, ChenLM, LinCY, ChaiKX (2008) The epidermal growth factor receptor (EGFR) is proteolytically modified by the Matriptase-Prostasin serine protease cascade in cultured epithelial cells. Biochim Biophys Acta 1783 : 896–903.
54. JinX, YagiM, AkiyamaN, HirosakiT, HigashiS, et al. (2006) Matriptase activates stromelysin (MMP-3) and promotes tumor growth and angiogenesis. Cancer Sci 97 : 1327–1334.
55. AhmedS, JinX, YagiM, YasudaC, SatoY, et al. (2006) Identification of membrane-bound serine proteinase matriptase as processing enzyme of insulin-like growth factor binding protein-related protein-1 (IGFBP-rP1/angiomodulin/mac25). Febs J 273 : 615–627.
56. BhattAS, Erdjument-BromageH, TempstP, CraikCS, MoasserMM (2005) Adhesion signaling by a novel mitotic substrate of src kinases. Oncogene 24 : 5333–5343.
57. SuzukiM, KobayashiH, KanayamaN, SagaY, SuzukiM, et al. (2004) Inhibition of tumor invasion by genomic down-regulation of matriptase through suppression of activation of receptor-bound pro-urokinase. J Biol Chem 279 : 14899–14908.
58. KangJY, Dolled-FilhartM, OcalIT, SinghB, LinCY, et al. (2003) Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer Res 63 : 1101–1105.
59. LeeSL, DicksonRB, LinCY (2000) Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem 275 : 36720–36725.
60. UstachCV, HuangW, Conley-LaCombMK, LinCY, CheM, et al. (2010) A novel signaling axis of matriptase/PDGF-D/ss-PDGFR in human prostate cancer. Cancer Res 70 : 9631–9640.
61. BergumC, ZorattiG, BoernerJ, ListK (2012) Strong expression association between matriptase and its substrate prostasin in breast cancer. J Cell Physiol 227 : 1604–1609.
62. SalesKU, MasedunskasA, BeyAL, RasmussenAL, WeigertR, et al. (2010) Matriptase initiates activation of epidermal pro-kallikrein and disease onset in a mouse model of Netherton syndrome. Nature genetics 42 : 676–683.
63. SchmidlinF, AmadesiS, DabbaghK, LewisDE, KnottP, et al. (2002) Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol 169 : 5315–5321.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 7
Nejčtenější v tomto čísle
- Wnt Signaling Interacts with Bmp and Edn1 to Regulate Dorsal-Ventral Patterning and Growth of the Craniofacial Skeleton
- Novel Approach Identifies SNPs in and with Evidence for Parent-of-Origin Effect on Body Mass Index
- Hypoxia Adaptations in the Grey Wolf () from Qinghai-Tibet Plateau
- DNA Topoisomerase 1α Promotes Transcriptional Silencing of Transposable Elements through DNA Methylation and Histone Lysine 9 Dimethylation in