
Strategies for Understanding and Reducing the and Hypnozoite Reservoir in Papua New Guinean Children: A Randomised Placebo-Controlled Trial and Mathematical Model
Ivo Mueller and colleagues conduct a randomized controlled trial of blood-only or blood - plus liver-stage malaria drugs to identify the contribution of relapses to the burden of P. vivax and P. ovale in children in Papua New Guinea, and model the potential impact of mass-drug administration to prevent recurrent P. vivax infections.
Published in the journal:
. PLoS Med 12(10): e32767. doi:10.1371/journal.pmed.1001891
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001891
Summary
Ivo Mueller and colleagues conduct a randomized controlled trial of blood-only or blood - plus liver-stage malaria drugs to identify the contribution of relapses to the burden of P. vivax and P. ovale in children in Papua New Guinea, and model the potential impact of mass-drug administration to prevent recurrent P. vivax infections.
Introduction
Renewed intensification of global malaria control efforts over the last 15 years have been successful in significantly reducing the global burden of malaria, with many countries in the Asia-Pacific and the Americas seeing a reduction of >90% in the number of clinical cases [1]. As a consequence, 34 countries are actively attempting to eliminate malaria, and many others are considering doing so in the near future [2]. In 2013, political leaders in Central American and East Asian countries, representing >60% of the global population, declared their intention to eliminate malaria in their regions by 2020 and 2030, respectively [3,4]. In parallel to the reduction in overall incidence, a pronounced shift in species composition has been observed, with Plasmodium vivax now the predominant Plasmodium species in the vast majority of countries outside Africa [2], accounting for 90%–100% of clinical cases in countries such as Guatemala, Brazil, Solomon Islands, and Vanuatu [1].
Despite a significant reduction in P. vivax malaria in the last 20 years, P. vivax has several biological characteristics that enable it to evade existing control and elimination efforts, which are mainly directed against P. falciparum blood stages [5,6]. First and foremost is the ability of P. vivax to relapse weeks, months, and years after a primary infection, via a poorly understood reactivation of dormant hypnozoite stages in the liver [7]. These stages cannot be detected with currently available diagnostic tools and are not cleared upon treatment with routinely administered anti-malarial drugs, unless primaquine (PQ)—a drug that requires at least 7–14 d of administration and can cause severe haemolysis in people with glucose-6-phosphate dehydrogenase (G6PD) deficiency—is added to the treatment [8].
In P. vivax–endemic regions, hypnozoites constitute a reservoir of diverse P. vivax strains that will cause blood-stage infections at a later point in time [9,10]. They are therefore likely not only to account for a high number of P. vivax blood-stage infections but also to contribute a high number of concurrently circulating parasite clones in the blood. Despite recent advances in the molecular detection and genotyping of P. vivax parasites [11], it is not yet possible to determine whether an infection detected in the blood of an individual originated from a new sporozoite inoculation or is a relapse from a hypnozoite. P. vivax produces gametocytes rapidly and continuously over the course of an infection [10], and even low-density infections are thus potentially infectious. If all clones, relapse-derived and newly acquired, produce gametocytes concurrently, these infections can potentially be transmitted together, and the chance for sexual recombination in the mosquito is greatly increased, thus contributing to the maintenance of high P. vivax genetic diversity even at low transmission levels [12–14].
P. vivax is thus considered one of the major challenges for elimination of malaria outside Africa [15]. Better data and tools are urgently required to estimate the P. vivax hypnozoite reservoir, quantify the relapse burden, better understand potential relapse triggers, and develop the most appropriate public health intervention strategies [15–17].
In malaria-endemic areas of Papua New Guinea (PNG), where four of the five human Plasmodium species coexist, P. vivax predominates as the cause of infection and illness in young children [18,19] and is gradually replaced by P. falciparum as the main cause of disease in older children and adults [20], although P. vivax infections remain common throughout childhood and into adulthood, with a prevalence of 13%–36% in cross-sectional surveys conducted in PNG between 2005 and 2010 [21–24]. P. ovale and P. malariae are much less common, with a 2010 survey revealing a prevalence of 0.1% and 1.3%, respectively (by quantitative real-time PCR [qPCR]), and are mostly observed in mixed-species infections [22,25]. The PNG standard anti-malarial treatment is artemether-lumefantrine (AL), which acts against the blood stage of the parasite but does not eliminate hypnozoites. In the absence of a nationwide, cost-effective strategy of screening for G6PD, PQ treatment for clearing liver stages, although recommended for G6PD-normal patients, is rarely given. Consequently, relapses are expected to contribute significantly to the high burden and limited seasonality of P. vivax in PNG children [18,26]. A previous study in PNG children aged 1–5 y observed that presumptive artesunate (7 d) and PQ (14 d, partially supervised) mass treatment to clear hypnozoites reduced the risk of P. vivax clinical episodes by 28% (p = 0.042) compared to only blood-stage treatment and by 33% (p = 0.015) compared to no treatment [27]. Although the study used a suboptimal treatment regimen, and thus substantially underestimated the hypnozoite burden, it did highlight the significant challenge relapses pose to successful control and eventual elimination of P. vivax malaria in PNG and provides a strong rationale for conducting a more comprehensive clinical trial with an in-depth molecular diagnostics component.
Intensified national control efforts have seen the prevalence of light microscopy (LM)–detectable P. vivax malaria parasites in the general population decrease from 17% in 2006 to 8% in 2010 [25] and to 0.5% in 2014 [28]. Despite these gains, a large reservoir of individuals infected with submicroscopic P. vivax persists. In a 2010 survey, P. vivax prevalence by qPCR was 12.8%. Of these infections, 89.6% were asymptomatic, 53.8% were submicroscopic, and 48.9% included P. vivax gametocytes [25]. Similarly high rates of persistent asymptomatic P. vivax infection and gametocyte carriage were also found in surveys in Thailand [29,30] and Brazil [31], which have seen substantial recent reductions in transmission.
Mass screening and treatment (MSAT) and mass drug administration (MDA) with artemisinin-based combination therapies have been advocated as important tools to reduce the asymptomatic P. falciparum reservoir [32,33]. These interventions are also likely to be of great importance for P. vivax elimination. Significant questions remain, however, none greater than how to best attack the undetectable hypnozoite reservoir.
To address these critical questions we have conducted a randomised double-blind placebo-controlled trial of a highly efficacious PQ treatment regimen in PNG children aged 5–10 y, using detailed molecular diagnostics to directly measure the contribution of relapses to the burden of P. vivax and P. ovale infection, disease, and transmission. By using these data in mathematical models, we further estimate the potential effect of MDA with treatment regimens that are part of first-line policy and regimens currently under investigation in clinical trials [34,35] on the prevention of recurrent P. vivax infections, providing critical evidence-based recommendations for policy-makers.
Methods
Ethics Statement
The protocol (S1 Text) received ethical clearance from the PNG Institute of Medical Research Institutional Review Board (0908), the PNG Medical Advisory Committee (09.11), and the Ethics Committee of Basel (237/11) and was conducted in full accordance with the Declaration of Helsinki. The study was retrospectively registered at ClinicalTrials.gov (NCT02143934) on 20 May 2014 due to the fact that it was designed and perceived by the investigating team as a treatment to re-infection cohort study where randomisation of PQ treatment was being done in order to modify exposure and allow a detailed investigation of relapses rather than as a trial to assess the efficacy of this specific treatment schedule. However, following further review, it was decided that the study did indeed fulfil the generally accepted definitions of a clinical trial, and the study was then retrospectively registered at ClinicalTrials.gov. All authors affirm that other trials involving PQ that they are involved in are registered at ClinicalTrials.gov (NCT01837992; NCT02364583).
Study Site, Design, and Participants
The study was conducted from 17 August 2009 to 20 May 2010 in five village clusters (13 hamlets) of the Albinama and Balif areas of Maprik District, East Sepik Province, PNG, where both P. falciparum and P. vivax are hyperendemic and P. vivax is responsible for the majority of malaria infection and disease in the first 3 y of life [18,22,36]. Malaria transmission is moderately seasonal, peaking in the early wet season from December to March [26]. Health services for the area are provided by the Albinama health sub-centre, the Balif aid post, and a system of village-based health workers operating in all study villages.
Between 17 August and 11 September 2009, 529 children aged 5 to 10 y whose parents provided written informed consent for their participation were screened for inclusion into this parallel double-blind placebo-controlled trial. Children were enrolled if they fulfilled the following criteria: (i) aged 5–10 y (± 3 mo in children without known date of birth, (ii) enrolled at selected elementary schools and permanent residents of the area, (iii) no disability, (iv) no chronic illness, (v) no known allergy to study drugs, (vi) haemoglobin > 50 g/l, (vii) no severe malnutrition (defined by the PNG national guidelines as weight-for-age nutritional Z score < 60th percentile), and (viii) no G6PD deficiency. The inclusion criteria were amended during the study to allow children who were aged 5–6 y but who were not yet enrolled in elementary school to participate in the study. This was necessary to ensure we adequately covered the desired age range of 5–10 y and reached the required sample size of 525 without expanding the geographical area. Five children were excluded on the basis of G6PD deficiency, and 524 were block randomised using a 1 : 1 allocation ratio to receive 20 d of directly observed treatment (DOT) over 4 wk (26 d) of either (i) chloroquine (CQ) (DOTs 1–3), AL (Coartem) (DOTs 11–13), and PQ (DOTs 1–20; 0.5 mg/kg) or (ii) CQ (DOTs 1–3), AL (DOTs 11–13), and placebo (PL) (DOTs 1–20) (Fig 1). This treatment regimen was deliberately chosen to maximise efficacy, and the dose of each drug was timed such that there would be minimal residual drug by day 0 of the follow-up period (baseline). In order to achieve this, a 4-wk wash-out period was required for CQ, and the 20 d of PQ DOTs were scheduled Monday to Friday of these 4 wk (Fig 1), for ease of direct supervision of every dose. AL was administered because there is documented CQ resistance in PNG, and the PNG national treatment guidelines for P. vivax are to use AL plus PQ. The administration of AL on DOTs 11–13 (days 15–17) was deliberate so that it wouldn’t interfere with CQ and so that drug levels would have reduced to zero by baseline. The intention was not to trial this unconventional drug regimen as a treatment for implementation, but rather to devise a maximally effective treatment to ensure radical cure in half of the cohort and thus allow a detailed investigation of relapses. Participants, field teams, and investigators were all blinded with respect to treatment allocation.

Randomisation, Blinding, and Treatment Allocation
After enrolment, children were randomly allocated to the PQ or PL treatment group using a pre-assigned list. Randomisation lists were prepared by an independent statistician using Microsoft Excel and consisted of ID assignments in blocks of six, each block comprising a list of the same six letters in random order. The independent statistician assigned three randomly selected letters to the PQ drug containers and the three other letters to the PL drug containers. The coding document was held by the statistician until completion of the trial. The PQ and PL tablets were identical in size, shape, and colour. The entire study team and principal investigators remained fully blinded for the entire study period.
Clinical Procedures and Follow-Up
Clinical assessment at enrolment included screening for symptoms of febrile illness, a detailed history of bednet use and recent illness/anti-malarial treatment, and collection of a finger-prick blood sample for assessment of G6PD deficiency using the visual, tube-based G6PD assay (Dojindo Laboratories, Japan), haemoglobin measurement, and later immunological and molecular studies. Children who were febrile were tested using a malaria rapid diagnostic test (RDT) (CareStart Malaria pLDH/HRP2 Combo; AccessBio, US); children with a positive test were treated with AL during DOTs 1–3. DOT 1 was administered at the end of the enrolment visit, with the subsequent 19 DOTs administered daily Monday–Friday over the subsequent 4 wk (Fig 1). All DOTs were supervised by a member of the clinical field team and were co-administered with food and well tolerated [37]. Safety monitoring was performed and reported previously [27]. No specific safety monitoring was conducted during post-intervention follow-up.
Three days after the final DOT (i.e., 4 wk after enrolment), a venous sample was collected and defined as baseline (time point day 0 of the follow-up period). Children were then actively monitored for the presence of febrile symptoms every fortnight for 8 mo. To ensure the capture of information related to any episodes of illness in the 2 wk between visits, passive surveillance measures were implemented at local health centres and aid posts and via the village health worker network. In all symptomatic children, Plasmodium spp. infection was initially confirmed using RDT and a 250-μl finger-prick blood sample was collected for confirmation of infection by LM and qPCR. Only symptomatic children who tested positive by RDT and/or LM were treated with AL. PQ was not re-administered for RDT - or LM-confirmed P. vivax malaria episodes during follow-up. All other illness episodes detected were referred to the local health centre and treated in accordance with PNG treatment guidelines. Finger-prick blood samples were also collected from all children every 2 wk for the first 12 wk and every 4 wk thereafter during the follow-up period (Fig 2). Children were considered lost to follow-up if they permanently relocated outside of the study area or withdrew from the study.
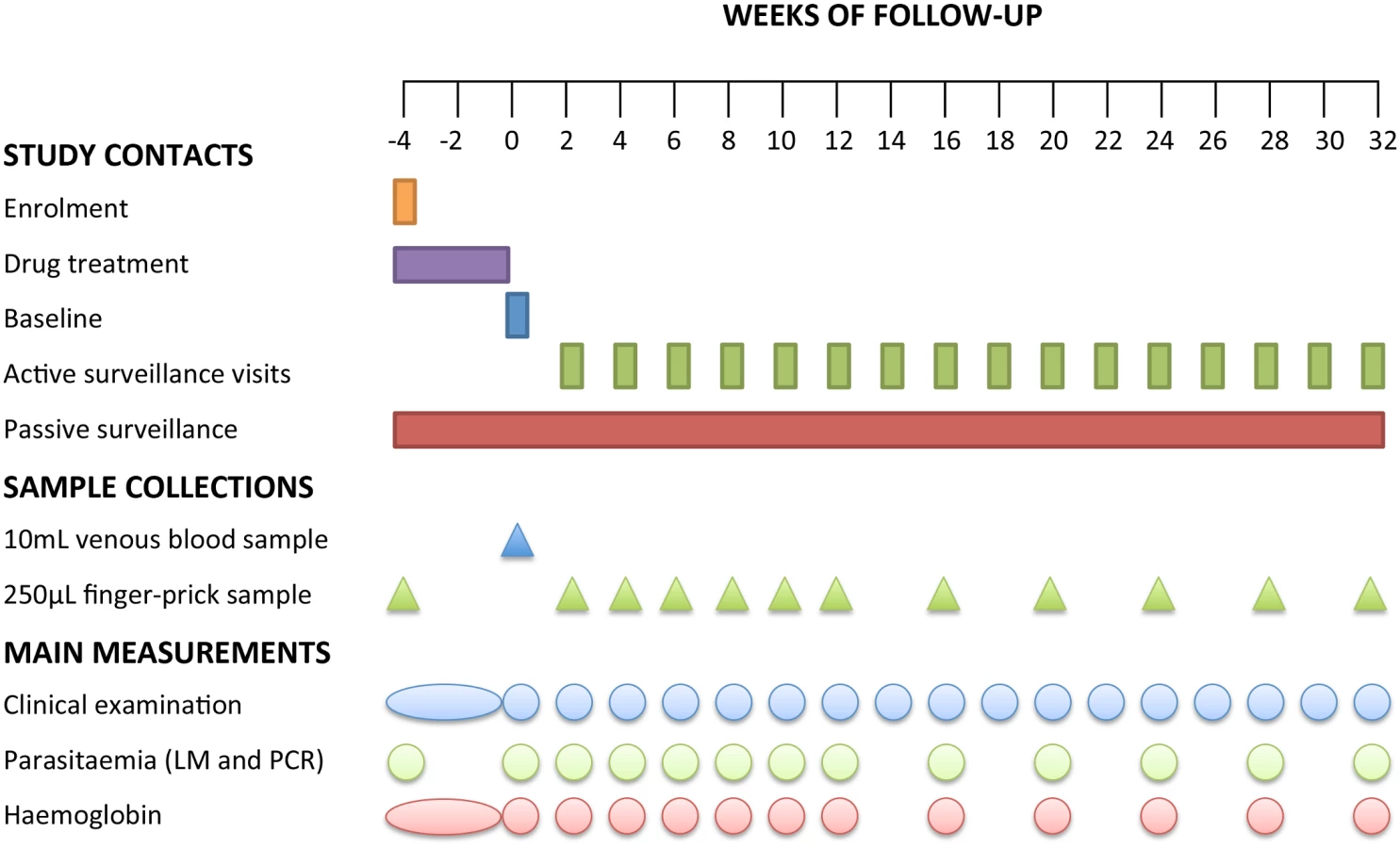
Laboratory Methods
All blood samples collected during active and passive surveillance were examined by LM and qPCR. Blood films were examined independently by two skilled microscopists, blind to allocated treatment, for 200 thick-film fields (1,000× magnification) before being declared Plasmodium-negative. Parasite density was calculated from the number of parasites per 200–500 leucocytes (depending on parasite density) and an assumed leucocyte density of 8,000/μl [38]. Slides discrepant for positivity/negativity, species determination, or density (>2 log difference) were adjudicated by a WHO-certified level 1 (expert) microscopist. Slides were scored as LM-positive for an individual Plasmodium species if the species was detected independently by at least two microscopists and/or if subsequent qPCR diagnosis confirmed the presence of the species. Densities were calculated as the geometric mean densities of all positive reads.
Venous blood samples were separated into plasma, peripheral blood mononuclear cells and red cell pellets, which were stored at −80°C, in liquid nitrogen, and at −20°C, respectively. Finger-prick blood samples were separated into plasma and red cell pellets and stored at −80 and −20°C, respectively. DNA was extracted using the FavorPrep 96-Well Genomic DNA Extraction Kit (Favorgen, Taiwan) from the red cell pellet fraction of all samples. Plasmodium spp. infections were detected using a generic qPCR to detect all four species, after which species-specific (P. falciparum, P. vivax, P. malariae, and P. ovale) qPCRs were performed on Plasmodium-positive samples [39,40]. The P. ovale PCR detects both P. ovale curtisi and P. ovale wallikeri. In addition, samples positive for P. vivax by qPCR were tested for gametocytes by Pvs25 quantitative reverse transcription PCR (qRT-PCR) [39], and individual P. vivax clones were genotyped by capillary electrophoresis using the molecular marker msp1F3 [41] to determine the number of genetically distinct P. vivax blood-stage clones acquired per individual per year at risk, i.e., the molecular force of blood-stage infection (molFOB) [42].
Statistical Analysis and Modelling
As the primary objective of the study was to determine relapse frequencies, the analysis was by modified intention to treat, excluding children who received fewer than 14 DOTs but including those that received 14–20 DOTs and had incomplete follow-up. For analysis purposes, a clinical episode of P. vivax or P. falciparum malaria was defined as febrile illness (current or previous 48 h) plus the presence of P. vivax or P. falciparum parasites by LM (any density). The primary trial endpoint was pre-defined as time to first P. vivax infection after baseline by qPCR. Secondary endpoints included time to first P. vivax infection after baseline by LM; time to first P. vivax clinical episode; time to first P. vivax gametocyte positivity; incidence rate of clinical P. vivax episodes; incidence rate of genetically distinct P. vivax infections (molFOB); incidence rate of P. vivax gametocyte positivity; time to first P. falciparum, P. malariae, and P. ovale infection by qPCR; and time to first P. falciparum clinical episode. A minimum sample size of 250 children per arm was calculated using hazard-rate-based calculations (log-rank tests) based on the effect (30% reduction in incidence risk in PQ group) observed in [27] with an α–error of 5% and a power of 80% and assuming a 30% increase in time to first infection following additional distributions of long-lasting insecticide-treated nets. Sample sizes were increased by 5% to account for children receiving fewer than 14 doses of PQ or PL.
Time to first Plasmodium infection or clinical episode and its association with treatment and covariates were modelled using Cox regression, and the proportional hazards (PH) assumption was checked using the test based on the Schoenfeld residuals. For these analyses, time at risk was censored on the last day before the first of two consecutively missed clinical follow-up visits, resulting in the censoring of 177 children (Fig 3). Kaplan-Meier estimates were computed for each endpoint by Plasmodium species and method of Plasmodium diagnosis. The log-rank test was used to test for differences between survival curves. In all survival analyses, children were considered “at risk” until they reached the endpoint of interest, withdrew, were lost to follow-up, were censored, or completed the study. Village membership was fitted as a fixed effect in the Cox PH model using the following equation:


Negative binomial regression (NB reg) models were used to calculate the incidence rate of clinical episodes, molFOB, and P. vivax gametocyte positivity. In these models, time at risk was calculated for individual children based on the number of attended versus missed visits. If a child was not seen for a consecutive time period of 108 d during the follow-up period, time at risk was censored at the last attended follow-up visit or passive surveillance contact. This resulted in 74 children being censored (44 in PL arm and 30 in PQ arm) rather than the 177 shown in Fig 3. In addition, for the molecular analyses, time at risk was reduced for children who were not seen for consecutive intervals of 42 d by subtracting the days of the missed intervals from the overall individual time at risk. As per PNG treatment guidelines, treatment during the follow-up period was given only if a clinical episode was detected. A potential competing risk could exist if treatment was administered for a clinical episode with one species before the first event for a heterologous species endpoint had occurred. However, since the incidence rate of clinical episodes was very low, this occurred very rarely. It should also be noted that the times at risk for the analyses of different endpoints were not adjusted for the post-treatment prophylactic effects of the aforementioned treatments. Clinical malaria episode incidence rate ratios (IRRs) were derived from models adjusted for treatment, age, and P. vivax positivity by PCR at enrolment, while the models for molFOB and P. vivax gametocyte positivity were further adjusted for village of residence. Differences between treatment groups at enrolment were investigated using chi-square and Fisher’s exact tests for categorical characteristics and Student’s t-test for normally distributed continuous variables. All tests were two-tailed, and the confidence level was set at 95%. All analyses were performed using R version 3.0.3. [43] and/or Stata version 12 [44].
The effects of MDA and MSAT programmes with anti-malarial drugs currently recommended as first-line treatment on the dynamics of P. vivax and P. falciparum transmission were investigated using a mathematical model. The model simulated the impact of a 3-d course of either dihydroartemisinin-piperaquine (DHA-PIP) or CQ to clear blood stages [45], and a 14-d PQ regimen to clear liver stages. The model also simulated the impact of tafenoquine plus CQ treatment, a highly promising short-course anti-relapse therapy that is currently undergoing phase 3 clinical trials [34]. A classical Ross-Macdonald model [46,47] was used to describe the qualitative dynamics of P. falciparum following treatment of “at risk” populations with drugs for clearing blood-stage infections. This model was extended to incorporate relapse infections of P. vivax and the effects of PQ treatment for the clearance of liver-stage hypnozoites [48]. In brief, individuals in a population can be susceptible to blood-stage infection with no liver-stage hypnozoites (S0), infected with blood-stage parasites but no hypnozoites (I0), susceptible to blood-stage infection but carrying hypnozoites (SL), or infected with both blood-stage parasites and hypnozoites (IL). Full details of the deterministic differential equations describing the mathematical model and parameter definitions are provided in S1 Table. The model was also implemented in a stochastic framework with population size 5,000 to capture stochastic variation and the potential for the elimination of transmission.
Details of all methods used in this study are given in S1–S3 Text, S1 Table, and S1 and S2 Figs, and all datasets are available in the Dryad repository: http://dx.doi.org/10.5061/dryad.m1n03 [49].
Results
Enrolment and Baseline Characteristics of Children
Of the 524 G6PD-normal children who were randomised to receive PQ/CQ/AL (PQ arm) or PL/CQ/AL (PL arm), a total of 504 children aged 4.8–10.5 y completed at least 14 d of DOT with PQ or PL and remained in the study at baseline (day 0 of follow-up) and were thus actively and passively monitored for 32 wk post-baseline (Fig 3).
No significant differences in demographic characteristics or infection status were observed at enrolment between the treatment groups (Table 1). At baseline (~4 wk after enrolment and commencement of drug treatment), four children were P. vivax–positive by qPCR (two each in the PL and PQ arms), two children in the PL arm were P. falciparum–positive, and one of these was also P. vivax–positive. These children therefore did not contribute to time at risk and were thus excluded from the respective analyses of time to first infection. No Plasmodium infections were detected by LM at baseline. During follow-up, 81.6% (2,308/2,827) of scheduled visits were attended by children in the PL arm, and 82.7% (2,248/2,717) in the PQ arm. The average number of all study contacts during follow-up did not differ between the treatment arms (PL arm: 14.0, PQ arm: 14.4, Student’s t-test, p = 0.25).

Risk of P. vivax Infection during Follow-Up
The time to first or only P. vivax blood-stage infection (as detected by qPCR) differed significantly between the two treatment arms: only 25.5% (63/247) of children who received PQ experienced at least one new P. vivax infection, compared to 65.0% (167/257) in the PL arm (Cox PH, p < 0.001; Table 2; Fig 4A). qPCR-positive, recurrent P. vivax blood-stage infections were detected rapidly in the PL arm, with 31.5% (80/254) of children infected by day 42 compared to only 8.2% (20/245) in the PQ arm (Fig 4A). As expected, LM-positive infections were less common in both arms and were observed later during follow-up (Fig 4B).
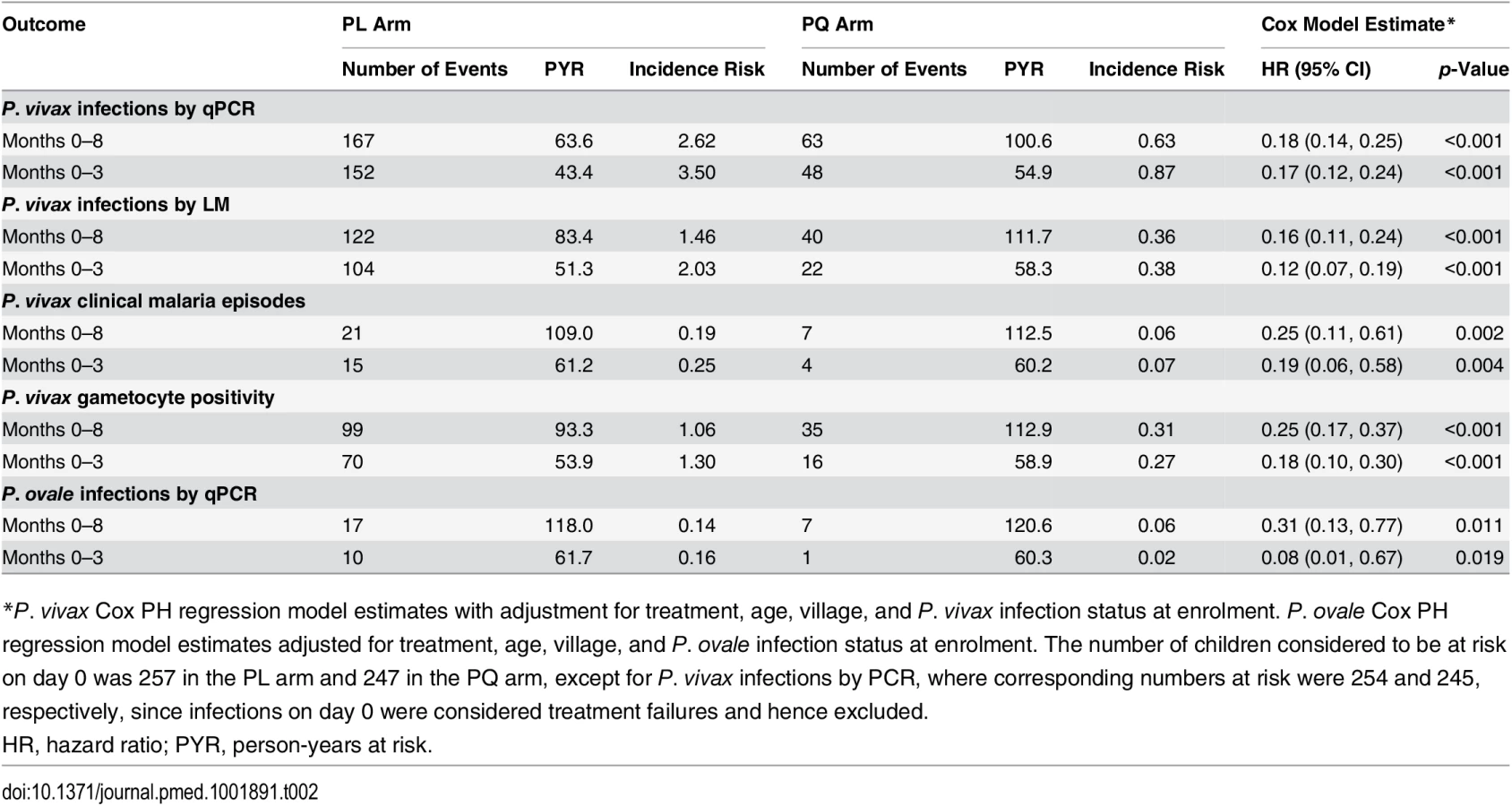

Clearance of liver stages through PQ treatment resulted in an 82%–84% reduction in the risk of recurrent P. vivax blood-stage infections diagnosed by qPCR (hazard ratio [HR] = 0.18 [95% CI 0.14, 0.25], Cox PH, p < 0.001) and LM (HR = 0.16 [95% CI 0.11, 0.24], Cox PH, p < 0.001) compared to PL (Table 2). This increased to an 83%–88% reduction in risk when only the first 3 mo of follow-up were considered (qPCR: HR = 0.17 [95% CI 0.12, 0.24], Cox PH, p < 0.001; LM: HR = 0.12 [95% CI 0.07, 0.19], Cox PH, p < 0.001; Table 2).
There was considerable heterogeneity in the prevalence and risk of P. vivax infection across the study villages. The risk of first P. vivax infection (by qPCR and LM) differed significantly among children living in different villages, with the highest risk in Bolumita (qPCR: HR = 4.70 [95% CI 3.14, 7.02], Cox PH, p < 0.001; LM: HR = 4.62 [95% CI 2.92, 7.30], Cox PH, p < 0.001, reference village Albinama) and Balanga (qPCR: HR = 2.33 [95% CI 1.53, 3.55], Cox PH, p < 0.001; LM: HR = 1.70 [95% CI 1.03, 2.81], Cox PH, p = 0.04, reference village Albinama). There was, however, no interaction between village and treatment effect. The risk of first P. vivax blood-stage infection diagnosed by qPCR after treatment was not significantly associated with age (qPCR: HR = 0.95 [95% CI 0.87, 1.03], Cox PH, p = 0.19); however, there was a 15% reduction in the risk of having at least one LM-patent P. vivax infection with each additional year of life (LM: HR = 0.85 [95% CI 0.77, 0.95], Cox PH, p = 0.003). As with village, there was no interaction between age and treatment effect.
When all P. vivax infections in a child were genotyped to identify new infections in the context of ongoing multiple clone infections, PQ treatment was associated with a 77% reduction in the incidence of genetically distinct P. vivax blood-stage clones (molFOB) compared to PL (PQ arm: molFOB = 1.62/y, PL arm: molFOB = 4.74/y, IRR = 0.23 [95% CI 0.18, 0.31], NB reg, p < 0.001; Table 3). The effect of the PQ treatment on molFOB was substantially larger in the first 3 mo (IRR = 0.21 [95% CI 0.15, 0.28], p < 0.001) than in months 4–8 (IRR = 0.34 [95% CI 0.24, 0.48], NB reg, p < 0.001; Table 3).

In addition, PQ treatment was associated with a 75% reduction in the hazard of becoming positive for P. vivax gametocytes by Pvs25 qRT-PCR compared to PL (HR = 0.25 [95% CI 0.17, 0.37], Cox PH, p < 0.001; Table 2). The incidence rate of P. vivax gametocytes (defined as the number of samples positive for Pvs25 qRT-PCR during follow-up) was more strongly reduced in the PQ arm compared to the PL arm in the first 3 mo (IRR = 0.18 [95% CI 0.11, 0.30], p < 0.001) than in the subsequent 5 mo (months 4–8: IRR = 0.37 [95% CI 0.24, 0.57], NB reg, p < 0.001; Table 3).
Irrespective of treatment group, clinical P. vivax malaria episodes were rare, with only 28 children experiencing one or more clinical P. vivax episodes during the 32-wk follow-up period (incidence risk: 0.19/child/y in PL arm and 0.06/child/y in PQ arm; Table 2). The clearance of hypnozoites by PQ treatment was associated with an 81% reduction compared to PL in the hazard of experiencing a P. vivax clinical episode of any density in the first 3 mo of follow-up (HR = 0.19 [95% CI 0.06, 0.58], Cox PH, p = 0.004; Table 2; Fig 4C) and a 75% reduction in the entire follow-up period (HR = 0.25 [95% CI 0.11, 0.61], Cox PH, p = 0.002; Table 2).
Risk of Non–P. vivax Malaria Infection during Follow-Up
Although the number of P. ovale infections diagnosed by PCR in either arm was low (incidence risk: both arms: 0.10 infections/child/y, PQ arm: 0.06, PL arm: 0.14), PQ treatment was associated with a 92% reduction compared to PL in the risk of P. ovale blood-stage infection diagnosed by qPCR in the first 3 mo of follow-up (HR = 0.08 [95% CI 0.01, 0.67], Cox PH, p = 0.019) and a 69% reduction in the entire 8-mo of follow-up period (HR = 0.31 [95% CI 0.13, 0.77], Cox PH, p = 0.011; Fig 4F; Table 2).
There was no significant association between PQ treatment and time to first (or only) P. falciparum blood-stage infection by qPCR (Cox PH, p = 0.104; Fig 4D; Table 4) or LM (Cox PH, p = 0.706; Table 4), and no effect of PQ treatment on time to first (or only) P. falciparum clinical episode during the 8 mo of follow-up (Cox PH, p = 0.333; Table 4). There was no significant association between PQ treatment and time to first (or only) P. malariae blood-stage infection by qPCR in the first 3 mo of follow-up (HR = 0.36 [95% CI 0.13, 1.02], Cox PH, p = 0.055) or during the entire follow-up period (HR = 0.55 [95% CI 0.24, 1.26], Cox PH, p = 0.157; Fig 4E; Table 4).

Implications for Malaria Control and Elimination Strategies
MSAT interventions target only individuals with detectable blood-stage parasitaemia. In the current cohort, 47.4% (239/504) of children had a P. vivax infection (single or mixed species) at enrolment, and another 12.9% (65/504) were positive with non–P. vivax blood-stage infections (Table 1). In the absence of PQ treatment, children with no patent infections at enrolment were significantly less rapidly re-infected with P. vivax (55.2% [58/105] became infected during 8 mo of follow-up) than those that had patent P. vivax (70.5% [79/112]) or P. falciparum/P. malariae/P. ovale infections (81.1% [30/37], log-rank test, p < 0.001; Fig 5; Table 5). In the PQ group, children with no patent infections were also less likely to be re-infected (17.9% [17/95]) than those in the other two groups (P. vivax patent infection: 31.7% [39/123], non–P. vivax patent infection: 25.9% [7/27], log-rank test, p = 0.0412; Fig 3; Table 5), indicating that children with no patent infections were more likely to live in low-transmission areas. PQ treatment was therefore equally efficient in preventing recurrent P. vivax infections in children with P. vivax, non–P. vivax, or no blood-stage infection at enrolment (Cox regression, adjusted for age and village of residence: likelihood ratio = 3.87, df = 2, p = 0.14 for treatment-by-infection status interaction).


The potential effect of MDA and MSAT with either blood-stage drugs only or blood - plus liver-stage drugs on the population prevalence of P. vivax and P. falciparum infections was further investigated using a basic stochastic transmission model. Administration of blood-stage drugs (DHA-PIP or CQ) was assumed to clear existing blood-stage infections and provide a 4-wk period of prophylactic protection against new infections, tafenoquine plus CQ was assumed to clear blood - and liver-stage infections and provide 8 wk of causal prophylactic protection (for full model details please refer to S1 Table). Fig 6 illustrates the predicted qualitative dynamics of P. falciparum and P. vivax transmission following two rounds of MDA (Fig 6A and 6B) or MSAT (Fig 6C and 6D) interventions with 80% coverage, separated by 6 mo. The interventions are predicted to cause a sharp decline in prevalence, followed by a gradual return to pre-intervention levels.

MDA is predicted to achieve much larger reductions in prevalence than MSAT, mostly due to the proportion of infections missed by the MSAT programme because of imperfect diagnostic sensitivity, but also because of the prophylactic protection in treated but uninfected individuals under the MDA programme.
The initial reductions achieved by each of the interventions against P. falciparum and P. vivax are the same; however, when only blood-stage drugs are administered, a rapid rebound in P. vivax prevalence is predicted due to the hypnozoite reservoir, which is left unaffected by the treatment. Notably, P. vivax levels are predicted to return to pre-intervention levels within 6 mo post-intervention for both the MDA and MSAT interventions (blue curves in Fig 6A and 6C), in contrast to P. falciparum, where, following an MDA programme, prevalence is predicted to remain below pre-intervention levels for a sustained period of time (Fig 6B).
In the stochastic transmission model, only the addition of 8-aminoquinolines to a regimen of blood-stage drugs targets the hypnozoite reservoir and prevents the rapid resurgence of P. vivax blood-stage infections caused by relapses. Consequently, interventions with PQ are shown to result in a sustained reduction of the P. vivax burden (red curves in Fig 6A and 6C). Especially in the case of MDA (red curve, Fig 6A), P. vivax parasite prevalence is predicted to remain very low (<10%) during the modelled 16 mo post-intervention. Tafenoquine is estimated to be more effective than PQ at reducing prevalence because of the higher level of efficacy and the longer duration of causal prophylaxis [34].
Discussion
By randomising children to receive either a blood - and liver-stage treatment of PQ/CQ/AL (PQ arm) or a blood-stage only treatment of PL/CQ/AL (PL arm), and thereby selectively removing hypnozoites from half of the children in the cohort, this study confirms that relapses from long-lasting liver stages account for four out of five P. vivax infections and three out of five P. ovale infections in PNG children aged 5–10 y living in an area with hyperendemic transmission. A transmission model estimated that MSAT with blood - and liver-stage treatment or MDA with blood-stage treatment alone would have only a transient effect on P. vivax transmission levels, while MDA with blood - and liver-stage treatment is predicted to be a highly effective strategy for P. vivax elimination.
The risk of having at least one PCR-detectable P. vivax infection during 8 mo of follow-up was reduced by 82% by PQ treatment. This reduction is substantially larger than the 44% reduction observed in an earlier study [27] that used a 30% lower dose of PQ combined with 7 d of artesunate, confirming that this earlier treatment regimen was not fully effective in preventing relapses. PQ also reduced the incidence of genetically distinct blood-stage infections (molFOB) [42] in the first 3 mo of follow-up by a similar amount (79%), indicating that relapses accounted for ~4/5 of all P. vivax infections in PNG children. Although the effect of the PQ treatment decreased with time since treatment, the incidence of new infections in the PQ arm was nevertheless still reduced by more than half (66%) after 3 mo of follow–up. This “wash-out” of the PQ effect is likely due to relapses activating rapidly in PNG [7] and the hypnozoite reservoir being replenished through continued exposure to new infected mosquito bites. The relatively sustained effect of the PQ treatment does however indicate that even in tropical P. vivax strains, a substantial proportion of hypnozoites remain dormant for three or more months.
Children who were not treated with PQ were also approximately four times more often positive for P. vivax gametocytes than children who were treated with PQ. Since even low-density P. vivax infections can infect mosquitoes [50–52], it is thus likely that relapsing infections are the primary source of P. vivax transmission.
PNG children acquire immunity to P. vivax rapidly [18], and, as shown in earlier studies in the same age group [20], clinical P. vivax episodes are rare. Nevertheless, PQ reduced the incidence of clinical P. vivax episodes by 69% in the first 3 mo after treatment, indicating that relapses can cause clinical episodes even in individuals with moderate levels of acquired immunity. Several studies have shown that relapses are often genetically distinct from the initial clinical episode [53,54]. These genetically diverse relapses may either be meiotic siblings [55] or originate from a previous (mosquito) bite and are likely to result in a higher risk of clinical illness in relapsing infection than that seen in malaria therapy patients infected twice with the same strain [10].
PQ treatment reduced the risk of PCR-detectable P. ovale infections by 92% in the first 3 mo of follow-up and by 69% in the entire follow-up period. This confirms that relapses account for a considerable portion of infections from this less prevalent species of relapsing malaria, and likely also sustain P. ovale transmission. This is relevant not only in PNG, but also in sub-Saharan Africa, where P. ovale prevalence can reach 4%–10% (by LM) in areas of west and central Africa [56]. We observed no significant association of PQ treatment with risk of PCR-detectable infections from the non-relapsing species P. falciparum. In the PQ arm, there was a non-significant (p = 0.055; only 24 total events) reduction in the risk of P. malariae infections in the first 3 mo of follow-up compared to the PL arm. Although P. malariae is not thought to form hypnozoites in the liver, chronic or relapsing infections of P. malariae up to 20 y after a person has left an endemic area have been documented [57,58]. Although it is currently not known how and where P. malariae infections can remain dormant for such extensive periods of time, our data suggest that such longer-lived P. malariae stages may be susceptible to PQ treatment. Larger clinical trials involving both symptomatic and asymptomatic P. malariae infections would be required to confirm the prevention of recurrent P. malariae infections by PQ treatment.
Given the very large contribution of relapses to the burden of P. vivax and P. ovale infection, illness, and transmission, it is essential that all P. vivax – and P. ovale–infected individuals receive therapy that is effective against both blood stages and relapses. PNG national standard treatment guidelines recommend treating confirmed P. vivax or P. ovale cases with 0.25 mg/kg PQ for 14 d; however, this recommendation is not being consistently implemented due to lack of access to point-of-care tests to screen for G6PD deficiency.
Interestingly, PQ was not only effective in preventing recurrent P. vivax infection in children who had PCR-detectable P. vivax infections but was equally effective in children with non–P. vivax infections and even those without any Plasmodium spp. infections at enrolment. All currently available malaria diagnostic tests identify only active blood-stage infections. They can thus not identify people who have P. vivax hypnozoites in their livers but are free of blood-stage infection. The rapid appearance of P. vivax infections and significant reduction in rates of recurrent infections in PQ-treated qPCR-negative children indicate that the number of PNG children without blood-stage infections who carry P. vivax hypnozoites is comparable to the number of children with P. vivax infections. The presence of active P. vivax blood-stage infection is thus a poor predictor for the risk of P. vivax relapse.
The rapid recurrence of P. vivax infections after treatment of P. falciparum infections has been observed in numerous P. falciparum in vivo drug efficacy trials, both in PNG [45,59] and elsewhere [60]. It has therefore been argued that anti-relapse therapy should be administered for all patients with malaria in regions of P. vivax co-endemicity [60]. Our results clearly support this recommendation and also extend it to asymptomatic parasite carriers.
With the renewed drive to eliminate malaria, the role of asymptomatic and/or submicroscopic infections in sustaining malaria transmission has become a major focus [15,61,62]. An increasing number of studies show that these infections contribute significantly to both P. falciparum and P. vivax transmission at all levels of endemicity [50–52,63,64]. As first noted by Robert Koch in 1900 during studies in then German New Guinea, the control of these infections is essential if elimination is to be achieved rapidly [65].
MSAT and MDA with artemisinin-based combination therapies are two interventions aimed at reducing the asymptomatic reservoir [32,66,67]. Although the simple model implemented in the present study agrees with the findings from more detailed P. falciparum models [67] that an MSAT programme with a highly sensitive diagnostic test such as qPCR can effectively reduce P. falciparum transmission, field trials have shown that MSAT with a less sensitive diagnostic test such as an RDT has limited or no effect on transmission [68–70]. As mass screening by PCR is difficult to implement, focalised MDA may be a more practical approach [71].
Our modelling predicted that MSAT will have only limited effectiveness for P. vivax even if conducted with a sensitive molecular diagnostic test and including an anti-liver-stage treatment since it will not target the blood-stage-negative population harbouring hypnozoites. MDA, on the other hand, is predicted to be highly effective in reducing the burden of future P. vivax infections but only if conducted with anti-blood - and anti-liver-stage treatment. Thus, effective control of P. vivax with anti-malarial drugs will require the inclusion of a treatment to attack the hypnozoite reservoir and will require mass administration regardless of the presence of blood-stage infections to target the undetectable parasite reservoir.
Currently, PQ is the only licensed drug with activity on the hypnozoite stage capable of preventing relapses [72]. However, due to its association with haemolysis in individuals with G6PD deficiency [72,73] and its long dosing schedule (up to 14 d), this drug is not in widespread use in many endemic areas, and WHO currently advises against PQ treatment without prior G6PD deficiency testing. In the absence of a reliable and affordable point-of-care G6PD test, the routine use of PQ for treatment thus remains a challenge in many endemic regions. The challenges of implementing MDA with PQ are even larger, although not insurmountable [74]. A new generation of G6PD tests are being developed [75,76], and the arrival of tafenoquine, a long-acting 8-aminoquinoline that can be given as a single dose [34,76], will not only make MDA logistically simpler, but our modelling also predicts that MDA with tafenoquine will be more effective than regimens using PQ. Nevertheless, the development of alternative anti-hypnozoite treatments remains an important research priority for the elimination of P. vivax [16].
The treatment schedule used in the present study was optimised for the best possible anti-hypnozoite activity. The results from the clinical trial are therefore not directly indicative of the effectiveness of an MDA programme with standard 14-d PQ dosing. As a consequence, we assumed a significantly lower PQ efficacy (75%) in our model than we found in our study. Similarly, our results refer to the fast-relapsing P. vivax strains that are found in relatively highly endemic areas in the southwest Pacific, southeast Asia, and parts of Amazonia [77]. Further studies in areas with lower transmission levels and longer relapse intervals will be required to determine the generalisability of our finding of relapses causing 80% of infections and to extend the model predictions to other geographical regions and transmission settings.
In conclusion, this study demonstrates that relapsing infections are the overwhelming source (i.e., ~80%) of not only P. vivax blood-stage infections in children but also clinical episodes, and that they contribute substantially to maintaining transmission. Given the very ambitious timelines set by political leaders of P. vivax–endemic regions and limited funding, it is essential that scarce resources available for both P. vivax research and elimination be optimally allocated. The development of novel anti-hypnozoite drugs and interventions that can specifically target the hypnozoite reservoir are of highest priority. However, by predicting that MSAT programmes will not be effective in reducing the burden of P. vivax in affected populations, the models presented here also suggest that it is not worthwhile investigating or implementing MSAT programmes in P. vivax–endemic countries. Instead, efforts should now be directed towards the development of approaches for MDA programmes targeting areas and risk groups with confirmed local transmission.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. World Health Organization. World malaria report 2014. Geneva: World Health Organization; 2014.
2. Cotter C, Sturrock H, Hsiang M, Liu J, Phillips A, Hwang J, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet. 2013;382 : 900–911. doi: 10.1016/S0140-6736(13)60310-4 23594387
3. Pan American Health Organization, World Health Organization. Central America and Hispaniola seek to eliminate malaria by 2025. 21 May 2013. Geneva: World Health Organization.
4. Asia Pacific Leaders Malaria Alliance. East Asia Summit adopts unprecedented regional malaria goal. 14 November 2014. Mandaluyong City (Philippines): Asia Pacific Leaders Malaria Alliance.
5. Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis. 2009;9 : 555–566. doi: 10.1016/S1473-3099(09)70177-X 19695492
6. Baird JK. Eliminating malaria—all of them. Lancet. 2010;376 : 1883–1885. doi: 10.1016/S0140-6736(10)61494-8 21035840
7. White NJ. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J. 2011;10 : 297. doi: 10.1186/1475-2875-10-297 21989376
8. Carson P, Flanagan C, Ickes C, Alving A. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science. 1956;124 : 484–485. 13360274
9. White NJ, Imwong M. Relapse. Adv Parasitol. 2012;80 : 113–150. doi: 10.1016/B978-0-12-397900-1.00002-5 23199487
10. Mckenzie FE, Jeffery GM, Collins WE. Plasmodium vivax blood-stage dynamics. J Parasitol. 2008;88 : 521–535.
11. Barry AE, Waltmann A, Koepfli C, Barnadas C, Mueller I. Uncovering the transmission dynamics of Plasmodium vivax using population genetics. Pathog Glob Health. 2015;109 : 142–152. doi: 10.1179/2047773215Y.0000000012 25891915
12. Liu Y, Auburn S, Cao J, Trimarsanto H, Zhou H, Gray K-A, et al. Genetic diversity and population structure of Plasmodium vivax in central China. Malar J. 2014;13 : 262. doi: 10.1186/1475-2875-13-262 25008859
13. Gray K-A, Dowd S, Bain L, Bobogare A, Wini L, Shanks GD, et al. Population genetics of Plasmodium falciparum and Plasmodium vivax and asymptomatic malaria in Temotu Province, Solomon Islands. Malar J. 2013;12 : 429. doi: 10.1186/1475-2875-12-429 24261646
14. Gunawardena S, Ferreira M, Kapilananda G, Wirth D, Karunaweera N. The Sri Lankan paradox: high genetic diversity in Plasmodium vivax populations despite decreasing levels of malaria transmission. Parasitology. 2014;141 : 880–890. doi: 10.1017/S0031182013002278 24533989
15. Lin JT, Saunders DL, Meshnick SR. The role of submicroscopic parasitemia in malaria transmission: what is the evidence ? Trends Parasitol. 2014;30 : 183–190. doi: 10.1016/j.pt.2014.02.004 24642035
16. malERA Consultative Group on Drugs. A research agenda for malaria eradication: drugs. PLoS Med. 2011;8:e1000402. doi: 10.1371/journal.pmed.1000402 21311580
17. World Health Organization. Plasmodium vivax control & elimination: development of global strategy and investment case. Geneva: World Health Organization; 2012.
18. Lin E, Kiniboro B, Gray L, Dobbie S, Robinson L, Laumaea A, et al. Differential patterns of infection and disease with P. falciparum and P. vivax in young Papua New Guinean children. PLoS ONE. 2010;5:e9047. doi: 10.1371/journal.pone.0009047 20140220
19. Senn N, Rarau P, Stanisic DI, Robinson L, Manong D, Salib M, et al. Intermittent preventive treatment for malaria in Papua New Guinean infants exposed to Plasmodium falciparum and P. vivax: a randomized controlled trial. PLoS Med. 2012;9:e1001195. doi: 10.1371/journal.pmed.1001195 22479155
20. Michon P, Cole-Tobian JL, Dabod E, Schoepflin S, Igu J, Susapu M, et al. The risk of malarial infections and disease in Papua New Guinean children. Am J Trop Med Hyg. 2007;76 : 997–1008. 17556601
21. Kasehagen LJ, Mueller I, McNamara DT, Bockarie MJ, Kiniboro B, Rare L, et al. Changing patterns of Plasmodium blood-stage infections in the Wosera region of Papua New Guinea monitored by light microscopy and high throughput PCR diagnosis. Am J Trop Med Hyg. 2006;75 : 588–596. 17038678
22. Mueller I, Widmer S, Michel D, Maraga S, McNamara DT, Kiniboro B, et al. High sensitivity detection of Plasmodium species reveals positive correlations between infections of different species, shifts in age distribution and reduced local variation in Papua New Guinea. Malar J. 2009;8 : 41. doi: 10.1186/1475-2875-8-41 19284594
23. Arnott A, Barnadas C, Senn N, Siba P, Mueller I, Reeder JC, et al. High genetic diversity of Plasmodium vivax on the north coast of Papua New Guinea. Am J Trop Med Hyg. 2013;89 : 188–194. doi: 10.4269/ajtmh.12-0774 23690553
24. Schultz L, Wapling J, Mueller I, Ntsuke PO, Senn N, Nale J, et al. Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar J. 2010;9 : 336. doi: 10.1186/1475-2875-9-336 21092231
25. Koepfli C, Robinson LJ, Rarau P, Salib M, Sambale N, Wampfler R et al. Blood-stage parasitaemia and age determine Plasmodium falciparum and P. vivax gametocytaemia in Papua New Guinea. PLoS ONE. 2015;10:e0126747. doi: 10.1371/journal.pone.0126747 25996916
26. Mueller I, Schoepflin S, Smith TA, Benton KL, Bretscher MT, Lin E, et al. Force of infection is key to understanding the epidemiology of Plasmodium falciparum malaria in Papua New Guinean children. Proc Natl Acad Sci U S A. 2012;109 : 10030–10035. doi: 10.1073/pnas.1200841109 22665809
27. Betuela I, Rosanas-Urgell A, Kiniboro B, Stanisic DI, Samol L, de Lazzari E, et al. Relapses contribute significantly to the risk of Plasmodium vivax infection and disease in Papua New Guinean children 1–5 years of age. J Infect Dis. 2012;206 : 1771–1780. doi: 10.1093/infdis/jis580 22966124
28. Hetzel M, Pulford J, Gouda H, Hodge A, Siba P, Mueller I. The Papua New Guinea National Malaria Control Program: primary outcome and impact indicators, 2009–2014. Goroka: Papua New Guinea Institute of Medical Research; 2014.
29. Mckenzie FE, Magill AJ, Forney JR, Lucas C, Erhart LM, Meara WPO, et al. Gametocytemia in Plasmodium vivax and Plasmodium falciparum infections. J Parasitol. 2006;92 : 1281–1285. 17304807
30. Douglas NM, Simpson JA, Phyo AP, Siswantoro H, Hasugian AR, Kenangalem E, et al. Gametocyte dynamics and the role of drugs in reducing the transmission potential of Plasmodium vivax. J Infect Dis. 2013;208 : 801–812. doi: 10.1093/infdis/jit261 23766527
31. Barbosa S, Gozze AB, Lima NF, Batista CL, Bastos MDS, Nicolete VC, et al. Epidemiology of disappearing Plasmodium vivax malaria: a case study in rural Amazonia. PLoS Negl Trop Dis. 2014;8:e3109. doi: 10.1371/journal.pntd.0003109 25166263
32. Okell LC, Griffin JT, Kleinschmidt I, Hollingsworth TD, Churcher TS, White MJ, et al. The potential contribution of mass treatment to the control of Plasmodium falciparum malaria. PLoS ONE. 2011;6:e20179. doi: 10.1371/journal.pone.0020179 21629651
33. Grueninger H, Hamed K. Transitioning from malaria control to elimination: the vital role of ACTs. Trends Parasitol. 2013;29 : 60–64. doi: 10.1016/j.pt.2012.11.002 23228225
34. Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK, et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet. 2014;383 : 1049–1058. doi: 10.1016/S0140-6736(13)62568-4 24360369
35. Hanboonkunupakarn B, Ashley EA, Jittamala P, Tarning J, Pukrittayakamee S, Hanpithakpong W, et al. Open-label crossover study of primaquine and dihydroartemisinin-piperaquine pharmacokinetics in healthy adult Thai subjects. Antimicrob Agents Chemother. 2014;58 : 7340–7346. doi: 10.1128/AAC.03704-14 25267661
36. Genton B, D’Acremont VV, Rare L, Baea K, Reeder JC, Alpers MP, et al. Plasmodium vivax and mixed infections are associated with severe malaria in children : a prospective cohort study from Papua New Guinea. PLoS Med. 2008;5:e127. doi: 10.1371/journal.pmed.0050127 18563961
37. Betuela I, Bassat Q, Kiniboro B, Robinson LJ, Rosanas-Urgell A, Stanisic D, et al. Tolerability and safety of primaquine in Papua New Guinean children 1 to 10 years of age. Antimicrob Agents Chemother. 2012;56 : 2146–2149. doi: 10.1128/aac.05566-11 22252800
38. Laman M, Laman M, Moore BR, Benjamin J, Padapu N, Tarongka N, et al. Comparison of an assumed versus measured leucocyte count in parasite density calculations in Papua New Guinean children with uncomplicated malaria. Malar J. 2014;13 : 145. doi: 10.1186/1475-2875-13-145 24739250
39. Wampfler R, Mwingira F, Javati S, Robinson L, Betuela I, Siba P, et al. Strategies for detection of Plasmodium species gametocytes. PLoS ONE. 2013;8:e76316. doi: 10.1371/journal.pone.0076316 24312682
40. Rosanas-Urgell A, Mueller D, Betuela I, Barnadas C, Iga J, Zimmerman PA, et al. Comparison of diagnostic methods for the detection and quantification of the four sympatric Plasmodium species in field samples from Papua New Guinea. Malar J. 2010;9 : 361. doi: 10.1186/1475-2875-9-361 21156052
41. Koepfli C, Schoepflin S, Bretscher M, Lin E, Kiniboro B, Zimmerman PA, et al. How much remains undetected? Probability of molecular detection of human Plasmodia in the field. PLoS ONE. 2011;6:e19010. doi: 10.1371/journal.pone.0019010 21552561
42. Koepfli C, Colborn KL, Kiniboro B, Lin E, Speed TP, Siba PM, et al. A high force of Plasmodium vivax blood-stage infection drives the rapid acquisition of immunity in Papua New Guinean children. PLoS Negl Trop Dis. 2013;7:e2403. doi: 10.1371/journal.pntd.0002403 24040428
43. R Development Core Team. R: a language and environment for statistical computing. Vienna: R Found Statistical Computing; 2014.
44. StataCorp. Stata release 12 [computer program]. College Station (Texas): StataCorp; 2011.
45. Karunajeewa HA, Mueller I, Senn M, Lin E, Law I, Gomorrai PS, et al. A trial of combination antimalarial therapies in children from Papua New Guinea. N Engl J Med. 2008;359 : 2545–2557. doi: 10.1056/NEJMoa0804915 19064624
46. Smith D, McKenzie F. Statics and dynamics of malaria infection in Anopheles mosquitoes. Malar J. 2004;3 : 13. 15180900
47. Smith DL, Battle KE, Hay SI, Barker CM, Scott TW, McKenzie FE. Ross, Macdonald, and a theory for the dynamics and control of mosquito-transmitted pathogens. PLoS Pathog. 2012;8:e1002588. doi: 10.1371/journal.ppat.1002588 22496640
48. White MT, Karl S, Battle KE, Hay SI. Modelling the contribution of the hypnozoite reservoir to Plasmodium vivax transmission. Elife. 2014;3 : 1–19. doi: 10.7554/eLife.04692
49. Robinson LJ, Wampfler R, Betuela I, Karl S, White MT, Li Wai Suen CSN, et al. Data from: Strategies for understanding and reducing the Plasmodium vivax and Plasmodium ovale hypnozoite reservoir in Papua New Guinean children: A randomised placebo-controlled trial and mathematical model. Dryad Digital Repository.
50. Coleman R, Kumpitak C, Ponlawat A, Maneechai N, Phunkitchar V, Rachapaew N, et al. Infectivity of asymptomatic Plasmodium-infected human populations to Anopheles dirus mosquitoes in western Thailand. J Med Entomol. 2004;41 : 201–208. 15061279
51. Sattabongkot J, Maneechai N, Rosenberg R. Plasmodium vivax: gametocyte infectivity of naturally infected Thai adults. Parasitology. 1991;102 : 27–31. 2038501
52. Bharti AR, Chuquiyauri R, Brouwer KC, Stancil J, Lin J, Llanos-cuentas A, et al. Experimental infection of the neotropical malaria vector Anopheles darlingi by human patient-derived Plasmodium vivax in the Peruvian Amazon. Am J Trop Med Hyg. 2007;75 : 610–616.
53. Imwong M, Boel ME, Pagornrat W, Pimanpanarak M, McGready R, Day NPJ, et al. The first Plasmodium vivax relapses of life are usually genetically homologous. J Infect Dis. 2012;205 : 680–683. doi: 10.1093/infdis/jir806 22194628
54. Chen N, Auliff A, Rieckmann K, Gatton M, Cheng Q. Relapses of Plasmodium vivax infection result from clonal hypnozoites activated at predetermined intervals. J Infect Dis. 2007;195 : 934–941. doi: 10.1086/512242 17330782
55. Bright AT, Manary MJ, Tewhey R, Arango EM, Wang T, Schork NJ, et al. A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl Trop Dis. 2014;8:e2882. doi: 10.1371/journal.pntd.0002882 24901334
56. Mueller I, Zimmerman PA, Reeder JC. Plasmodium malariae and Plasmodium ovale—the “bashful” malaria parasites. Trends Parasitol. 2007;23 : 278–283. doi: 10.1016/j.pt.2007.04.009 17459775
57. White NJ. Malaria. In: Cook GC, Zumla AI, editors. Manson’s tropical diseases. 21st ed. Philadelphia: W. B. Saunders; 2003. pp. 1205–1296.
58. Siala E, Khalfaoui M, Bouratbine A, Hamdi S, Hili K, Aoun K. Relapse of Plasmodium malariae malaria 20 years after living in an endemic area. Presse Med. 2005;34 : 371–372. 15859572
59. Laman M, Moore BR, Benjamin JM, Yadi G, Bona C, Warrel J, et al. Artemisinin-naphthoquine versus artemether-lumefantrine for uncomplicated malaria in Papua New Guinean children: an open-label randomized trial. PLoS Med. 2014;11:e1001773. doi: 10.1371/journal.pmed.1001773 25549086
60. Douglas NM, Nosten F, Ashley EA, Phaiphun L, van Vugt M, Singhasivanon P, et al. Plasmodium vivax recurrence following falciparum and mixed species malaria: risk factors and effect of antimalarial kinetics. Clin Infect Dis. 2011;52 : 612–620. doi: 10.1093/cid/ciq249 21292666
61. Karl S, Laman M, Koleala T, Ibam C, Kasian B, Drewei NN, et al. Comparison of three methods for detection of gametocytes in Melanesian children treated for uncomplicated malaria. Malar J. 2014;13 : 1–8. doi: 10.1186/1475-2875-13-319
62. Okell LC, Bousema T, Griffin JT, Ouédraogo AL, Ghani AC, Drakeley CJ. Factors determining the occurrence of submicroscopic malaria infections and their relevance for control. Nat Commun. 2012;3 : 1237. doi: 10.1038/ncomms2241 23212366
63. Schneider P, Bousema J, Gouagna L, Otieno S, van de Vegte-Bolmer M, Omar S, et al. Submicroscopic Plasmodium falciparum gametocyte densities frequently result in mosquito infection. Am J Trop Med Hyg. 2007;76 : 470–474. 17360869
64. Ouédraogo AL, Bousema T, Schneider P, de Vlas SJ, Ilboudo-Sanogo E, Cuzin-Ouattara N, et al. Substantial contribution of submicroscopical Plasmodium falciparum gametocyte carriage to the infectious reservoir in an area of seasonal transmission. PLoS ONE. 2009;4:e8410. doi: 10.1371/journal.pone.0008410 20027314
65. Koch R. Professor Koch’s investigations on malaria: fourth report to the Colonial Department of the German Colonial Office. Br Med J. 1900;1 : 1597–1598. 20759083
66. Griffin JT, Hollingsworth TD, Okell LC, Churcher TS, White M, Hinsley W, et al. Reducing Plasmodium falciparum malaria transmission in Africa: a model-based evaluation of intervention strategies. PLoS Med. 2010;7:e1000324. doi: 10.1371/journal.pmed.1000324 20711482
67. Crowell V, Briët OJT, Hardy D, Chitnis N, Maire N, Di Pasquale A, et al. Modelling the cost-effectiveness of mass screening and treatment for reducing Plasmodium falciparum malaria burden. Malar J. 2013;12 : 4. doi: 10.1186/1475-2875-12-4 23286228
68. Tiono AB, Ouédraogo A, Ogutu B, Diarra A, Coulibaly S, Gansané A, et al. A controlled, parallel, cluster-randomized trial of community-wide screening and treatment of asymptomatic carriers of Plasmodium falciparum in Burkina Faso. Malar J. 2013;12 : 79. doi: 10.1186/1475-2875-12-79 23442748
69. Tiono AB, Ouédraogo A, Diarra A, Coulibaly S, Soulama I, Konaté AT, et al. Lessons learned from the use of HRP-2 based rapid diagnostic test in community-wide screening and treatment of asymptomatic carriers of Plasmodium falciparum in Burkina Faso. Malar J. 2014;13 : 30. doi: 10.1186/1475-2875-13-30 24467946
70. Halliday KE, Okello G, Turner EL, Njagi K, Mcharo C, Kengo J, et al. Impact of intermittent screening and treatment for malaria among school children in Kenya: a cluster randomised trial. PLoS Med. 2014;11:e1001594. doi: 10.1371/journal.pmed.1001594 24492859
71. Mosha JF, Sturrock HJW, Greenhouse B, Greenwood B, Sutherland CJ, Gadalla N, et al. Epidemiology of subpatent Plasmodium falciparum infection: implications for detection of hotspots with imperfect diagnostics. Malar J. 2013;12 : 221. doi: 10.1186/1475-2875-12-221 23815811
72. Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39 : 1336–1345. doi: 10.1086/424663 15494911
73. Beutler E. G6PD deficiency. Blood. 1994;84 : 3613–3636. 7949118
74. Kondrashin A, Baranova AM, Ashley EA, Recht J, White NJ, Sergiev VP. Mass primaquine treatment to eliminate vivax malaria: lessons from the past. Malar J. 2014;13 : 51. doi: 10.1186/1475-2875-13-51 24502194
75. Von Seidlein L, Auburn S, Espino F, Shanks D, Cheng Q, McCarthy J, et al. Review of key knowledge gaps in glucose-6-phosphate dehydrogenase deficiency detection with regard to the safe clinical deployment of 8-aminoquinoline treatment regimens: a workshop report. Malar J. 2013;12 : 112. doi: 10.1186/1475-2875-12-112 23537118
76. Kim S, Nguon C, Guillard B, Duong S, Chy S, Sum S, et al. Performance of the CareStartTM G6PD deficiency screening test, a point-of-care diagnostic for primaquine therapy screening. PLoS ONE. 2011;6:e28357. doi: 10.1371/journal.pone.0028357 22164279
77. Battle KE, Karhunen MS, Bhatt S, Gething PW, Howes RE, Golding N, et al. Geographical variation in Plasmodium vivax relapse. Malar J. 2014;13 : 144. doi: 10.1186/1475-2875-13-144 24731298
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2015 Číslo 10
- Alternativní léčebné možnosti u hypercholesterolemie při intoleranci statinů
- Vliv kombinace nutraceutik na remodelaci levé komory srdeční u osob s metabolickým syndromem
- Nutraceutika a jejich ovlivnění mírného kardiometabolického rizika
- Princip účinku medu v léčbě chronických i infikovaných ran
- Superoxidovaný roztok a jeho využití v léčbě ran
Nejčtenější v tomto čísle
- Effect of Health Risk Assessment and Counselling on Health Behaviour and Survival in Older People: A Pragmatic Randomised Trial
- Using Qualitative Evidence in Decision Making for Health and Social Interventions: An Approach to Assess Confidence in Findings from Qualitative Evidence Syntheses (GRADE-CERQual)
- Monitoring Pharmacologically Induced Immunosuppression by Immune Repertoire Sequencing to Detect Acute Allograft Rejection in Heart Transplant Patients: A Proof-of-Concept Diagnostic Accuracy Study
- The REporting of studies Conducted using Observational Routinely-collected health Data (RECORD) Statement