
Genomics of Divergence along a Continuum of Parapatric Population Differentiation
A variety of evolutionary forces influence the genomic landscape of divergence during ecological speciation. Here we characterize the evolution of genomic divergence patterns based on 60 fully sequenced three-spined stickleback genomes, contrasting lake and river populations that differ in parasite abundance. Our comparison of the size and abundance of divergent regions in the genomes across a continuum of population differentiation suggests that selection and the hitchhiking effect on neutral sites mainly contributes to the observed heterogeneous patterns of genomic divergence. Additional divergent regions of the genome can be explained by a local reduction of gene flow. Our description of genomic divergence patterns across a continuum of population differentiation combined with an analysis of molecular signatures of evolution highlights how adaptation shapes the differentiation of sticklebacks in freshwater habitats.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004966
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004966
Summary
A variety of evolutionary forces influence the genomic landscape of divergence during ecological speciation. Here we characterize the evolution of genomic divergence patterns based on 60 fully sequenced three-spined stickleback genomes, contrasting lake and river populations that differ in parasite abundance. Our comparison of the size and abundance of divergent regions in the genomes across a continuum of population differentiation suggests that selection and the hitchhiking effect on neutral sites mainly contributes to the observed heterogeneous patterns of genomic divergence. Additional divergent regions of the genome can be explained by a local reduction of gene flow. Our description of genomic divergence patterns across a continuum of population differentiation combined with an analysis of molecular signatures of evolution highlights how adaptation shapes the differentiation of sticklebacks in freshwater habitats.
Introduction
During ecological speciation, divergence along the genome has been observed to be heterogeneous in numerous taxonomic groups [e.g., [1–4]]. Typically, the average genome-wide divergence is low, interspersed with regions of exceptional differentiation. However, studies describing divergence patterns across the genome have found regions of exceptional differentiation to be either numerous and small [4] or few and large [5, 6], the latter sometimes referred to as ‘genomic islands’. A variety of explanations have been proposed for the observed heterogeneity in genomic divergence, including stochastic processes such as genetic drift, but also deterministic mechanisms such as locus-specific reduction of gene flow in the vicinity of genes causing reproductive isolation, hitchhiking around selected variants, or variation in recombination and mutation rates [7]. Generally, genetic drift, population expansion, migration, and other demographic events affect the whole genome, whereas natural selection modified by local environmental differences impact only those regions of the genome that affect the respective phenotypes and fitness.
It is not known whether or not genomic patterns such as the variation of divergence and recombination along the genome tend to follow a predictable evolutionary trajectory as populations proceed along a speciation continuum [7]. We investigated the early phase of divergence using lake-river stickleback population pairs varying in their degree of genetic differentiation. If divergence patterns are driven by locus-specific effects of gene flow and divergent selection, the extent of divergence is expected to be more localized than widespread, in line with the “island view” [6]. These regions might hold “speciation genes” maintaining reproductive isolation between species including genes underlying a fitness reduction in hybrids [8]. Furthermore, “divergence hitchhiking”, the accumulative effect of selectively advantageous loci, predicts a positive correlation between genomic divergence and island size progression [9]. An alternative explanation posits that the lack of differentiation across most of the genome is due to shared ancestral polymorphism rather than ongoing gene flow [10, 11], whereas regions of high differentiation represent regions influenced by selection at linked sites [12]. Such a hitchhiking pattern may be caused by both advantageous (positive selection) and deleterious alleles (background selection). Therefore, if adaptation alone (assuming some degree of geographic separation) shapes the genomic landscape, population genetic processes unrelated to the extent of overall genomic differentiation govern divergence patterns. Disentangling such alternative scenarios is a crucial yet challenging step in understanding the genomics of divergence, especially in parapatry where the current and historic extent of migration and gene flow contribute to the overall genomic patterns.
We tested predictions inherent to the different scenarios explaining genomic patterns of divergence using whole-genome sequencing data of replicated population pairs of three-spined sticklebacks varying in their degree of genetic differentiation. Five population pairs were sampled from connected lakes and rivers from the United States (Us), Canada (Ca), Norway (No), and from two sites in Germany (G1 and G2; Fig. 1 and S1 Table). As ice sheets covered these regions during the last glaciation, these populations represent recent colonization events (~12 000 years ago). Both lake and river populations are derived from marine ancestors that became landlocked during de-glaciation, and in which ecotype differentiation between watersheds has occurred repeatedly. Some phenotypic traits such as feeding morphology [13], brain development [7], and parasite resistance [14] seemingly differentiated in parallel with habitat (i.e. lake and river) suggestive of local adaptation. Furthermore, experimental studies have shown evidence for local adaptation to lake and river habitats mediated by parasites [15]. Hence, contrasting the differentiation between populations from distinct ecosystems permits us to study the onset of divergence, which might eventually lead to complete reproductive isolation (i.e. speciation). Here, we scan genomic divergence patterns and evaluate differences and commonalities across a wide geographic sampling of parapatric population pairs to uncover the relative importance and interaction of evolutionary factors like drift, selection, and recombination during adaptive divergence.

Results and Discussion
Lake and river population pairs
One consistent difference between lake and river habitats is that lake fish posses a higher parasites diversity than parapatric river fish. From previous work on three-spined sticklebacks, lakes and rivers in Northern Germany are known to harbour distinct parasite communities [14, 16]. Despite the relatively low sample size for individual locations in this study (n = 12–17), this ecological difference between lakes and rivers is here confirmed on a broader geographic scale (Fig. 1). From each of the ten sampled populations, six stickleback genomes were sequenced using a combination of paired-end and mate-pair libraries on the Illumina HiSeq platform to an average genomic coverage of 26-fold (S2 Table). Instead of sequencing many individuals with low coverage, a small number of genomes per population was chosen to be sequenced to high coverage. This approach takes advantage of the greater resolution of single nucleotide polymorphisms (SNPs) and copy number variations (CNVs; evaluated in greater detail in a companion paper [17]) plus increased genotype accuracy within each individual to decipher the divergence mechanisms acting towards an apparent repeated differentiation between lake and river fish. Besides evaluating allele frequencies, the high individual sequence coverage permits us to infer haplotypes and examine recombination patterns. After stringent quality filtering, we accessed 297,437,667 bp from the 20 autosomes (380,547,835 bp). SNP density varied from 3 to 10 SNPs per kilobase (kb) within each population (S3–S4 Tables). For each of the five parapatric comparisons, pairwise genome-wide averages of divergence (FST) ranged from 0.10 to 0.28, disclosing a varying degree of differentiation in the ascending order of Us, G2, No, G1, and Ca (Table 1). The parapatric pairs emerge as repeated independent differentiation events (neighbor joining tree, Fig. 1A) except for the German populations, despite belonging to different draining systems (North Sea versus Baltic Sea). Due to low land levels and historically varying water levels, water bodies and connections across Northern Germany have most likely fluctuated over time. Thus the two lake and river population pairs in Germany (G1 and G2) might have been originally connected. Because of this, G1 and G2 share some postglacial history, common ancestral variation, and divergence while currently the two water systems are physically separated. Specifically, studies on the German system have proposed parasite communities as a promising candidate mediating divergent selection, pointing out their role in local adaptation [15, 18]. As a further global perspective of this hypothesis, we find a signal of isolation-by-adaptation (partial mantel test: r = 0.622, P = 0.0007) shown by a significant association of genome-wide FST and parasite community (jaccard distance of parasite sums across individuals, counts were 4th square root transformed) while correcting for geographic distance (geodetic distance between GPS coordinates of each sampling location). As we detected isolation-by-adaptation at a spatial scale beyond which gene flow occurs, this signal might be most likely caused by a loose linkage between locally adapted loci and the genome-wide neutral regions [19]. This result suggests a role of parasites for the local adaptation of freshwater stickleback populations.

Repeated divergence involves distinct genomic regions
Spatial heterogeneity along the genome was analyzed between parapatric populations by applying a genome scan approach, which averaged genetic divergence (FST) in 10 kb and 100 kb non-overlapping windows across the 20 autosomes (Fig. 2). The shape of the distribution of FST values across the genome qualitatively matches a skewed Poisson distribution, suggestive of divergence with gene flow (S1 Fig.) [9]. The pronounced right tail of the distributions aided the identification of outlier windows, which are significantly different from the genome-wide average. Outlier windows were detected for each population pair as the top 1% of the empirical distribution in addition to being significantly differentiated compared to a random permutation of markers across windows, applying a false discovery rate (FDR) of 0.01. Using the exact same approach comparing marine and freshwater populations, regions known to be under strong divergent selection such as Eda and Atp1a1 were detected as outliers demonstrating the robustness and reliability of the applied methods (details see Methods). Across all five parapatric lake-river comparisons, we identified a total of 1,530 extreme 10 kb outlier windows, in which 47 are shared between at least two of the five population pairs, a proportion that is slightly more than expected by chance (10,000 permutations of random sampling gave on average 28 overlaps, one-tailed P = 0.0006), but none of the windows were shared across all five population pairs. Although we found a weak positive correlation of FST along the genome between the five lake and river ecotype pairs (Fig. 2 and S2 Fig.), there is a negative correlation of FST among the 1,530 outlier windows (Pearson correlation ranging from r = -0.2531 to -0.1064, all P<10-4). These results indicate that outlier windows in one population pair are often windows of low FST in the other population pairs. Hence, outlier windows are not the same across the different population pairs. Annotations for all genes overlapping common outlier windows can be found in S5 Table. None of these outlier windows overlapped with those detected in a previous lake and river comparison of different stickleback populations on the Haida Gwaii archipelago [20]. Thus outliers of exceptional differentiation appear to be locally specific for lake-river ecotypes on a wide geographic scale as well as on a narrow scale [20, 21]. This is in contrast to earlier comparisons between marine and freshwater sticklebacks where few loci are repeatedly found under divergent selection on a global scale [22, 23]. Our results are in line with the notion that the repeated differentiation between derived freshwater stickleback populations occurs as a response to different ecological pressures specific to their local environment [24]. This might reflect locally specific parasite communities, aside from the general trend of an increase in parasite diversity in lakes compared to rivers. However, genomic diversification seem to be an inevitable consequence following the dispersal across habitats, reinforcing the concept that local adaptation is a major contributor to the evolution of species.

Patterns of divergence across a continuum of differentiation
In order to further understand processes shaping the heterogeneity of genomic divergence, we evaluated if divergence is widespread or localized along the genome. Divergence hitchhiking predicts a trend towards an increase in size of divergent regions with overall population differentiation [8, 19]. Conversely, if size was largely determined by the strength and duration of selection, the size of divergent regions would be independent of overall population differentiation. To test these predictions in our dataset, we exploited our comprehensive sequencing resolution to identify precise borders and dimensions of regions of exceptional differentiation. Amongst the 1,530 outlier windows, adjacent outlier windows were combined into 794 continuous outlier “regions” of exceptional differentiation estimated to the nearest 1 kb (S6 Table). The size of a region of exceptional differentiation was determined utilizing barrier strength (b, ref [25]) to contrast local divergence to the genome-wide average. We found a high degree of size heterogeneity among divergent regions within and across population pairs, with no evidence that the size of these regions increases with higher levels of genome-wide differentiation (Table 1, S3 Fig.). This also holds true when recombination rates are taken into account (see below). Therefore, the genomic pattern of divergence observed across a continuum of population differentiation suggests that selection at linked sites drives the observed pattern rather than the interplay of gene flow and divergent selection, consistent with the perspective of geographically specific local adaptation. However, additional factors such as soft sweeps resulting from adaptation based on standing genetic variation might also contribute to the observed patterns, further complicating interpretations.
To further explore if the observed divergence patterns are indeed facilitated by selection and not induced by drift alone, we investigated fine-scale linkage patterns and their effects on genomic heterogeneity across a populations. For each population, we estimated the realized population-scaled recombination rates (ρ/Θ) along the genome. Both a local reduction of gene flow mediated by divergent selection and selection with the hitchhiking of linked neutral sites are predicted to produce a negative correlation between FST and recombination rate [12, 26], however this association would be unlikely mediated by drift alone. In addition, divergence hitchhiking predicts that over time, linkage will extend along the genome and eventually encompass large tracts of the genome [27]. In our study, realized recombination rates in regions of exceptional differentiation were often significantly reduced compared to genome-wide estimates (Fig. 3). We found that genome-wide recombination rates tended to decrease with increasing overall differentiation (Fig. 3). However, realized recombination rates in divergent regions are not significantly correlated with genome-wide differentiation, adding to the growing lack of empirical evidence for divergence hitchhiking [28]. These results suggest that either actual recombination rates coincide regions of the genome, which become divergent, or selection drives local reductions in realized recombination rates. The coalescent-based population recombination rates (4Ner) estimated in this study are simultaneously affected by the variation in genomic structure within and across populations, which may influence actual recombination rates, as well as by selection. Hence, selection might have locally reduced realized recombination rates in certain genomic regions or actual recombination has been reduced due to the intrinsic genomic structural variations thereby promoting genomic divergence. Previous studies evaluating large-scale map-based recombination patterns in sticklebacks have also found a correlation between recombination and divergence, suggesting that genome structure, via its influence on recombination, is important in understanding patterns of genomic differentiation [29, 30]. Here, the low correlation in divergence (FST) between different population pairs (Fig. 2) suggests that local factors specific to each population pair drive genomic differentiation, and that population specific selection reduces realized recombination, particularly if genomic structure is conserved across populations. However, it is possible that genome structure is not so strongly conserved across these geographically distant pairs. Structural variations such as inversions and CNVs have been shown to be abundant within stickleback populations [31]. A companion paper [17] highlights the prevalence of CNVs among and between the populations studied here, in which CNVs tend to also be population specific. These findings indicate that genome structure might be more variable than expected, and therefore might hold potential for promoting genomic differentiation in a population specific manner. We cannot here distinguish between selection-induced influences on realized recombination rates, and actual variation in recombination rates due to differences in genome structure and resultant effects on patterns of genomic differentiation. Further understanding of genome structure’s influence on recombination rates, and its variability within and across populations, will be crucial for disentangling the combined influences of selection and recombination on patterns of genomic variation.
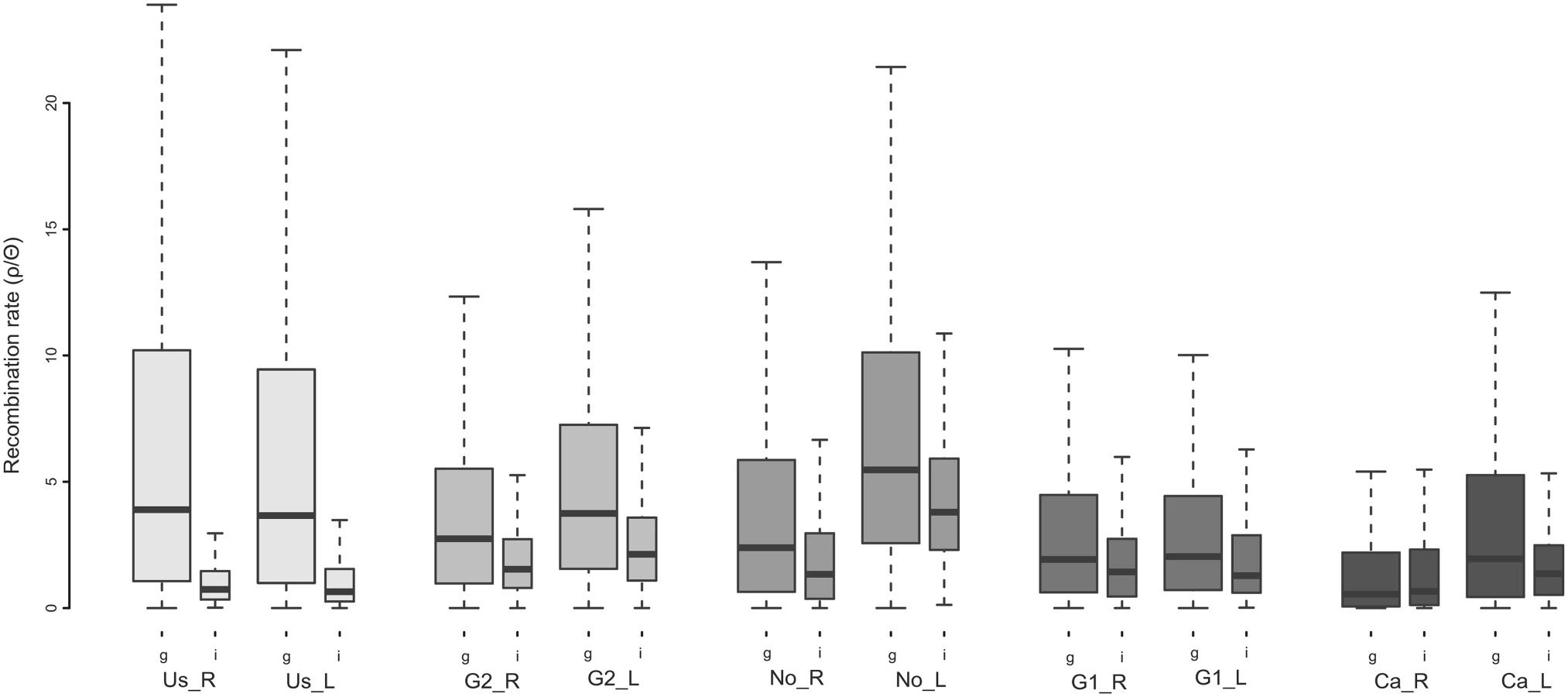
Molecular signatures of selection in divergent genomic regions
Relative divergence (FST) in regions with low levels of recombination might be misleadingly interpreted as conclusive evidence for a local reduction of gene flow. For this reason, measurements of absolute divergence such as Dxy have been suggested as a complement to more reliably identified regions of locally reduced gene flow [10, 12, 32]. However, absolute divergence measurements are unreliable statistics for nascent populations and in non-equilibrium situations during population differentiation. Hence, we aim to disentangle different mechanisms shaping regions of exceptional differentiation by assessing selective sweep signatures in one or both populations of each parapatric pair. Utilizing the base pair resolution of our whole genome sequence data, we evaluated allele frequency spectra to differentiate between molecular signatures of selection among individual regions of exceptional differentiation. In divergent regions differentiated due to a local restriction of gene flow mediated by selection, the spectrum is not expected to be affected locally and should reveal a signature of neutral evolution [12]. The opposite is true for regions resulting from selection with hitchhiking at linked sites, which causes a characteristic skew of the spectrum. An excess of rare alleles is expected in a population experiencing a selective sweep [33], or in both populations in the case of background selection [34]. Distortions in the allele frequency spectrum were calculated for each population as Tajima’s D (TD) across the genome in 100 kb windows and in each region of exceptional differentiation. Genome-wide averages of TD varied from 0.0385 to 0.5936 suggesting predominantly neutral evolution across the genome with no indication for an excess of low frequency polymorphism in any of the populations. TD values within regions of exceptional differentiation were shifted towards negative values except for the Alaskan river (Us_R, Fig. 4A). These negative shifts of TD are consistent with selection as a major mechanism responsible for localized divergent regions along the genome.

In order to quantify the relative contribution of different mechanisms shaping the genomics of speciation, we partitioned individual regions of exceptional differentiation into four mutually exclusive categories with different molecular signatures of evolution based on contrasting local TD values to the genome-wide average (Table 1 and Fig. 4B–F). The minority of divergent regions is consistent with background selection (12%, TD reduced in both populations, Fig. 4B), whereas adaptation seems to shape most of the divergent regions (48%), consistent with the influential role of selection. Divergent regions with signals of positive selection (TD reduced in one of the two populations) should harbor those genes responsible for local adaptation. Genes in divergent regions with a signature of positive selection in lakes (Fig. 4C) were overrepresented with functions involved in structural molecule activity (18 out of 260 annotated genes, P = 0.0018), while genes in divergent regions with signals of positive selection in rivers (Fig. 4D) were overrepresented with functions involved in G-protein coupled receptor activity (15 out of 105, P = 0.0038), antiporter activity (6 out of 36, P = 0.0280), and drug transmembrane transporter activity (4 out of 8, P = 0.0367), suggesting functions in environmental response. Divergent regions with neutral TD patterns (TD in both populations similar to genome-wide average, Fig. 4E) potentially harbor genes restricting gene flow. Despite the prominent occurrence of neutral TD patterns among divergent regions (35%), we found no functional overrepresentation of genes within those regions (S6 Table). This indicates that a variety of different genes and functions might be involved in reproductive isolation, but the current state of gene annotations does not allow drawing compelling conclusions. Overall, the variety of molecular signatures of selection found in divergent regions suggests that different evolutionary processes shape regions of exceptional differentiation. We acknowledge that our approach of strictly categorizing regions based on thresholds simplifies a complex situation, in which various factors most likely interact to shape genomic divergence. However, our analysis suggests that different processes have different impacts across the genome, with selection being a probably major contributor. Therefore, the effects of a local reduction of gene flow and local adaptation are mutually compatible and probably act in concert to shape the genomic landscape of divergence between differentiating parapatric stickleback populations.
Conclusion
We presented multiple lines of evidence for the role of adaptation shaping the genomic divergence patterns between lake-river populations of three-spined sticklebacks. Aside from adaptive processes, stochastic variation in coalescent times and variable mutation rates could further contribute to the observed heterogeneity of genomic divergence [35]. In particular, demographic history such as colonization events (population range expansions) might lead to a substantial variation in allele frequencies across the genome, possibly mimicking the patterns of adaptive hitchhiking [36]. Here, we have chosen the genome-wide average as proxy of the underlying demographic history and the effect of random drift on these populations, as detailed demographic information is scarce. Today, fish migration from the sampled rivers flowing into lake habitats is possible while migration in the opposite direction is likely constrained by physical barriers (S1 Table). However, as freshwater systems have been subject to recurrent water-level changes during de-glaciation, the spatial context at different stages of population divergence might have fluctuated over the years affecting demographic history of the populations. Due to pronounced local differences and variable genomic patterns across the sampled continuum of genetic population differentiation we conclude that the main mode of contemporary divergence between parapatric three-spined sticklebacks is associated with population-specific local adaptation. This is potentially partially mediated by differences in the parasite, as we also found a corresponding signature of isolation by adaptation. Furthermore, our fine-scale examinations of molecular evolution suggest that some heterogeneity of genomic divergence is also the result of locus-specific differences in gene flow mediated by divergent selection. Our study has taken an important step towards deciphering the underlying mechanisms responsible for the genomic patterns during speciation, one of the fundamental enigmas in evolutionary biology.
Materials and Methods
Sampling and data processing
Three-spined stickleback fish were caught from five pairs of lakes and rivers in North America and Northern Europe (S1 Table and Fig. 1). Between 12 and 17 fish were screened for macroparasites following established procedures [14]. Both Shannon diversity indices for each population and jaccard distance between populations were estimated on the basis of 4th square root transformed parasite counts. Muscle tissue from six sampled individuals from each location was used for DNA extraction (using a Qiagen DNA Midi Kit following the manufacturer’s protocol for high molecular weight DNA) and Illumina sequencing following previous methods [31]. To capture natural variation present in the wild, we randomly picked individual fish for sequencing (albeit targeting equal sex ratio per population and similar fish sizes across populations), thus without pre-selection of any particular morphological or parasitological characteristics. For each individual, two paired-end libraries (100bp reads, average insert size of 140bp and 300bp) and a mate-pair library (50bp reads, average insert gap of 3kb) were produced, achieving an average depth of coverage of 26x (S2 Table). Data is deposited in the European Nucleotide Archive (PRJEB5198). Raw sequence data was processed and filtered following previous procedures [31] and mapped against the three-spined stickleback reference genome [22] from Ensembl version 68 [37] with BWA (Burrows-Wheeler Aligner) software [38].
Mapped reads were further filtered and processed utilizing the Picard toolkit following previous procedures [31]. SNPs and indels were called with GATKv1.6 [39, 40] using concordant SNP calls from SAMtools v0.1.18 [41] for variant recalibration. Phasing and imputation was performed with BEAGLE v3.1 [42]. VCFtools [43] was utilized for processing genotypes. Positions overlapping with ‘N’s and repeat-masked regions from the Ensembl annotations (version 68) were removed from the final genotype file. Furthermore, variants within 10bp of an indel or indicating copy number variation were also excluded. Copy number variable (CNV) regions were identified by deviations in expected read depth with the software CNVnator [44]. More details on the CNV analysis are given in a companion paper submitted by Chain et al. The following analyses were performed on the 20 autosomes, spanning 380,547,835 sites in the reference genome. After removing masked sites and CNV region and imputing genotypes across 60 individuals, 297,437,667 sites were reliably genotyped and used for estimating population genetics parameters.
Validation of genotypes
We used Illumina’s Golden Gate platform for cross checking genotypes from SNP sites distributed across the genome. Each chromosome held on average 9 (range 2–21) markers and the total of 183 loci were mostly interspersed by at least 50 kb. We found a high overall concordance (98% in 12,041 comparable sites) between genotype calls from the Golden Gate assay and our sequencing pipeline.
Population genetics
The population genetics estimators of nucleotide diversity (π and Θ) and Tajima’s D (TD) were calculated with VCFtools v0.1.11 [43] for each of the 10 populations (S3 Table), in addition to the relative divergence (Weir and Cockerham FST) and absolute divergence (Dxy [45]) estimated for each of the 5 parapatric lake-river pairs (S4 Table). Numbers of polymorphic sites per population and per population pair are reported in S3–S4 Tables. To illustrate the relationship amongst all sampled populations, we utilized a set of 1,074,467 intergenic autosomal polymorphic loci to estimate pairwise divergence (Weir and Cockerham FST) and built a neighbor joining tree. To gain support for the tree topology we randomly down sampled this dataset 100 times to 100,000 loci. For the genome scan, FST was calculated on the full dataset that was further filtered for minor allele frequencies below 25% across each pairwise comparison excluding uninformative polymorphism [46]. This way we evaluated the divergence between parapatric population pairs on the basis of 691,957 to 1,227,732 sites across the 20 autosomes. Population genetics estimators were averaged across the genome (20 autosomes) in non-overlapping windows to ensure statistical independence of windows. We used window sizes of 10 kb and 100 kb and confirmed that results are qualitatively the same. Diversity estimates have been corrected for the number of sites for which genotypes are available.
Divergent regions of exceptional differentiation
Outlier windows were determined by combining an empirical approach with a permutation approach. First, windows above the top 1% of the empirical distribution were identified as putative outlier windows. Second, we applied a permutation approach in which loci across the genome were permuted 1,000,000 times and window estimates of FST were tested against permutations holding the same amount of variable sites. Putative outlier windows from this permutation approach were identified after adjusting for a FDR of 0.01. Our final set of outlier windows consisted of those windows that were significant outliers in both approaches. All statistical procedures and visualizations were implemented in R [47]. Outlier window positions were compared across the five replicated lake-river comparisons. To evaluate how many overlapping outlier windows were expected by chance, windows were permutated 10,000 times utilizing bedtools [48].
To approximate the size of regions of exceptional differentiation more in detail, adjacent outlier windows were combined to form larger contiguous divergent regions of extreme differentiation. In each resulting candidate region, the locus of maximal divergence was determined as a starting point, in which outward steps of 1 kb windows were binned to estimate barrier strength (b, ref [25]). Margins of divergent regions showing extreme differentiation were determined when b dropped below 1 (genome-wide average) in two consecutive 1 kb bins. This resulted in divergent regions of exceptional differentiation with distinct sizes estimated to the nearest 1 kb. Divergent regions with sequence coverage (sequence information accessible, see details above) spanning less than 50% of their length were excluded from subsequent analyses. Average sizes of about 50 kb are independent of the initial window size used but specific values reported here are based on the 10 kb window size approach (Table 1).
Effect of sample size on power
We acknowledge that estimates of FST based on allele frequencies can vary depending on samples size [49]. To reduce variation of estimates between populations we kept the samples size constant at 12 alleles per populations. Additionally, our analysis did not rely on per site estimates but instead on averages of FST over larger regions (see above). We evaluated the effect of sample size on our power to describe genomic patterns, detect outlier windows, and define divergent regions in the three following ways. (i) We tested the accuracy of our FST estimates at individual loci by comparing them to estimates based on a larger sample size. The 183 loci used for validating the genotypes (see above) were also used to genotype a larger population sample (n = 26–59 per population) to validate allele frequencies and resulting FST estimates. For all population pairs, the FST estimates based on the sequencing approach with 6 individuals per population (12 alleles) had a significant positive correlation with the FST estimates from the Golden Gate assay using at least 26 individuals (Pearson correlation, r = 0.85, P< 10-16, df = 241, S4 Fig.). (ii) We tested the consistency of window FST estimates across the whole range of potential FST values by jack-knifing samples (S5 Fig.). On average, jack-knifed values (comparing 10 alleles per population) had 95% confidence intervals of 0.039 up to a maximum of 0.175. Windows with high FST values (>0.75) had even narrower confidence intervals (average of 0.027 and maximum of 0.088). These results support the notion that pronounced differences (“near-” and “post-fixation”) can be more reliably detected using our sample sizes than more settled differences (“pre-fixation” regime). (iii) We tested our ability to detect known candidate genes, which highly differentiate between marine and freshwater populations. For this we utilized previous sequencing data available for a marine population from Denmark [31]. Our genome scan based on FST estimates averaged across 10 kb windows reliably detected windows overlapping ATP1a1 [50], a well known candidate gene for physiological adaptation to osmotic differences on linkage group I, in all 6 pairwise European marine-freshwater comparisons (S6 Fig.). Eda, the major gene (linkage group IV) underlying the reduction of lateral plate number frequently observed in freshwater populations [51], was detected in 5 out of the 6 pairwise European marine-freshwater comparisons (S7 Fig.). As expected G1_L, a lake population showing phenotypic variation at this trait did not show significant differentiation in the Eda region, in which two of the six sequenced individuals were fully plated and carried the same haplotype as the fully plated marine fish. This is in line with a simulation demonstrating that sampling 12 haplotypes yields between 67–95% power compared to a gold standard, while notably, sampling fewer individuals has the greatest impact in the “pre-fixation” regime (a beneficial allele is starting to rise in one population) compared to “near-fixation” and “post-fixation” regimes (a beneficial allele is nearly or completely fixed in one population) [52].
Molecular signature of selection in divergent regions
To assess the molecular signature of selection in regions of exceptional differentiation, shifts in the allele frequency spectrum were evaluated utilizing TD. TD in these regions was compared to the genome-wide average of each respective population. A 5% threshold was applied to classify divergent regions into four mutually exclusive categories: background selection if TD dropped below the threshold in both parapatric populations, adaptation in lake or river if TD dropped below the threshold only in the respective population, and reduced gene flow if TD appeared neutral (not below the threshold). Comparing the utilization of population specific thresholds for each pairwise comparison with the utilization of the same overall averaged threshold for all populations resulted in minor differences in absolute numbers of regions in different categories. Furthermore, these differences did not affect qualitative changes with respect to the functional annotation of different categories, nor the proportion of different categories across the five parapatric population pairs.
Recombination rates
Direct measures of fine-scale population recombination rates (ρ = 4Ner) were obtained with LDhat [53, 54] from patterns of genetic variation for each population separately. We filtered highly localized breakdowns of linkage disequilibrium (values of ρ above 100 between adjacent SNPs), as those are most likely artifacts, possibly due to local misassembly of the reference genome or clusters of erroneous SNPs [55]. Resulting recombination rate estimates were averaged over each 10 kb window and over each divergent region with exceptional differentiation, and corrected by the population specific mutation rate (Θ = 4Neµ) estimated as an average across all autosomes.
Annotation
Regions overlapping with gene annotations from version 68 of Ensembl were identified using intersectBed of bedtools [48]. Annotations for shared outlier windows and divergent regions are reported in S5–S6 Tables. To determine enrichment of functional classes of genes among regions, topGO [56] was used with a universe of autosomal genes, and significance was determined at the 0.05 level using FDR adjusted p values to correct for multiple testing.
Ethical statement
This study was performed according to the requirements of the German Protection of Animals Act (Tierschutzgesetz) and was approved by the ‘Ministry of Energy, Agriculture, the Environment and Rural Areas’ of the state of Schleswig-Holstein, Germany (reference number: V 312–72241.123–34). Wild sticklebacks were caught using minnow traps or hand nets. Before dissection, the fish were anesthetized with MS222 and sacrificed by an incision into the brain followed by immediate decapitation, and every effort was made to minimize suffering. No further animal ethics committee approval was needed. The species used in this study are not endangered or protected in any of the populations studied.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Hanikenne M, Kroymann J, Trampczynska A, Bernal M, Motte P, et al. (2013) Hard Selective Sweep and Ectopic Gene Conversion in a Gene Cluster Affording Environmental Adaptation. PLoS Genet 9: e1003707. doi: 10.1371/journal.pgen.1003707 23990800
2. Sadier, A, Viriot, L, Pantalacci, S, Laudet, V The ectodysplasin pathway: from diseases to adaptations. Trends Genet.
3. Soria-Carrasco V, Gompert Z, Comeault AA, Farkas TE, Parchman TL, et al. (2014) Stick insect genomes reveal natural selection’s role in parallel speciation. Science 344 : 738–742. doi: 10.1126/science.1252136 24833390
4. Renaut S, Grassa CJ, Yeaman S, Moyers BT, Lai Z, et al. (2013) Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nat Commun 4 : 1827. doi: 10.1038/ncomms2833 23652015
5. Via S, Conte G, Mason-Foley C, Mills K (2012) Localizing FST outliers on a QTL map reveals evidence for large genomic regions of reduced gene exchange during speciation-with-gene-flow. Mol Ecol 21 : 5546–5560. doi: 10.1111/mec.12021 23057835
6. Turner TL, Hahn MW, Nuzhdin SV (2005) Genomic islands of speciation in Anopheles gambiae. PLoS Biol 3: e285. doi: 10.1371/journal.pbio.0030285 16076241
7. Seehausen O, Butlin RK, Keller I, Wagner CE, Boughman JW, et al. (2014) Genomics and the origin of species. Nat Rev Genet 15 : 176–192. doi: 10.1038/nrg3644 24535286
8. Wu CI, Ting CT (2004) Genes and speciation. Nat Rev Genet 5 : 114–122. doi: 10.1038/nrg1269 14735122
9. Feder JL, Egan SP, Nosil P (2012) The genomics of speciation-with-gene-flow. Trends Genet 28 : 342–350. doi: 10.1016/j.tig.2012.03.009 22520730
10. Noor MAF, Bennett SM (2009) Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity 103 : 439–444. doi: 10.1038/hdy.2009.151 19920849
11. White BJ, Cheng C, Simard F, Costantini C, Besansky NJ (2010) Genetic association of physically unlinked islands of genomic divergence in incipient species of Anopheles gambiae. Mol Ecol 19 : 925–939. doi: 10.1111/j.1365-294X.2010.04531.x 20149091
12. Nachman MW, Payseur BA (2012) Recombination rate variation and speciation: theoretical predictions and empirical results from rabbits and mice. Philos Trans R Soc Biol Sci 367 : 409–421. doi: 10.1098/rstb.2011.0249
13. Berner D, Roesti M, Hendry AP, Salzburger W (2010) Constraints on speciation suggested by comparing lake-stream stickleback divergence across two continents. Mol Ecol 19 : 4963–4978. doi: 10.1111/j.1365-294X.2010.04858.x 20964754
14. Kalbe M, Wegner KM, Reusch TBH (2002) Dispersion patterns of parasites in 0+ year three-spined sticklebacks: a cross population comparison. J Fish Biol 60 : 1529–1542. doi: 10.1111/j.1095-8649.2002.tb02445.x
15. Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012) Rapid and adaptive evolution of MHC genes under parasite selection in experimental vertebrate populations. Nat Commun 3 : 621. doi: 10.1038/ncomms1632 22233631
16. Wegner KM, Reusch TBH, Kalbe M (2003) Multiple parasites are driving major histocompatibility complex polymorphism in the wild. J Evol Biol 16 : 224–232. doi: 10.1046/j.1420-9101.2003.00519.x 14635861
17. Chain FJJ, Feulner PGD, Panchal M, Eizaguirre C, Samonte IE, et al. (2014) Extensive copy-number variation of young genes across stickleback populations. PLoS Genet 10: e1004830. doi: 10.1371/journal.pgen.1004830 25474574
18. Eizaguirre C, Lenz TL, Traulsen A, Milinski M (2009) Speciation accelerated and stabilized by pleiotropic major histocompatibility complex immunogenes. Ecol Lett 12 : 5–12. doi: 10.1111/j.1461-0248.2008.01247.x 19087108
19. Nosil P, Funk DJ, Ortiz-Barrientos D (2009) Divergent selection and heterogeneous genomic divergence. Mol Ecol 18 : 375–402. doi: 10.1111/j.1365-294X.2008.03946.x 19143936
20. Deagle BE, Jones FC, Chan YGF, Absher DM, Kingsley DM, et al. (2012) Population genomics of parallel phenotypic evolution in stickleback across stream-lake ecological transitions. Proc R Soc Biol Sci Ser 279 : 1277–1286. doi: 10.1098/rspb.2011.1552
21. Roesti M, Hendry AP, Salzburger W, Berner D (2012) Genome divergence during evolutionary diversification as revealed in replicate lake—stream stickleback population pairs. Mol Ecol 21 : 2852–2862. doi: 10.1111/j.1365-294X.2012.05509.x 22384978
22. Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, et al. (2012) The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484 : 55–61. doi: 10.1038/nature10944 22481358
23. Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, et al. (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet 6: e1000862. doi: 10.1371/journal.pgen.1000862 20195501
24. Kaeuffer R, Peichel CL, Bolnick DI, Hendry AP (2012) Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback Evolution 66 : 402–418.
25. Barton N, Bengtsson BO (1986) The barrier to genetic exchange between hybridising populations. Heredity 57 : 357–376. doi: 10.1038/hdy.1986.135 3804765
26. Keinan A, Reich D (2010) Human population differentiation is strongly correlated with local recombination rate. PLoS Genet 6: e1000886. doi: 10.1371/journal.pgen.1000886 20361044
27. Feder JL, Gejji R, Yeaman S, Nosil P (2012) Establishment of new mutations under divergence and genome hitchhiking. Philos Trans R Soc Biol Sci 367 : 461–474. doi: 10.1098/rstb.2011.0256
28. Feder JL, Nosil P (2010) The efficacy of divergence hitchhiking in generating genomic islands during ecological speciation. Evolution 64 : 1729–1747. doi: 10.1111/j.1558-5646.2009.00943.x 20624183
29. Roesti M, Moser D, Berner D (2013) Recombination in the threespine stickleback genome—patterns and consequences. Mol Ecol 22 : 3014–3027. doi: 10.1111/mec.12322 23601112
30. Hohenlohe PA, Bassham S, Currey M, Cresko WA (2012) Extensive linkage disequilibrium and parallel adaptive divergence across threespine stickleback genomes. Philos Trans R Soc Biol Sci 367 : 395–408. doi: 10.1098/rstb.2011.0245
31. Feulner PGD, Chain FJJ, Panchal M, Eizaguirre C, Kalbe M, et al. (2013) Genome-wide patterns of standing genetic variation in a marine population of three-spined sticklebacks. Mol Ecol 22 : 635–649. doi: 10.1111/j.1365-294X.2012.05680.x 22747593
32. Cruickshank TE, Hahn MW (2014) Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol 23 : 3133–3157. doi: 10.1111/mec.12796 24845075
33. Smith JM, Haigh J (1974) The hitch-hiking effect of a favourable gene. Genetics Res 23 : 23–35. doi: 10.1017/S0016672300014634
34. Charlesworth B, Morgan MT, Charlesworth D (1993) The effect of deleterious mutations on neutral molecular variation. Genetics 134 : 1289–1303. 8375663
35. Cutter AD, Payseur BA (2013) Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet 14 : 262–274. doi: 10.1038/nrg3425 23478346
36. Hofer T, Ray N, Wegmann D, Excoffier L (2009) Large allele frequency differences between human continental groups are more likely to have occurred by drift during range expansions than by selection. Annals of Human Genetics 73 : 95–108. doi: 10.1111/j.1469-1809.2008.00489.x 19040659
37. Flicek P, Amode MR, Barrell D, Beal K, Brent S, et al. (2011) Ensembl 2011. Nucleic Acids Res 39: D800–D806. doi: 10.1093/nar/gkq1064 21045057
38. Li H, Ruan J, Durbin R (2008) Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18 : 1851–1858. doi: 10.1101/gr.078212.108 18714091
39. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303. doi: 10.1101/gr.107524.110 20644199
40. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43 : 491–498. doi: 10.1038/ng.806 21478889
41. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079. doi: 10.1093/bioinformatics/btp352 19505943
42. Browning BL, Browning SR (2009) A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Gen 84 : 210–223. doi: 10.1016/j.ajhg.2009.01.005
43. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, et al. (2011) The variant call format and VCFtools. Bioinformatics 27 : 2156–2158. doi: 10.1093/bioinformatics/btr330 21653522
44. Abyzov A, Urban AE, Snyder M, Gerstein M (2011) CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21 : 974–984. doi: 10.1101/gr.114876.110 21324876
45. Nei M (1987) Molecular Evolutionary Genetics. New York: Columbia UP.
46. Roesti M, Salzburger W, Berner D (2012) Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol Biol 12 : 94. doi: 10.1186/1471-2148-12-94 22726891
47. R_Development_Core_Team (2011) R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
48. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 : 841–842. doi: 10.1093/bioinformatics/btq033 20110278
49. Waples R (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89 : 438–450. doi: 10.1093/jhered/89.5.438
50. McCormick SD (2001) Endocrine control of osmoregulation in teleost fish1. Am Zool 41 : 781–794. doi: 10.1668/0003-1569(2001)041%5B0781:ECOOIT%5D2.0.CO;2
51. Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G Jr., Dickson M, et al. (2005) Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307 : 1928–1933. doi: 10.1126/science.1107239 15790847
52. Udpa N, Ronen R, Zhou D, Liang J, Stobdan T, et al. (2014) Whole genome sequencing of Ethiopian highlanders reveals conserved hypoxia tolerance genes. Genome Biol 15: R36. doi: 10.1186/gb-2014-15-2-r36 24555826
53. McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, et al. (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304 : 581–584. doi: 10.1126/science.1092500 15105499
54. Auton A, McVean G (2007) Recombination rate estimation in the presence of hotspots. Genome Res 17 : 1219–1227. doi: 10.1101/gr.6386707 17623807
55. Auton A, Fledel-Alon A, Pfeifer S, Venn O, Ségurel L, et al. (2012) A fine-scale chimpanzee genetic map from population sequencing. Science 336 : 193–198. doi: 10.1126/science.1216872 22422862
56. Alexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22 : 1600–1607. doi: 10.1093/bioinformatics/btl140 16606683
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates