
Discovery of CTCF-Sensitive Cis-Spliced Fusion RNAs between Adjacent Genes in Human Prostate Cells
Genes are considered the units of hereditary information; thus, neither genes nor their encoded products are expected to mingle with each other unless in some disease situations. However, the genes are not alone in the genome. Genes have neighbors, some close, some far. With RNA-seq, many fusion RNAs involving neighboring genes are being identified. However, little is done to validate and characterize the fusion RNAs. Using one prostate cell line and a discovery pipeline for cis-splicing between adjacent genes (cis-SAGe), we found 16 new such events. We then developed a set of rules based on the characteristics of these fusion RNAs, and applied them to 20 random neighboring gene pairs. Four turned out to be true. The majority of the fusions are found in cancer cells, as well as in non-cancer cells. These results suggest that the genes are “leaky”, and the fusions are not limited to cancer cells.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1005001
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005001
Summary
Genes are considered the units of hereditary information; thus, neither genes nor their encoded products are expected to mingle with each other unless in some disease situations. However, the genes are not alone in the genome. Genes have neighbors, some close, some far. With RNA-seq, many fusion RNAs involving neighboring genes are being identified. However, little is done to validate and characterize the fusion RNAs. Using one prostate cell line and a discovery pipeline for cis-splicing between adjacent genes (cis-SAGe), we found 16 new such events. We then developed a set of rules based on the characteristics of these fusion RNAs, and applied them to 20 random neighboring gene pairs. Four turned out to be true. The majority of the fusions are found in cancer cells, as well as in non-cancer cells. These results suggest that the genes are “leaky”, and the fusions are not limited to cancer cells.
Introduction
The traditional thinking is that gene fusions and their fusion products are caused by chromosomal rearrangement at the DNA level. However, a few isolated reports of RNA trans-splicing provide support for additional mechanisms for fusion RNA production in humans [1–4]. In prostate cancer, the SLC45A3-ELK4 fusion has recently gained attention because of its biomarker potential [5–7]. Interestingly, the fusion RNA is neither a product of chromosomal rearrangement, nor is it generated via RNA trans-splicing. With the two parental genes located next to each other, and transcribing the same strand, SLC45A3-ELK4 is produced by a mechanism of read-through, or cis-splicing between adjacent genes (cis-SAGe) [7,8].
Traditionally, true cis-SAGe events have been considered very rare in mammalian systems. In recent years, several studies using the EST database and RNA-sequencing approaches have identified fusion RNAs involving neighboring genes, which were named “transcription-mediated gene fusions”, “tandem chimerism” and “conjoined genes” by various groups [9,10]. However, no effort has been made to characterize these fusions in terms of their generating mechanism. Theoretically, fusions involving same-strand neighboring genes could be products of interstitial DNA deletion, RNA trans-splicing, or cis-SAGe. As a result, it is still unclear whether other examples of cis-SAGe truly exist, and how widespread the phenomenon actually is.
In this study, we first noticed a high percentage of potential cis-SAGe fusion RNAs in prostate cancer, as well as in their matching normal cases. We then used LNCaP cells as a model, and by integrating RNA sequencing data and CTCF silencing, we identified close to one hundred fusions. Bioinformatics analyses and experimental evidence support 16 of these fusions as true cis-SAGe events that are similar to SLC45A3-ELK4. By characterizing the 16 parental gene pairs, we developed a set of rules, which were then used to identify novel fusion RNAs between neighboring genes. Interestingly, most of these fusion RNAs are also present in non-cancerous cells, indicating that cis-SAGe is not unique to cancer cells.
Results
Transcriptome analyses revealed high percentages of fusion RNAs involving neighboring genes
RNA-seq and various software tools have been used to identify fusion events in prostate cancer [5,11–13]. We used SOAPfuse [14] to analyze RNA-sequencing data [15] from 14 pairs of matched prostate normal and cancer samples. Over 300 fusion events were observed in both the normal and cancer groups. We categorized these fusion RNAs into three groups: fusions involving parental genes located on different chromosomes (INTERCHR), fusions involving neighboring genes transcribing the same strand (INTRACHR-SS-0GAP), and other fusions with parental genes on the same chromosome (INTRACHR-OTHER) (Fig. 1A). On average, around 30% of chimeric RNAs are in the category of INTRACHR-SS-0GAP (33% in cancer, 29% in normal groups), candidates for cis-SAGe. Interestingly, the normal and cancer pairs tend to have similar percentages of these fusions (ranging from 9% to 80%) (Pearson’s R = 0.6) (Fig. 1B). As these normal samples are adjacent histologically “normal” tissues from prostate cancer patients, they are not true non-neoplasia. We then analyzed RNA sequencing data from four non-cancer donors’ prostate tissues [16]. The percentage of INTRA-SS-0GAP fusions were also close to 30% (33%, 33%, 25% and 38%) (S1 Fig.). These findings suggest that cis-SAGe may be a more frequent event, and that it occurs both in normal and cancer cells. To gain a better understanding of the process, we decided to use the LNCaP prostate cancer cell line as a model.

Transcriptome sequencing of human prostate cancer LNCaP cells
Previously, we showed an inverse correlation between the fusion RNA SLC45A3-ELK4 expression and transcription factor CTCF binding to the insulators located at the two parental gene boundaries [7]. Consistent with a negative role in regulating cis-SAGe, silencing CTCF resulted in an induction of SLC45A3-ELK4 fusion expression in LNCaP cells (Fig. 1C). In contrast, CTCF has been shown to facilitate the juxtaposition of interchromosomal regions [17], thus facilitating trans-splicing events. Indeed, silencing CTCF resulted in a down-regulation of a trans-spliced fusion RNA JAZF1-JJAZ1 [7], suggesting that changes in CTCF expression can have opposite effects on a fusion RNA depending on its generating mechanism. We reasoned that more cis-SAGe fusions could be uncovered based on their response to CTCF silencing.
RNA extracted from LNCaP cells transfected with siRNA against CTCF (siCTCF), or negative control siRNA (si-) were processed, and sequenced by two different companies using the Illumina Hi-seq platform to generate two output sequences: paired-end 50-nucleotide and 101-nucleotide in read length (S1 Table.). Nearly 100 million and 50 million raw reads were yielded from each sample respectively. We used FastQC to confirm the quality of the raw fastq sequencing data, and SOAPfuse software to detect fusion transcripts. To identify cis-SAGe events, we applied the following criteria outlined in Fig. 1D: 1) fusion RNAs involving neighboring genes transcribing the same strand (INTRACHR-SS-0GAP); 2) absence of interstitial DNA deletion in-between the two fused exons; 3) presence of CTCF binding site at gene boundaries; 4) chimeric RNAs induced by siCTCF; and 5) presence of intergenic transcript.
Classifications of the fusion RNAs
In order to identify more fusion events, we combined the reads from 50 bp read-length with those of 101 bp read-length. A total of 95 fusions (64 unique parental gene pairs) were identified from the si—and siCTCF samples (Fig. 2A, S2 Fig., and S2 Table.). Illustrated by Circos plots, it is obvious that the majority of the fusions are intrachromosomal (Fig. 2B). Out of the 95 fusions, 56 are composed of exons belonging to the INTRACHR-SS-0GAP category. There are only 13 interchromosomal fusions. The remaining 26 are fusions joining genes on the same chromosome on different strands (DS), or with more than one gene in between (INTRACHR-OTHER) (Fig. 2C). cBioPortal query on TP53 showed only 14.7% alteration in TCGA in the prostate cancer set. Consistently, LNCaP cells contain wild type TP53 [18]. This could be the reason for the relatively lower incidence of interchromosomal fusions.

We chose 71 fusions (including 48 INTRACHR-SS-0GAP, 12 INTERCHR and 11 INTRACHR-OTHER) to validate by RT-PCR. The PCR products were gel-purified, and sequenced by Sanger sequencing. A total of 62 were confirmed (one example of each category in S3 Fig., S4 Fig., and S5 Fig.). Interestingly, a higher percentage of the INTRACHR-SS-0GAP was confirmed by this method (95.8%), compared with 90.9% for INTRACHR-OTHER, and 50% for INTERCHR. We then focused on the 46 INTRACHR-SS-0GAP fusions that are composed of unique parental gene pairs as candidate cis-SAGe fusions.
Further validation for cis-SAGe fusion RNAs
Immediate neighboring genes. When viewed on the hg19 Assembly of UCSC Genome Browser, we found that 8 out of the 46 candidate pairs have other gene transcripts in between (one example in S6A Fig.), raising concerns about whether or not they are truly immediate neighboring genes (Table 1). To avoid complications, we then selected the remaining 38 pairs to continue the validation.

Absence of interstitial deletion. As one common mechanism for generating INTRACHR-SS fusions is the deletion of the sequence in-between the fused exons, we examined the copy number variation in LNCaP cells deposited on GEO database (GSM947411). We did not find convincing evidence for interstitial deletions of the genomic DNA in-between the fused exons of any of the candidates (Fig. 3A and Table 1), supporting mechanisms other than chromosomal deletion.
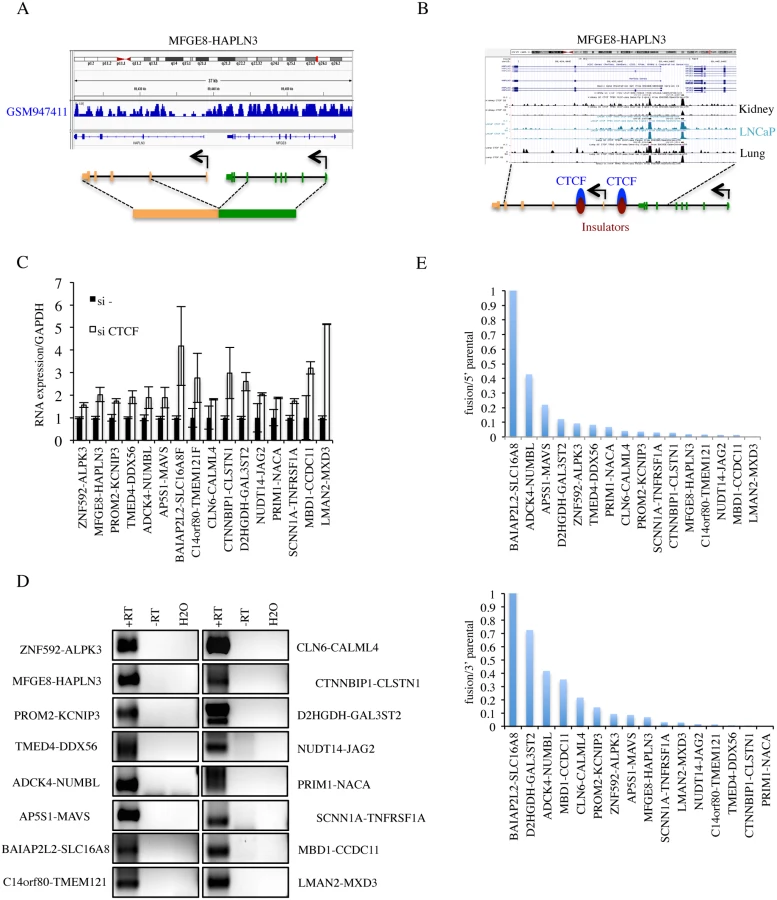
CTCF binding in between parental genes. We used two methods to evaluate the evidence for CTCF binding in-between the 38 pairs of parental genes: 1) visual examination of CTCF binding sites generated by ENCODE on the UCSC genome browser (Fig. 3B and Table 1), and 2) searching for CTCF binding site on the Insulator Database, CTCFBSDB. For fusions CHCHD10-VPREB3F, EIF3K-ACTN4, DTD2-HEATR5A, and VAMP1-CD27-AS1 (S6B Fig.), there was no evidence of CTCF binding in-between the parental genes. Among these, CHCHD10-VPREB3F and VAMP1-CD27-AS1 were eliminated based on the “neighboring genes” criteria. After the above three steps of elimination, 36 fusions that have at least one CTCF binding site in-between the parental genes were left for further validation.
Induced by siCTCF. To evaluate the induction by CTCF silencing, we performed qRT-PCR for the remaining 36 candidates (examples shown in Fig. 3C). We found that 16 fusions could be induced by silencing CTCF, 5 down-regulated and 16 unchanged (fold change greater than or equal to 1.5) (S7 Fig. and Table 1).
Detection of intergenic transcripts. cis-SAGe is essentially alternative splicing between the exons of neighboring genes. Therefore, transcripts in-between the two genes that are involved in cis-SAGe should be present. To further confirm the 16 candidate fusions that are up-regulated with siCTCF, we used RT-PCR to detect intergenic transcripts. DNase treatment eliminated most, if not all, DNA contaminates in the RNA samples manifested by the absence of signal in the “no reverse transcriptase” control (examples in S8 Fig.). Intergenic transcripts were detected in all 16 candidates. To further confirm the generating mechanism, we used antisense primers of downstream parental genes for reverse transcription, and detected by PCR all 16 intergenic transcripts (Fig. 3D). These results argue against the possibility that the intergenic transcripts detected are produced by antisense transcripts. For one fusion, CLN6-CALML4, we could also amplify and sequence confirm the primary transcript spanning from the last exon of CLN6 to the first exon of CALML4 (S9 Fig.).
We hypothesized that these intergenic transcripts are more likely to be induced by siCTCF like the cis-SAGe fusions. Indeed, qRT-PCR results showed that 12 out of the 16 had obvious induction when CTCF was silenced (S10 Fig.).
Relative expression of the fusion to parental genes. To gain insight on the relative level of the fusion RNAs to the parental genes expression, we converted the number of junction reads of the fusion RNAs to FKPM (fragments per kilobase of transcript per million fragments sequenced). Five fusions contribute to a significant portion of the total 5′ parental gene expression (>10%). Similarly, seven fusions are above 10% of the total expression of 3′ parental genes (Fig. 3E).
Characterization of the cis-SAGe fusions
We noticed that the FKPMs of these 32 parental genes range from 0 to 122 (Fig. 4A). Considering the highest FKPM in the RNA-seq is above 2800, it is thus unlikely that the cis-SAGe fusions are non-specific side products, all due to overwhelming quantities of parental gene transcripts. The expression of several fusions contributes significant portions of parental genes (Fig. 3E), further supporting the argument. In addition, the fact that all of these 16 fusion RNAs were induced by siCTCF, yet many parental genes were not, suggests that the cis-SAGe events are actively regulated by additional mechanisms other than their parental genes’ expression.

Previous analyses reported that in the human genome, roughly 25,000 genes are composed of over 200,000 exons, with a typical gene containing 8.7 exons, and an average exon length of 174.5bp [19]. We used Genes and Gene Prediction Tracks from hg19 Assembly to download gene features, including exon number. The density of genes containing certain numbers of exons are plotted in Fig. 4B. In the genome, a high percent of genes have a single exon (12%). Another peak represents genes with about five exons (~ 8%). The overall distribution of exon numbers of the cis-SAGe parental genes is different from this whole genome analysis, especially for the 5′ parental genes (p = 2.445e-06) (S11 Fig.). Notably, none of the cis-SAGe parental genes are single-exonic.
In order to investigate whether the distance between the neighboring genes plays a role in the generation of cis-SAGe fusions, we analyzed the distribution of the distances between neighboring genes transcribing the same strand within the genome. The median distance between such neighboring genes in the genome is around 54 Kb (Fig. 4C). We found a strong statistical difference between the 16 pairs of cis-SAGe parental genes and the whole genome analyses, as the longest distance out of the 16 pairs is less than 30kb (p = 2.703e-05).
It is known that longer introns tend to facilitate alternative splicing, as RNA pol II has a higher chance of pausing on longer introns [20]. We compared the intron size involved in cis-SAGe fusions (for 5′ genes, the intron after the fused exon; for 3′ genes, the intron before the fused exon) with the hg19 genome (Fig. 4D). No statistical difference was noted (p = 0.7769, and 0.1353 for 5′ and 3′ genes respectively).
We then used CisFinder [21] to identify potential DNA motifs in the fused exons and immediate introns. No consistent motif in the introns, or in the 5′ and 3′ exons was found in the 16 pairs.
To determine whether a certain exon is favored for a cis-SAGe event, the positions of the exons immediately next to the fusion junction site, relative to the parental genes were plotted (Fig. 4E). No obvious “hot exonic position” was observed for the 5′ parental genes, but strikingly, more than 68% of fusion junctions have been observed occurring at the second exon of the 3′ parental genes (11 out of 16) (Fig. 4E). If the participation of the exons are totally random, the chances of having 11 out of the 16 fusions involving the 2nd exon of the 3′ gene is very low, illustrated by a simulation test (p = 1.718378e-05) (S12 Fig.). On the other hand, the bias towards shorter intergenic distance shown earlier does support the frequent usage of the 2nd exon, as it is the closest exon that has a splicing acceptor sequence. However, arguing against a totally non-specific model, there are three fusions that use the first exon of their 3′ parental genes (TMED4-DDX56, NUDT14-JAG2, and PRIM1-NACA), and no common splicing acceptor site (AG or AC), which account for over 99.98% of known splicings [22], was found in the sequence before these first exons.
Considering that the distance between the spliced exons may be one key factor, we went back to examine the distribution of 5′ exon positions counting backward. Impressively, we found that 50% of the fusions actually used the second-to-last exon of the 5′ parental genes (p<0.001)(Fig. 4E). When the top combinations of 5′ and 3′ exon usage were plotted, we noticed a strong bias towards the “2–2”, that is “second-to-last” of the 5′ gene fused to the second exon of the 3′ gene (Fig. 4F).
Expression of the cis-SAGe fusion RNAs in prostate cell lines and clinical cases
Out of the 16 fusion RNAs, five fusions have their junction sites fall into the UTR region, thus not changing protein-coding sequence (NR) (Fig. 5A). In six fusions, the protein coding sequence of the 3′ genes use a different reading frame than the 5′ gene (out-of-frame). In the remaining five, the reading frame of the 3′ gene is the same as the 5′ gene (in-frame). The number of in-frame fusions versus. that of the out-of-frame fusions (5 vs. 6) is higher than random (1 vs. 2), but the small number prevents further generalization at this stage. We then fractionated LNCaP cells, and measured the relative amount of these 16 fusion RNAs in the nuclear versus cytoplasmic fractions. Interestingly, the NR and out-of-frame fusions (10/11) were enriched in the nuclear fraction (nuclear/cytoplasma >1), indicating potential non-coding roles. In contrast, four out of five in-frame fusions showed more or equal amounts in the cytoplasmic fraction, possibly functioning as traditional protein coding mRNAs (Fig. 5A).

For one fusion RNA, ADCK4-NUMBL, we were able to obtain two siRNAs that specifically target the fusion transcript (Fig. 5B). They both significantly knocked down the fusion RNA, and had no obvious effect on the ADCK4 parental transcript. si-AN2 caused a slight reduction of the NUMBL parental transcript, whereas si-AN1 even caused an induction of NUMBL. We didn’t notice any obvious change in cellular proliferation rate, but cell motility was significantly reduced when LNCaP cells were transfected with these siRNAs (Fig. 5C)
To further validate these cis-SAGe fusions in other systems, we performed RT-PCR for the final 16 fusions in LNCaP, PC3, a castration-resistant prostate cancer cell line, and RWPE-1, a non-cancer prostate epithelial cell line (Fig. 5D). The majority of the fusion RNAs can be detected in two or more lines. ZNF592-ALPK3 and LMAN2-MXD3 were only detectable in RWPE-1 cells, but hardly detectable in the two cancer cell lines. In contrast, three fusions, PROM2-KCNIP3, BAIAP2L2-SLC16A8, and D2HGDH-GAL3ST2 had higher expression in the two cancer cell lines than in RWPE-1 cells.
We also performed STAR alignment [23] of the RNA-seq data from 14 clinical prostate cancer cases [15]. With IGV analyses [24], 11 out of the 16 cis-SAGe fusions were also found in this dataset (Fig. 5E). Using this method, we found 12 fusions in the matched normal group. Of note, most fusions were not cancer specific.
Identification of novel cis-SAGe candidate RNAs
Next, we wondered whether or not the rules generalized from the 16 fusions could lead us to discover novel cis-SAGe fusions. We summarized the following four rules for cis-SAGe fusions (Fig. 6A): 1) neighboring genes transcribing the same strand; 2) with an intergenic distance less than 30kb; 3) 5′ genes actively transcribing and 4) favoring a configuration of the second-to-last exon in the 5′ fused to the second exon in the 3′ parental genes. According to hg19 genome assembly, there are 9478 pairs of neighboring genes transcribing in the same strand, and that are within a 30kb distance. We then downloaded RNA-seq data for prostate samples [25], and selected 20 random pairs with the 5′ genes expressed in prostate (FPKM>1). Using primers annealing to the second-to-last exon in the 5′ and the second exon in the 3′ gene, we successfully amplified four pairs in LNCaP cells. Sanger sequencing revealed an exact exon-exon splicing pattern (Fig. 6B). All four pairs were also detected in RWPE-1, or LHS, both non-cancer prostate epithelial cell lines (Fig. 6C).

Discussion
Traditionally, fusion RNAs containing exons of neighboring genes have been considered rare in mammalian cells, with only a handful of examples experimentally identified [26]. However, in our initial analyses of paired—end RNA-sequencing data from the 14 pairs of normal and prostate cancer cases, we found a high percentage of fusion RNAs that are candidates for cis-SAGe (averaging around 30%). This observation is consistent with other in silico analyses and sequencing efforts [9,10,27]. However, the majority of the reported fusion RNAs have not been validated, and their generating mechanisms remain unknown. In fact, some other studies have attributed many such chimeras to experimental artifacts [28], raising questions about whether these fusions are even real. In our previous study, we reported SLC45A3-ELK4 as the first verified cis-SAGe event [7]. Here, we applied a set of criteria and identified 16 additional fusions that are generated by this cis-SAGe mechanism.
CTCF is a highly conserved zinc finger protein that binds to insulator sequences in the genome [29]. Insulators between the neighboring genes act as boundaries to protect a gene against the encroachment of adjacent, inactive, condensed chromatin, or against the activating influence of distal enhancers associated with other genes [30]. CTCF plays diverse regulatory functions, including transcriptional activation/repression, insulation, imprinting, and X chromosome inactivation [29]. Here we manipulated CTCF levels to enhance certain cis-SAGe events. To estimate that 25% of the fusions (16 out of a total number of 64 (unique parental gene pairs)) could be considered as cis-SAGe events may still be an underestimation. It is very likely that some cis-SAGe events are not regulated by CTCF. It is also possible that not all cis-SAGe events have CTCF binding in the experimental conditions we used, and/or are not induced by siCTCF. Thus, the study is aimed to discover CTCF-sensitive cis-SAGe events. Of note, this is also an artificial system created only to enhance some cis-SAGe signals in a cell line system. cis-SAGe is likely to be a complex event regulated by multiple factors. At this moment, the role of CTCF in regulating global cis-SAGe events in clinical prostate cancer is not clear.
It has been reported that CTCF could also facilitate the formation of chromosomal translocation by bringing distant genes into close proximity [31,32]. As we noted before, silencing CTCF resulted in a reduced expression of a trans-spliced chimera JAZF1-JJAZ1 in endometrial cells [7]. The discarded fusions for this study are likely to be candidates for RNA trans-splicing or chromosomal rearrangement, especially the ones that are down-regulated with siCTCF.
Neither statically significant trends in intron size, nor consistent motifs were found in the final 16 fusions, possibly due to the small number of the these cis-SAGe events. However, the parental genes tend to be multi-exonic, and we found a strong preference for shorter intergenic distance. This may partially explain the biased involvement of the second exon in the 3′ parental gene, and the second-to-last exon in 5′ gene. Even though this configuration only applies to about half of the final 16 fusions, we were able use this rule to discover four novel fusion RNAs in 20 randomly selected neighboring genes. Interestingly, these four fusions were also found in at least one non-cancer prostate cell line.
Traditionally, fusion RNAs were thought to be uniquely expressed in cancer cells, and sought after as ideal biomarkers. Some of the fusions we found here did show differential expression between prostate cancer cells and non-cancerous cells, and are potential biomarkers. However, the finding of many new chimeras in normal clinical samples, as well as in non-cancerous cell lines suggests that these events also happen physiologically. For cis-SAGe fusions, this means that genes are more “leaky” than we previously thought.
Materials and Methods
Cell culture, siRNA knockdown and transfection
Prostate cancer cell lines LNCaP (androgen-dependent) were grown in RPMI1640 (Hyclone) media supplemented with 10% FBS and 1% 100 x Pen/Strep (Hyclone). siRNA against luciferase gene was used as control siRNA [33]. CTCF siRNA was purchased from Invitrogen and as described before [7]. The two siRNAs against ADCK4-NUMBL targeting sequences were TCCGCCCTTGGTTTCAAAG, and GGGUCCGCCCTTGGTTTCA. siRNA transfection was carried out using Lipofectamine RNAiMAX (Life Technologies) following the manufacturer’s protocols. Cellular fractionation was carried out according to manufactures’ protocol (NE-PER Nuclear and Cytoplasmic Extraction Kit, Thermo).
Wound healing assay
LNCaP cells transfected with si-negative control, or siRNAs against ADCK4-NUMBL were cultured for 3 days to obtain 80–90% monolayer confluency. A wound was created by scraping the cells using a 10ul plastic pipette tip, and the medium was replaced with fresh medium. Images were captured immediately after the scratch and six hours later. Cell migration was qualitatively assessed by the size of the wounds at the end of the experiment.
RNA extraction and sequencing
Cells transfected with si - or siCTCF were harvested 3 days after transfection. The RNA was extracted with TRIzol reagent (Life Technologies) following the manufacturer’s instruction. To assure the high quality RNA for next generation sequencing, RNA was further cleaned using the RNeasy kit (Qiagen). The mRNA in total RNA was converted into a library of template molecules suitable for subsequent cluster generation using the reagents provided in the Illumina TruSeq TM RNA Sample Preparation Kit. Millions of unique clusters on flow cells were loaded into the Hiseq 2000 platform and processed for RNA sequencing.
Data analysis for RNA-seq data
The two samples, the negative control and siCTCF samples were sequenced by the Illumina Hi-seq platform with paired ends to reach 100 million reads with 50bp read lengths (HudsonAlpha Insitute, Huntsville, AL), or 50 million reads with 101bp read lengths (Axeq, Seoul, Korea). To check the quality of the raw data, the software FastQC was used. The deep sequencing data was mapped to Human genome version hg19, and analyzed using the SOAPfuse software [14]. The identified chimeric RNAs were presented using Circos as previously described [34]. STAR align was used to identify the final 16 fusions in the clinical prostate cancer samples downloaded from the European Bioinformatics Institute.
RT-PCR and Sanger sequencing
Fusion candidates from SOAPfuse analyzed RNA-seq data were validated at the RNA level by real-time PCR. Quantitative RT-PCR was performed using the ABI Step One Plus real time PCR system (Applied Biosystems) following the manufactures’ instructions. We designed specific primer pairs (S3 Fig.) for the fusion candidates, intergenic transcripts and randomly selected 20 pairs, with each primer targeting one parental gene. Following RT-PCR and gel electrophoresis, all purified bands were sent for Sanger sequencing.
Visualization
Multi Experiment Viewer (Mev, 4.9 version) software suite was used to generate the heat map containing the visualization of the expression levels of parental genes that are involved in the 16 fusions [35]. IGV analysis was used to visualize fusions in the clinical samples as described before [24].
Statistical analyses
For intergenic distance, exon numbers, and intron size, the Kolmogorov-Smirnov test [36] was used to decide if the distributions of the 16 pair parental genes were different from those of the whole human genome, by calculating the maximum distance between the sample and population empirical/cumulative distribution function (cdf). In each case, two hypotheses were tested, H0: the distribution of the 32 parental genes follows a normal human genome distribution versus H1: it does not follow the specified distribution.
To test if the total number of fusions use exon2 of the 3′ parental genes is statistically significant, we ran a simulation, in which 10,000 random samples of fusions for the 16 3′ parental genes’ exons were created. We then counted the total number of exon2 fusions in each simulation and plotted the results, creating an approximate binomial distribution of sample size n = 16. We then calculated the probability underneath the binomial distribution in which there are 11 or more exon2 fusions, Pr (number of exon2> = 11), and found the probability to be 1.718378e-05. Thus concluding that getting 11 exon2 fusions is significant.
Data access
The Raw and processed RNA-sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE63487.
The prostate clinical datasets from 14 patients were downloaded from EBI (European Bioinformatics Institute) (http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-567) [15].
Supporting Information
Zdroje
1. Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, et al. (1991) Scrambled exons. Cell 64 : 607–613. 1991322
2. Li H, Wang J, Mor G, Sklar J (2008) A Neoplastic Gene Fusion Mimics Trans-Splicing of RNAs in Normal Human Cells. Science 321 : 1357–1361. doi: 10.1126/science.1156725 18772439
3. Zaphiropoulos PG (2012) Genetic variations and alternative splicing: the Glioma associated oncogene 1, GLI1. Frontiers in genetics 3 : 119. doi: 10.3389/fgene.2012.00119 22833753
4. Velusamy T, Palanisamy N, Kalyana-Sundaram S, Sahasrabuddhe AA, Maher CA, et al. (2013) Recurrent reciprocal RNA chimera involving YPEL5 and PPP1CB in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America 110 : 3035–3040. doi: 10.1073/pnas.1214326110 23382248
5. Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, et al. (2009) Transcriptome sequencing to detect gene fusions in cancer. Nature 458 : 97–101. doi: 10.1038/nature07638 19136943
6. Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, et al. (2009) SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res 69 : 2734–2738. doi: 10.1158/0008-5472.CAN-08-4926 19293179
7. Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, et al. (2012) Chimeric Transcript Generated by cis-Splicing of Adjacent Genes Regulates Prostate Cancer Cell Proliferation. Cancer Discovery 2 : 598–607. doi: 10.1158/2159-8290.CD-12-0042 22719019
8. Kumar-Sinha C, Kalyana-Sundaram S, Chinnaiyan AM (2012) SLC45A3-ELK4 Chimera in Prostate Cancer: Spotlight on cis-Splicing. Cancer Discovery 2 : 582–585. doi: 10.1158/2159-8290.CD-12-0212 22787087
9. Akiva P, Toporik A, Edelheit S, Peretz Y, Diber A, et al. (2006) Transcription-mediated gene fusion in the human genome. Genome Res 16 : 30–36. 16344562
10. Nacu S, Yuan W, Kan Z, Bhatt D, Rivers CS, et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genomics 4 : 11. doi: 10.1186/1755-8794-4-11 21261984
11. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, et al. (2011) The genomic complexity of primary human prostate cancer. Nature 470 : 214–220. doi: 10.1038/nature09744 21307934
12. Beillard E, Ong SC, Giannakakis A, Guccione E, Vardy LA, et al. miR-Sens—a retroviral dual-luciferase reporter to detect microRNA activity in primary cells. RNA 18 : 1091–1100. doi: 10.1261/rna.031831.111 22417692
13. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. (2005) Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310 : 644–648. 16254181
14. Jia W, Qiu K, He M, Song P, Zhou Q, et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol 14: R12. doi: 10.1186/gb-2013-14-2-r12 23409703
15. Ren S, Peng Z, Mao JH, Yu Y, Yin C, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res 22 : 806–821. doi: 10.1038/cr.2012.30 22349460
16. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13 : 397–406. doi: 10.1074/mcp.M113.035600 24309898
17. Ong CT, Corces VG (2009) Insulators as mediators of intra - and inter-chromosomal interactions: a common evolutionary theme. Journal of biology 8 : 73. doi: 10.1186/jbiol165 19725934
18. Scott SL, Earle JD, Gumerlock PH (2003) Functional p53 increases prostate cancer cell survival after exposure to fractionated doses of ionizing radiation. Cancer Res 63 : 7190–7196. 14612513
19. Koralewski TE, Krutovsky KV Evolution of exon-intron structure and alternative splicing. PLoS One 6: e18055. doi: 10.1371/journal.pone.0018055 21464961
20. de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, et al. (2003) A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12 : 525–532. 14536091
21. Sharov AA, Ko MS (2009) Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res 16 : 261–273. doi: 10.1093/dnares/dsp014 19740934
22. Burset M, Seledtsov IA, Solovyev VV (2000) Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res 28 : 4364–4375. 11058137
23. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 : 15–21. doi: 10.1093/bioinformatics/bts635 23104886
24. Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29 : 742–749. doi: 10.1038/nbt.1914 21804560
25. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & cellular proteomics: MCP 13 : 397–406. doi: 10.1038/ncomms7071 25608249
26. Wang K, Ubriaco G, Sutherland LC (2007) RBM6-RBM5 transcription-induced chimeras are differentially expressed in tumours. BMC Genomics 8 : 348. 17908320
27. Kannan K, Wang L, Wang J, Ittmann MM, Li W, et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci U S A 108 : 9172–9177. doi: 10.1073/pnas.1100489108 21571633
28. Houseley J, Tollervey D Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One 5: e12271. doi: 10.1371/journal.pone.0012271 20805885
29. Phillips JE, Corces VG (2009) CTCF: master weaver of the genome. Cell 137 : 1194–1211. doi: 10.1016/j.cell.2009.06.001 19563753
30. Burgess-Beusse B, Farrell C, Gaszner M, Litt M, Mutskov V, et al. (2002) The insulation of genes from external enhancers and silencing chromatin. Proc Natl Acad Sci U S A 99 Suppl 4 : 16433–16437. 12154228
31. Handoko L, Xu H, Li G, Ngan CY, Chew E, et al. (2011) CTCF-mediated functional chromatin interactome in pluripotent cells. Nature genetics 43 : 630–638. doi: 10.1038/ng.857 21685913
32. Ong CT, Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nature reviews Genetics 15 : 234–246. doi: 10.1038/nrg3663 24614316
33. Machida YJ, Chen Y, Machida Y, Malhotra A, Sarkar S, et al. (2006) Targeted comparative RNA interference analysis reveals differential requirement of genes essential for cell proliferation. Mol Biol Cell 17 : 4837–4845. 16957053
34. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, et al. (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19 : 1639–1645. doi: 10.1101/gr.092759.109 19541911
35. Saeed AI, Sharov V, White J, Li J, Liang W, et al. (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34 : 374–378. 12613259
36. Nguyen CD, Carlin JB, Lee KJ Diagnosing problems with imputation models using the Kolmogorov-Smirnov test: a simulation study. BMC Med Res Methodol 13 : 144. doi: 10.1186/1471-2288-13-144 24252653
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates