
Investigating the Causal Relationship of C-Reactive Protein with 32 Complex Somatic and Psychiatric Outcomes: A Large-Scale Cross-Consortium Mendelian Randomization Study
Using genetic instruments, Behrooz Z. Alizadeh and colleagues examine the hypothesis that increased CRP levels play a causal role in common somatic and psychiatric conditions.
Published in the journal:
. PLoS Med 13(6): e32767. doi:10.1371/journal.pmed.1001976
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001976
Summary
Using genetic instruments, Behrooz Z. Alizadeh and colleagues examine the hypothesis that increased CRP levels play a causal role in common somatic and psychiatric conditions.
Introduction
Emerging evidence suggests that persistent dysregulation of the inflammatory response is linked to a plethora of complex somatic and neuropsychiatric disorders [1–18]. Epidemiological studies have shown that C-reactive protein (CRP), a well-studied biomarker of inflammation, is associated with and exhibits reliable predictive value for cardiovascular disease [19,20], type 2 diabetes [21], immunity-related disorders such as inflammatory bowel disease (IBD) [22], rheumatoid arthritis [23], and all-cause mortality [20,24]. Nevertheless, the evidence for a causal involvement of CRP in these outcomes from traditional experimental or observational studies remains controversial [25,26], fueling the debate surrounding whether CRP contributes to the chain of causality in disease mechanisms [27]. The use of genetically informed instrumental variables (IVs), termed Mendelian randomization (MR), is a complementary approach to epidemiological observations and allows investigation of whether the effect of an exposure (i.e., CRP level) on observed outcome phenotypes is likely to be causal [28].
Recent large-scale MR studies, focusing mainly on cardiovascular disease and metabolic traits, failed to show a causal association between CRP level and these outcomes (S1 Table). This has led to the notion that elevated CRP levels do not causally contribute to these traits and disorders. However, these studies used a single CRP-associated single nucleoid polymorphism (SNP) or a very limited set of CRP-associated SNPs (S1 Table). Common SNPs serving as proxies for CRP level represent only a small effect on CRP level per se and thus require a large enough sample size to detect causal effects on the outcome. Moreover, most studies have generally included a limited range of common complex diseases, often not more than two or three outcomes, or they have been performed in a single or small population, yielding inadequate study power (S1 Table). In other words, the evidence for a causal relationship between CRP and a broad range of common traits or diseases remains inconclusive. This is mostly due to the lack of well-powered MR studies that use optimally informative genetic IVs for CRP. Here, we sought to comprehensively examine the hypothesis that genetically determined CRP level directly contributes to common somatic and psychiatric outcomes. To optimize IV power, we applied a MR approach using summary statistics from large-scale genome-wide association study (GWAS) consortia of 32 somatic and psychiatric phenotypes for the four CRP variants representing 98% of the common variation in the CRP gene and for the largest known set of independent SNPs known to be associated with CRP. We further aimed to confirm the identified association between CRP and schizophrenia using a CRP polygenic risk score (CRPPRS) from individual-level genotype data from the largest consortium of schizophrenia to date. We performed an in silico pathway analysis (see Discussion) to provide insights into the possible mechanism underlying the observed association of CRP level with schizophrenia.
Methods
Study Design and Rationale
The present MR study consists of two key components. First, we used established gene variants associated with CRP level and combined them to build two genetic risk scores (GRSs) for CRP. The first GRS consisted of only four SNPs in the CRP gene (GRSCRP) selected from the largest recent MR study of CRP [29], and the second consisted of 18 SNPs that were associated with CRP level at a genome-wide significance level in the largest GWAS for CRP to date (GRSGWAS) [30]. Second, we obtained summary association statistics from GWAS consortia for a panel of 32 common somatic and psychiatric outcomes (Table 1). BPP and BZA selected the studies, and contacted each consortium with a standardized request for study data, including the name of the study or consortium, the number of cases and controls, the number of available CRP SNPs for GRSCRP and GRSGWAS, and the estimated effect for each SNP (or its proxy) on outcome, i.e., the per allele regression coefficient with standard error or the odds ratio (OR) and corresponding 95% confidence interval. Data were available for 32 different outcomes in five broad disease/trait classes (autoimmune/inflammatory, cardiovascular, metabolic, neurodegenerative, and psychiatric), including at least 1,566, and up to 184,305, participants per outcome from populations of European ancestry (Table 1). These outcomes were selected based on the following two inclusion criteria: (i) the outcome having been associated with CRP level in epidemiological studies and (ii) availability of large meta-GWAS analyses for the outcome (Table 1).

Genetic Instruments
Weak IVs yielding insufficient statistical power may have hampered estimation of causal effects of CRP on the outcomes in previous analyses (S1 Table). Our MR approach, by using GWAS data and combining multiple independent SNPs into a GRS (i.e., IV), has the potential to greatly increase power. The selected SNPs have been described elsewhere [30,55,56] and are further detailed in S2–S4 Tables. These IVs were used to test the combined effect of the associations of CRP-level-influencing alleles with the outcomes. Our approach was implemented in such a way that the effects of both independent SNPs in the CRP gene (GRSCRP) [55,56] (S1 Methods) and independent SNPs known to be genome-wide significantly associated with CRP levels (GRSGWAS) [30], as well as pleiotropic effects of SNPs, could be discriminated [57]. Pleiotropy exists if CRP SNPs influence exposures (risk factors) other than CRP level and therefore violate one of the key MR assumptions.
Statistical Analysis
All analyses were done using the GRS function implemented in the grs.summary module of the R package Genetics ToolboX (version 2.15.1 for Windows). The grs.summary module approximates the regression of an outcome onto an additive GRS, using only single SNP association summary statistics extracted from GWAS results. The method is described in more detail elsewhere [58]. In brief, we performed MR analyses using GRS IVs in two steps. First, we used four individual CRP gene SNPs (i.e., IVs) associated with CRP level [56,59] (S2 and S3 Tables) to create a weighted GRS, named GRSCRP, corresponding to the joint effect of the four SNPs within the CRP gene [55]. We extracted ω (the estimated coefficient, or weight) for individual SNPs from association results reported by the CRP Coronary Heart Disease Genetics Collaboration (CCGC) [29,55]; ω represents a one-unit (in mg/l) increase of the natural log of CRP level (lnCRP) per dose of the coded allele. The four tagging SNPs represent 98% of the common variation in the CRP gene, assuming a minor allele frequency of ≥0.05 and an r2 threshold of ≥0.8, and aggregately explain ~2% of the total variation (i.e., phenotypic variance) in serum CRP level in populations of European descent [55,59]. Second, we constructed a multilocus GRS, named GRSGWAS, that combined 18 SNPs associated with serum CRP level at a genome-wide significance level (p < 5×10−8; S2 and S3 Tables), derived from a large meta-GWAS analysis of CRP conducted by the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) Consortium [30]. This multilocus GRS explains approximately ~5% of the total variation in serum CRP level [30].
We integrated ω for each CRP SNP from the reference data of CCGC [55] or meta-analysis of GWASs [30] for CRP level with the summary association statistics extracted from the GWAS consortium data for each outcome (S1 Data; S2 Methods). This MR approach using meta-GWAS summary statistics data is equivalent to an inverse-variance-weighted meta-analysis and has previously been validated in comparison to individual-level data [57,60]. To estimate the causal effect of CRP level on an outcome, we obtained the β values (estimated effects from regression analysis) for the effects of CRP SNPs on the outcome, with standard errors, seβ, from the corresponding GWAS results. Where no summary statistics for a CRP SNP in the GRS IVs were available in the look-up dataset, we chose the proxy SNP that had the highest linkage disequilibrium with the initial SNP (r2 > 0.9 in HapMap release 22; S3 Table). If several proxy SNPs had the exact same r2 value, we chose the proxy nearest to the original SNP in the instrument. Separate regressions of outcomes on GRSs were performed to calculate αIV estimators (i.e., causal IV estimators) for each outcome. Correspondingly, the value of a GRS is the sum of the ω values multiplied by the allele dosage (i.e., 0, 1, or 2) for each CRP SNP in the CCGC or in the CHARGE Consortium data [30,55]. For uncorrelated SNPs, when maximizing the likelihood function, the αIV value and its standard error, seα, can be approximated with the formula α ≅ (Σω × β × seβ−2) /(Σω2 × seβ−2), with seα ≅ √1/ Σω2 × seβ−2. lnCRP was used as the outcome in reference studies [30,55], so in obtaining the ω values (i.e., effect sizes) for each of the CRP SNPs, a unit increase in lnCRP equals a 10 symmetric percentage (s%) increase in CRP level, which corresponds to a unit change in the level of a continuous outcome or logit of risk estimate (i.e., beta coefficient) for a dichotomous outcome [61]. The αIV value (i.e., causal estimate) for each CRP SNP is, therefore, presented for each outcome as corresponding to a 10-s% increase in actual CRP level. During the course of this study, an updated, larger GWAS dataset for coronary artery disease (CAD) became publicly available (CARDIoGRAMplusC4D Consortium, release 2015 [41]); we therefore redid the analysis for CAD using the release 2015 data.
To assess which SNPs might have violated the key MR assumption regarding pleiotropy, we performed goodness-of-fit tests to correct both GRSs for the heterogeneity of their corresponding SNPs’ effects on each outcome. Heterogeneity, which indicates the potential presence of pleiotropy, was measured using the Q statistic and was considered statistically significant at a conservative uncorrected p-value of <0.05. Although heterogeneity could be an indicator of pleiotropy, there are other factors that could introduce heterogeneity in the analyses. Even though the adjustments for heterogeneity that we have made could be overconservative, we have used this method in order to minimize false positives. After stepwise removal of SNPs with potential pleiotropic effects, we repeated the analyses until significant heterogeneity was no longer observed.
To further ensure the strength of these two GRSs as IVs, we generated an F-statistic for each outcome. We used variance in lnCRP explained by each set of CRP SNPs (2% and 5%, respectively, for GRSCRP and GRSGWAS) to calculate the F-statistic using the formula F-statistic = [R2 × (n − 1 − K)]/[(1 − R2) × K], where R2 represents the proportion of variability in CRP level that is explained by the GRS, n represents sample size, and K represents the number of IVs included in model (i.e., for this study K = 1) [62]. As a rule of thumb, an F-value above ten indicates that a causal estimate is unlikely to be biased due to weak instruments [57].
Multiple Testing
The present study included 32 independent sample sets. For each sample set, we did one statistical test, for which a global nominal significance level of ≤0.05 was considered as satisfactory to derive conclusions. The need for correction for multiple testing is debatable. Nevertheless, to ensure the validity of our conclusions, we took a conservative approach and applied a Bonferroni-corrected significance threshold calculated as 0.05 divided by 32 (i.e., 0.0016). We considered a statistical test with an observed p-value more than 0.05 as a definitely nonsignificant result, i.e., no association; an observed p-value equal to or less than 0.05 as nominally significant evidence for a potential, but yet to be confirmed, causal association; and an observed p-value equal to or less than 0.0016 as statistically significant evidence for a causal association.
CRP Polygenic Risk Score and Schizophrenia Using Individual-Level Data
In an ancillary follow-up study, inspired by comments by the editors and the reviewers, we aimed further to determine whether GRSGWAS was causally associated with schizophrenia using individual-level data retrieved from the Psychiatric Genomics Consortium (PGC) schizophrenia dataset (S3 Methods) [54]. This dataset consisted of 36 independent cohorts with a combined 25,629 cases and 30,976 controls for which we had ethics approval (S4 Methods). Three family-based samples of European ancestry (1,235 parent–affected offspring trios) were excluded from our analysis. To evaluate whether the observed protective causal association between GRSGWAS and schizophrenia was persistent, we investigated whether the CRPPRS was also protectively associated with schizophrenia. Briefly, CRPPRS values were calculated for each individual by summing the total effect of the SNP dosages by their effect size. In addition to the 18 genome-wide significant CRP SNPs, we grouped subthreshold CRP-associated SNPs at the following p-value thresholds: 1 × 10−4, 0.001, 0.01, 0.05, and 0.1. Standardized CRPPRS values were tested for association with schizophrenia case status in each cohort with adjustment for ten principal components (PCs). A fixed effects inverse-variance-weighted meta-analysis was performed across all 36 cohorts to obtain the overall effect size estimate as explained in S4 Methods and elsewhere [63]. The variance in schizophrenia case status explained by CRPPRS was estimated using the deviation in Nagelkerke’s pseudo-R2 between a null model (which included ten PCs) and the full model (which included GRS in addition to the ten PCs), calculated in R using the Functions for Medical Statistics Book with Some Demographic Data (fmsb) R package (S3 Methods). Similar to previous studies, the statistical significance of CRPPRS values was estimated based on their logistic regression coefficient [64], and reported CRPPRS ORs correspond to a 1-SD increase in CRPPRS [65].
Results
Using GRSCRP, we first tested whether a CRP-gene-determined increase in lnCRP was associated with each outcome. In Table 2, the causal effects of lnCRP estimated for each outcome are summarized. We found no heterogeneity in the IV analyses (pheterogeneity ≥ 0.11 for all outcomes), and GRSCRP was a strong instrument (F ≥ 31). IV analyses provided nominal evidence for potential causal relationships of lnCRP with risk of Crohn disease (OR 0.78 [95% CI 0.65–0.94]; p < 0.009), psoriatic arthritis (1.45 [1.04–2.04]; p < 0.03), and schizophrenia (0.90 [0.82–0.99]; p < 0.03), and with an increase in systolic blood pressure (SBP) (mean increase 1.23 mm Hg per 10-s% increase in CRP level [95% CI 0.45–2.01]; p < 0.002) and diastolic blood pressure (DBP) (0.70 [0.20–1.19]; p < 0.006). GRSCRP showed no significant effect on any of the other outcomes (Table 2; S1 Fig).

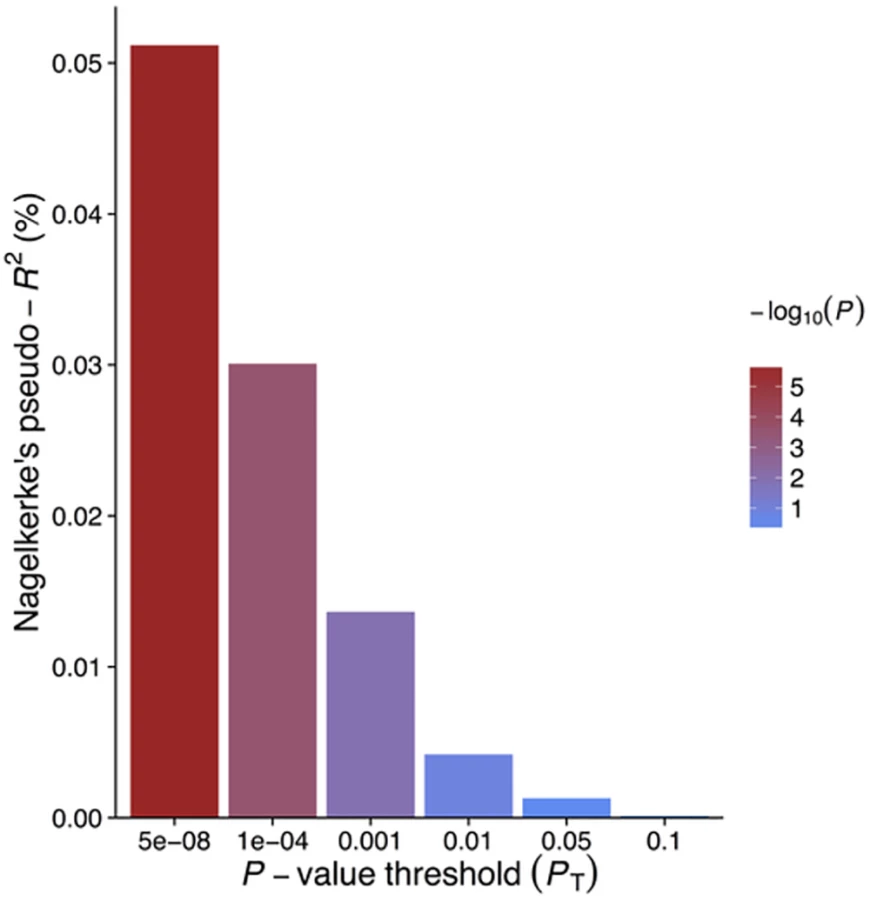
GRSGWAS showed a statistically significant protective effect of lnCRP on the risk of schizophrenia (per 10-s% increase in CRP level, OR 0.86 [95% CI 0.79–0.94]; p < 0.0010) (Figs 1 and S1; Table 3). In a follow-up analysis using the individual-level PGC data, we found that a GRS incorporating the same 18 CRP SNPs used to construct the GRSGWAS was again significantly associated with a lower risk of schizophrenia (OR 0.96 [95% CI 0.94–0.98]; p < 1.72 × 10−6). This signal persisted when we included all SNPs meeting a less stringent p-value threshold of 1 × 10−4 (OR 0.97 [95% CI 0.95–0.99]; p < 2.45 × 10−4). At less stringent p-value thresholds, less variance was explained by the logistic model, and the protective effect of CRP risk scores became less significant, but across all p-value thresholds, the direction of the effect was consistently protective (Figs 2 and 3). To ensure that the association between risk alleles for CRP and schizophrenia was not driven by a small number of genome-wide significant SNPs, we performed a leave-one-out sensitivity analysis of the 18 genome-wide SNPs. In the 18 sets of 17 SNPs, the variance explained (Nagelkerke’s pseudo-R2) ranged from 0.012% to 0.034%, with p-values ranging from 9.3 × 10−5 to 1.6 × 10−2, suggesting that the protective effect observed between risk alleles for CRP and schizophrenia was not driven by a small number of SNPs with large effects.
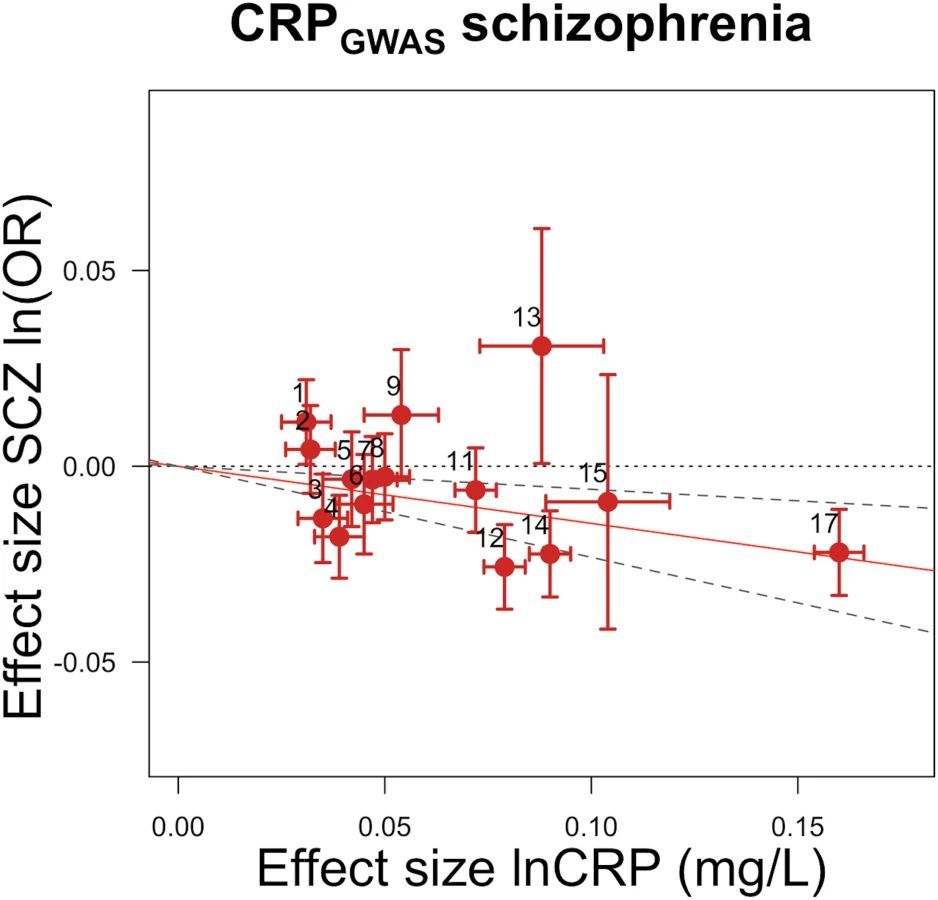


GRSGWAS also showed moderate but nominally significant effects of lnCRP on the risk of IBD (OR 0.85 [95% CI 0.74–0.98]; p < 0.03), Crohn disease (0.81 [0.70–0.94]; p < 0.005), psoriatic arthritis (1.36 [1.00–1.84]; p < 0.049), knee osteoarthritis (1.17 [1.01–1.36]; p < 0.04), and bipolar disorder (1.21 [1.05–1.40]; p < 0.007), while its effect was statistically significant for CAD (0.88 [0.84–0.94]; p < 2.4 × 10−5) (Table 3; Figs 4 and S1). GRSGWAS revealed a nominally significant effect of lnCRP on blood pressure: an increase of 0.72 (95% CI 0.11–1.34; p < 0.02) and 0.45 (0.06–0.84; p < 0.02) mm Hg in SBP and DBP, respectively (Table 3; S1 Fig). Likewise, a genetically determined 10-s% increase in CRP level was nominally associated with a 0.01 ml/min/1.73 m2 (95% CI 0.003–0.02; p < 0.005) higher estimated glomerular filtration rate from serum creatinine (eGFRcr), a 0.01 g/dl (0.0004–0.02; p < 0.04) higher serum albumin level, and a 0.03 g/dl (0.008–0.05; p < 0.009) higher serum protein level. The remaining outcomes tested for causal associations using GRSGWAS did not reach statistical significance, though the corresponding GRSGWAS proved to be a strong IV, with F-values ≥ 82 (Table 3; S1 Fig).

Using GRSGWAS, there was no significant evidence of heterogeneity of the effect size for knee osteoarthritis, bipolar disorder, schizophrenia, or SBP, while the heterogeneity test was statistically significant for psoriatic arthritis, IBD, Crohn disease, CAD, DBP, eGFRcr, serum albumin, and serum protein. These heterogeneities in the effects of GRSGWAS may be attributable to pleiotropic effects of the SNPs used to build the GRSGWAS. We subsequently performed a stepwise removal of SNPs from GRSGWAS until no significant heterogeneity remained (Table 4). This adjustment in the GRSGWAS resulted in the removal of three SNPs from the GRSGWAS for IBD (in GCKR, IRF1, and PTPN2), five SNPs from the GRSGWAS for Crohn disease (in GCKR, IL6R, IRF1, PABPC4, and PTPN2), one SNP from the GRSGWAS for psoriatic arthritis (in IRF1), three SNPs for CAD (in APOC1, HNF1A, and IL6R), one SNP from the GRSGWAS for DBP (in PABPC4), two SNPs from the GRSGWAS for eGFRcr (in LEPR and GCKR), six SNPs from the GRSGWAS for serum albumin level (in APOC1, BCL7B, GCKR, PPP1R3B, PTPN2, and IRF1), and one SNP from the GRSGWAS for serum protein level (in GCKR). After removal of these variants from the GRSGWAS, we found no statistically significant (at p < 0.0016) association between genetically increased lnCRP level and any of these outcomes (Table 4). However, the effect estimate of CRP on DBP, serum albumin, and psoriatic arthritis showed nominal association at p < 0.05. For example, for DBP, 17 SNPs remained in the GRSGWAS and yielded a slightly lower causal estimate (compared to the values before adjustment) of a 0.39 (95% CI −0.01 to 0.78) mm Hg increase in DBP per 10-s% increase in lnCRP level, with a nominal significance of p < 0.05.

Likewise, we hypothesized that the fact that GRSGWAS showed a nonsignificant effect of CRP on celiac disease, ulcerative colitis, rheumatoid arthritis, type 1 diabetes, and type 2 diabetes can be to some extent explained by the significant heterogeneity observed for these outcomes (Table 3). The stepwise adjustment in the GRSGWAS resulted in the removal of two SNPs from the GRSGWAS for celiac disease (in PABPC4 and PTPN2), one SNP from the GRSGWAS for ulcerative colitis (in GCKR), five SNPS from the GRSGWAS for rheumatoid arthritis (in HNF4A, IL6R, SALL1, NLRP3, and PTPN2), one SNP from the GRSGWAS for type 1 diabetes (in PTPN2), and one SNP from the GRSGWAS for type 2 diabetes (in APOC1). After adjusting for heterogeneity, the association of GRSGWAS with these outcomes remained statistically nonsignificant (Table 4).
Discussion
In this large-scale cross-consortium MR study of 32 complex outcomes, we found evidence for a potential protective causal relationship between elevated CRP level and schizophrenia in both genetic IVs (i.e., GRSCRP and GRSGWAS) and confirmed this protective relationship in follow-up analyses using individual-level genotype data from the schizophrenia GWAS. We also found a statistically significant association of CRP level with CAD, and nominally significant evidence for a predisposing causal association of CRP level with IBD, Crohn disease, psoriatic arthritis, knee osteoarthritis, SBP, DBP, eGFRcr, serum albumin level, serum protein level, and bipolar disorder, using GRSGWAS as an IV. However, after adjustment for heterogeneity, neither GRS showed a significant effect (at p < 0.0016) of CRP level on any of these outcomes, including CAD, nor on the 20 other common somatic and psychiatric outcomes we investigated, including celiac disease, ulcerative colitis, psoriasis (all types), rheumatoid arthritis, systemic lupus erythematous, systemic sclerosis, type 1 and 2 diabetes, stroke (all types), body mass index, chronic kidney disease, amyotrophic lateral sclerosis, Alzheimer disease, Parkinson disease, autism, and major depressive disorder.
CRP Protection against Schizophrenia
Strikingly, as opposed to the current literature and previous inconclusive small-scale studies [66–68], our findings suggest that genetically elevated levels of CRP are not predisposing but in fact protective for schizophrenia. The significant causal protective role of CRP for schizophrenia was consistent in both IVs using summary statistics, i.e., GRSCRP and GRSGWAS. When incorporating 18 genome-wide CRP-associated SNPs using individual-level data, we confirmed a modest, but significant, protective effect of CRP level for schizophrenia. This signal persisted when we included all SNPs meeting a less stringent p-value threshold of 1×10−4. Notably, the leave-one-out sensitivity analysis revealed that the genetic overlap between CRP level and schizophrenia we observed at genome-wide and 1×10−4 significance thresholds was not driven by a few major SNPs. In contrast, others have previously shown that CRP levels are significantly elevated in patients with schizophrenia [69,70], with a recent meta-analysis concluding that the association between elevated CRP and schizophrenia is indeed robust [71]. Given that clinical studies report elevated CRP levels in schizophrenia, one would expect to find that alleles for elevated CRP would confer an increased risk for schizophrenia. The fact that we found the completely opposite effect—in a cohort of over 25,000 cases and 30,000 controls—should give one pause when deriving clinical meaning from these results. Our observation that a genetically determined marginal increase in the level of CRP is likely to be protective for schizophrenia may fuel the debate about whether the observed CRP elevation in schizophrenia is a by-product of the pathogenesis of schizophrenia or directly contributing to clinical manifestations of the disorder [6]. Our finding may also point out potential biases in previous studies regarding the causes of elevated CRP levels in patients with schizophrenia, such as reverse causality and/or pleiotropic effects within chosen instruments.
The exact mechanism for how elevated CRP levels are linked to schizophrenia requires a well-defined experimental analysis. In addition to CRP variants, other recent studies have identified several inflammatory genetic variants associated with schizophrenia and bipolar disorder, which include variants in the major histocompatibility complex (MHC) region on Chromosome 6p21 [72]—harboring many cytokine genes [54,73–76]—and in the IL10 promoter [77], TNF promoter [78], IL1B [79], and C4 [80].
Biological Annotation
Following comments made by the reviewers, we explored the possible underlying pathways that may explain the potential protective causal association between CRP and schizophrenia. We performed a follow-up in silico functional pathway analysis using a previously reported approach [81] as summarized in S5 Methods and S4–S13 Tables. In brief, our results show that pathways associated with the interferon response are significantly enriched amongst genes harbored by CRP loci and their associated expression quantitative trait loci (eQTLs) and that there are differentially expressed genes between schizophrenia cases and controls. Previous studies showed that the induction of T cell IFN cytokine release stimulates microglia and astrocytes to facilitate glutamate clearance in neuronal cells without evoking inflammatory mediators [82,83]. One could speculate that CRP-interferon pathways may induce neuroprotection by contributing to glutamate clearance, leading to the protection of neurons against the oxidative stress associated with an excess of glutamate [84,85], and thereby offering a protective effect against schizophrenia.
CRP GRSGWAS Association with Bipolar Disorder
As for bipolar disorder, we found a nominal effect of a 1.21-fold increase in risk for bipolar disorder with a 10-s% increase in CRP level. Though this nominal predisposing effect needs to be confirmed, our finding corroborates epidemiological observations suggesting that elevated CRP is associated with the disease and supports a potential causal influence of general inflammation in bipolar disorder [86]. We note that, though it may be biologically sensible, this result failed to pass multiple testing correction. Confirmation by replication in independent cohorts, functional follow-up analyses, or the use of a stronger CRP GRSGWAS in upcoming studies is required to draw a definitive conclusion.
CRP GRSGWAS Association with Blood Pressure and Hypertension
We found nominally significant evidence for an up to ~0.70-mm Hg increase in blood pressure with a 10-s% increase in CRP level and no evidence of heterogeneity for SBP. Additionally, there was nominally borderline significance for a causal association between CRP and DBP after adjustment for heterogeneity. These nominally significant findings, on the one hand, are in line with numerous epidemiological studies that have highlighted an association between elevated CRP and an increased risk of hypertension. For instance, one study found an association between CRP loci and hypertension in Asian individuals [87]. An additional line of support for a possible causal association of CRP and blood pressure comes from an experimental study in which an increase in CRP gene expression in mice, and subsequently CRP protein levels, led to a rise in SBP particularly [88]. Moreover, an ex vivo study by Zhou et al. showed that combining IL6 treatment and mechanical strain leads to a consistent increase in CRP expression at the protein and mRNA levels in smooth muscle cells [89]. Both inflammatory factors and local mechanical strains are abundant in blood vessels and are well-known risk factors for high blood pressure. Our finding did not reach a statistically significant level after correction for multiple testing; thus, it may echo previous MR studies that have failed to find a causal relationship between CRP level and blood pressure or hypertension in Europeans [90,91]. However, our systematic literature review showed that previous studies had some limitations (S1 Table). For instance, no study used a refined GWAS set of 18 CRP-associated SNPs; instead, they tested single or a limited set of CRP SNPs. Using such instruments might have led to biased estimates as their corresponding effects on CRP levels have been found to be small [30,57]. A combination of weak instruments and small sample sizes might have led to type II error [28,57] and hence to a conclusion of no causal association between CRP and blood pressure traits in previous studies. When all of the evidence is taken together, a direct link between CRP and blood pressure remains to be elucidated, though our nominal associations between GRSCRP and GRSGWAS and blood pressure do add to a line of findings from experimental studies suggesting a potential causal relationship between CRP and blood pressure.
CRP GRSGWAS Association with Osteoarthritis
Our nominally significant finding that CRP might be a potential causal factor for knee osteoarthritis (using GRSGWAS) should be interpreted with caution. In line with our findings, we have previously shown that levels of CRP were higher in women with early radiological knee osteoarthritis (i.e., Kellgren-Lawrence grade 2+) and in women whose disease progressed [92]. Additionally, another study showed that genetically elevated CRP levels contribute to osteoarthritis severity [93]. However, other studies have found contrasting results [71,72,94]. One systematic review provided evidence that the relationship between CRP and osteoarthritis does exist but is dependent on body mass index [95]. It remains to be further investigated whether weight gain over the lifetime mediates the potential causal association between genetically elevated CRP and knee osteoarthritis.
CRP GRSGWAS Shows No Association with Other Remaining Outcomes
The present study was able to calculate nominal causal estimates for IBD, Crohn disease, psoriatic arthritis, CAD, eGFRcr, serum albumin level, and serum protein level using CRP GRSGWAS, but the estimates were altered by removal of SNPs from GRSGWAS based on heterogeneity tests, resulting in nominal or nonsignificant associations. These outcomes appeared therefore to have heterogeneity in the causal estimates, suggesting that these observed estimates were biased, likely due to pleiotropic effects of CRP loci. These results corroborate negative findings of previous studies (S1 Table), suggesting that a causal role of CRP in these traits and diseases is unlikely.
Methodological Concerns and Advantages
Pleiotropic biases in Mendelian randomization analyses using CRP GRSGWAS
A detailed evaluation of pleiotropic SNPs in our study showed that the method applied to identify heterogeneity sources was able to indicate and exclude several already known pleiotropic loci from the GRSGWAS IV. For instance, the use of a SNP in IL6R (rs4129267), amongst others, resulted in heterogeneity of effects on CAD risk. The same variant contributed to heterogeneity of effects for Crohn disease in our study, and it has been shown that this SNP is associated with levels of biomarkers other than CRP [56]. Further, a MR study found that IL6R SNPs, specifically the nonsynonymous SNP rs8192284, are associated with CAD risk and CRP levels [96]. Our selected IL6R SNPs, namely rs4537545 and rs4129267, are in extremely high linkage disequilibrium with rs8192284 (r2 ≥ 0.96 for both SNPs in HapMap data, CEU population). Carriers of the risk allele of rs8192284 have higher CRP, IL6, and fibrinogen levels [96]. Fibrinogen is also a well-known risk factor for CAD. Therefore, it is unclear so far which biomarker(s) mediates the effect of IL6R SNPs on CAD. Besides the IL6 locus, APOC1 and PABPC4 have been indicated as pleiotropic in three out of 32 our investigated outcomes, and PTPN2 and GCKR in six. With this information taken together, we were able to disentangle at least part of the pleiotropy regarding the causal estimates of CRP for outcomes. Again, we found no significant association of CRP GRSGWAS with IBD, Crohn disease, psoriatic arthritis, CAD, eGFRcr, serum albumin level, and serum protein level after adjustment for heterogeneity.
Using summary statistics of large-scale consortia
It is of utmost interest whether the observed effect of CRP as a risk predictor for human disease is causal, and thus whether reduction of CRP levels will lower the risk of disease. Here, we investigated the causality of CRP in 32 phenotypes by leveraging very large sample sizes collected by GWAS consortia, an approach that was much better powered than most previous MR studies. We found that genetically elevated CRP levels approximated by powerful instruments did not appear to contribute directly to most of the studied somatic and psychiatric outcomes. Our findings are consistent with previous MR studies reporting null associations of genetically elevated CRP levels with inflammation-related outcomes including CAD [56,59,97], type 2 diabetes [98], high body mass index [99], Alzheimer disease, and depression [100]. All previous MR studies were substantially limited to a single or a few outcomes, used only SNPs in the CRP gene, or had sample sizes much smaller than that of the present study (S1 Table). In addition to these studies, the current GWAS data do not corroborate epidemiological observations suggesting that elevated CRP levels are associated with amyotrophic lateral sclerosis [101], Alzheimer disease [102], Parkinson disease [103], and major depressive disorder [104]. Furthermore, patients with immunity-related disorders frequently have a very high CRP level (as high as 100 mg/l) due to their disease status. Our findings may therefore more favorably indicate reverse causality. Taken together, these results show that CRP is highly unlikely to contribute causally to most of the major common somatic and neuropsychiatric outcomes that were investigated in the present study, with the possible exception of schizophrenia.
Strength of instrumental variables
The results presented in Table 2 show that our GRSCRP is not a weak instrument, as indicated by its high F-values owing to the large sample sizes of available outcomes from GWASs for the phenotypes under study. The strength of our instrument increased considerably in all disease classes when we used variants of multiple loci associated with CRP in GWASs. However, the variants comprising the CRP GRSGWAS explain on average only a moderate ~5% of the total variance in baseline CRP levels [30]. Moreover, the possibility of effect modification by nongenetic CRP-related factors on the outcomes remains to be investigated. We may be able to create even stronger instruments based on ongoing efforts to identify additional variation influencing CRP levels. Even if larger sample sizes and stronger instruments can be realized, the overwhelming lack of causal effects observed for most outcomes in our study implies that therapies targeted at lowering CRP will not directly result in decreased risk of the investigated outcomes, or in better symptom management [105,106].
Using summary statistics instead of individual-level data
Here we used summary association statistics obtained from previously conducted meta-GWASs in order to maximize our study power. One may argue this may induce bias compared to when one uses individual-level data. Nevertheless, previous studies showed high agreement in results from MR methods using GWAS summary data and individual-level data [60,107]; Furthermore, our analyses of individual-level data for schizophrenia led to the same conclusion as our analyses using summary statistics data, confirming the robustness of our methodological approach.
Other potential sources of bias
An important rationale for MR is that the gene variants do not change over time and are inherited randomly. Thus, the genetic variants are considered free from confounding and reverse causation [108]. However, one cannot completely control for the possibility of confounding of genotype–intermediate phenotype–disease associations. For instance, there could be a confounding effect by ethnic/racial group (i.e., population stratification), but this is unlikely to be a major problem in most situations [108]. In the present study, we included summary statistics data from highly credible results of meta-GWASs. All the original meta-GWASs corrected for population stratification in cohort-level analyses and at meta-GWAS level.
Another caveat of MR is that developmental compensation might occur, through a genotype being expressed during fetal development that in turn buffers the effects of either environmental or genetic factors, a process called canalization [108,109]. Therefore, buffering mechanisms could hamper the associations between genetic variants and the outcome of interest. As opposed to this, a lifetime exposure to a risk factor may enhance its effects on the disease [109]. However, it is not clear to what extent genetically determined small changes in any given exposure would be sufficient to induce compensation [108].
All 32 of the meta-GWASs from which instrument summary estimates were taken were performed in individuals of European descent in Europe and the US and included thousands of samples for each outcome (S1 Table), which was also the case for our previous CRP meta-GWAS from which we chose the CRP-associated SNPs to calculate GRSGWAS. Therefore, the results of this MR study are applicable to individuals of European descent and are not necessarily generalizable to other ethnic groups.
Conclusion
We showed that elevated CRP levels driven by genetic factors are causally associated with protection against schizophrenia, suggesting that CRP may be one important puzzle piece that leads to an improved understanding of the pathogenesis of schizophrenia. We observed nominal evidence that genetically elevated CRP is causally associated with SBP, DBP, knee osteoarthritis, and bipolar disorder. Based on current GWAS data, we cannot verify any causal effect of CRP on the other 27 common somatic and neuropsychiatric outcomes investigated in the present study. Therefore, disease-associated rise in CRP levels may be a response to the disease process rather than a cause for these 27 outcomes. This implies that interventions to lower CRP levels are unlikely to result in decreased risk for the majority of common complex outcomes.
Supporting Information
Zdroje
1. Stockinger B. Immunology: cause of death matters. Nature. 2009;458 : 44–45. doi: 10.1038/458044a 19262664
2. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369 : 1627–1640. doi: 10.1016/S0140-6736(07)60750-8 17499605
3. Bruunsgaard H, Pedersen M, Pedersen BK. Aging and proinflammatory cytokines. Curr Opin Hematol. 2001;8 : 131–136. 11303144
4. Cesari M, Penninx BWJH, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, et al. Inflammatory markers and cardiovascular disease (The Health, Aging and Body Composition [Health ABC] Study). Am J Cardiol. 2003;92 : 522–528. 12943870
5. Couzin-Frankel J. Inflammation bares a dark side. Science. 2010;330 : 1621. doi: 10.1126/science.330.6011.1621 21163993
6. Fan X, Goff DC, Henderson DC. Inflammation and schizophrenia. Expert Rev Neurother. 2007;7 : 789–796. doi: 10.1586/14737175.7.7.789 17610386
7. Grimaldi MP, Vasto S, Balistreri CR, di Carlo D, Caruso M, Incalcaterra E, et al. Genetics of inflammation in age-related atherosclerosis: its relevance to pharmacogenomics. Ann N Y Acad Sci. 2007;1100 : 123–131. doi: 10.1196/annals.1395.010 17460170
8. Hanson DR, Gottesman II. Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet. 2005;6 : 7. doi: 10.1186/1471-2350-6-7 15707482
9. Johnson TE. Recent results: biomarkers of aging. Exp Gerontol. 2006;41 : 1243–1246. doi: 10.1016/j.exger.2006.09.006 17071038
10. Kiecolt-Glaser JK, Glaser R. Depression and immune function: central pathways to morbidity and mortality. J Psychosom Res. 2002;53 : 873–876. 12377296
11. Kiecolt-Glaser JK, McGuire L, Robles TF, Glaser R. Emotions, morbidity, and mortality: new perspectives from psychoneuroimmunology. Annu Rev Psychol. 2002;53 : 83–107. doi: 10.1146/annurev.psych.53.100901.135217 11752480
12. Lynch MA, Mills KHG. Immunology meets neuroscience—opportunities for immune intervention in neurodegenerative diseases. Brain Behav Immun. 2012;26 : 1–10. doi: 10.1016/j.bbi.2011.05.013 21664452
13. Meyer U, Schwarz MJ, Müller N. Inflammatory processes in schizophrenia: a promising neuroimmunological target for the treatment of negative/cognitive symptoms and beyond. Pharmacol Ther. 2011;132 : 96–110. doi: 10.1016/j.pharmthera.2011.06.003 21704074
14. Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005;17 : 485–495. doi: 10.1080/02646830500381930 16401547
15. Sansoni P, Vescovini R, Fagnoni F, Biasini C, Zanni F, Zanlari L, et al. The immune system in extreme longevity. Exp Gerontol. 2008;43 : 61–65. doi: 10.1016/j.exger.2007.06.008 17870272
16. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444 : 860–867. doi: 10.1038/nature05485 17167474
17. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454 : 436–444. doi: 10.1038/nature07205 18650914
18. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9 : 46–56. doi: 10.1038/nrn2297 18073775
19. Emerging Risk Factors Collaboration, Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375 : 132–140. doi: 10.1016/S0140-6736(09)61717-7 20031199
20. Emerging Risk Factors Collaboration, Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367 : 1310–1320. doi: 10.1056/NEJMoa1107477 23034020
21. Wang X, Bao W, Liu J, Ouyang Y-Y, Wang D, Rong S, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2013;36 : 166–175. doi: 10.2337/dc12-0702 23264288
22. Henriksen M, Jahnsen J, Lygren I, Stray N, Sauar J, Vatn MH, et al. C-reactive protein: a predictive factor and marker of inflammation in inflammatory bowel disease. Results from a prospective population-based study. Gut. 2008;57 : 1518–1523. doi: 10.1136/gut.2007.146357 18566104
23. Rhodes B, Merriman ME, Harrison A, Nissen MJ, Smith M, Stamp L, et al. A genetic association study of serum acute-phase C-reactive protein levels in rheumatoid arthritis: implications for clinical interpretation. PLoS Med. 2010;7:e1000341. doi: 10.1371/journal.pmed.1000341 20877716
24. Ridker PM. High-sensitivity C-reactive protein as a predictor of all-cause mortality: implications for research and patient care. Clin Chem. 2008;54 : 234–237. doi: 10.1373/clinchem.2007.099465 18223130
25. Dehghan A, Kardys I, de Maat MPM, Uitterlinden AG, Sijbrands EJG, Bootsma AH, et al. Genetic variation, C-reactive protein levels, and incidence of diabetes. Diabetes. 2007;56 : 872–878. doi: 10.2337/db06-0922 17327459
26. Lee CC, Adler AI, Sandhu MS, Sharp SJ, Forouhi NG, Erqou S, et al. Association of C-reactive protein with type 2 diabetes: prospective analysis and meta-analysis. Diabetologia. 2009;52 : 1040–1047. doi: 10.1007/s00125-009-1338-3 19326095
27. Danesh J, Pepys MB. C-reactive protein and coronary disease: is there a causal link? Circulation. 2009;120 : 2036–2039. doi: 10.1161/CIRCULATIONAHA.109.907212 19901186
28. Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27 : 1133–1163. doi: 10.1002/sim.3034 17886233
29. C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC), Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, et al. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. doi: 10.1136/bmj.d548 21325005
30. Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123 : 731–738. doi: 10.1161/CIRCULATIONAHA.110.948570 21300955
31. Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42 : 295–302. doi: 10.1038/ng.543 20190752
32. Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42 : 1118–1125. doi: 10.1038/ng.717 21102463
33. Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43 : 246–252. doi: 10.1038/ng.764 21297633
34. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41 : 199–204. doi: 10.1038/ng.311 19169254
35. Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet. 2010;42 : 991–995. doi: 10.1038/ng.689 20953188
36. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42 : 508–514. doi: 10.1038/ng.582 20453842
37. Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358 : 900–909. doi: 10.1056/NEJMoa0707865 18204098
38. Radstake TRDJ, Gorlova O, Rueda B, Martin J-E, Alizadeh BZ, Palomino-Morales R, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42 : 426–429. doi: 10.1038/ng.565 20383147
39. Bradfield JP, Qu H-Q, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7:e1002293. doi: 10.1371/journal.pgen.1002293 21980299
40. Ramos YFM, Metrustry S, Arden N, Bay-Jensen AC, Beekman M, de Craen AJM, et al. Meta-analysis identifies loci affecting levels of the potential osteoarthritis biomarkers sCOMP and uCTX-II with genome wide significance. J Med Genet. 2014;51 : 596–604. doi: 10.1136/jmedgenet-2014-102478 25057126
41. Nikpey M, Goel A, Won H, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47 : 1121–1130. doi: 10.1038/ng.3396 26343387
42. International Consortium for Blood Pressure Genome-Wide Association Studies, Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478 : 103–109. doi: 10.1038/nature10405 21909115
43. International Stroke Genetics Consortium (ISGC), Wellcome Trust Case Control Consortium 2 (WTCCC2), Bellenguez C, Bevan S, Gschwendtner A, Spencer CCA, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012;44 : 328–333. doi: 10.1038/ng.1081 22306652
44. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42 : 937–948. doi: 10.1038/ng.686 20935630
45. Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42 : 579–589. doi: 10.1038/ng.609 20581827
46. Köttgen A, Pattaro C, Böger CA, Fuchsberger C, Olden M, Glazer NL, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42 : 376–384. doi: 10.1038/ng.568 20383146
47. Franceschini N, van Rooij FJA, Prins BP, Feitosa MF, Karakas M, Eckfeldt JH, et al. Discovery and fine mapping of serum protein loci through transethnic meta-analysis. Am J Hum Genet. 2012;91 : 744–753. doi: 10.1016/j.ajhg.2012.08.021 23022100
48. Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9 : 986–994. doi: 10.1016/S1474-4422(10)70197-6 20801717
49. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43 : 429–435. doi: 10.1038/ng.803 21460840
50. International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin U-M, et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377 : 641–649. doi: 10.1016/S0140-6736(10)62345-8 21292315
51. Weiss LA, Arking DE, Gene Discovery Project of Johns Hopkins & the Autism Consortium, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461 : 802–808. doi: 10.1038/nature08490 19812673
52. Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43 : 977–983. doi: 10.1038/ng.943 21926972
53. Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18 : 497–511. doi: 10.1038/mp.2012.21 22472876
54. Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511 : 421–427. doi: 10.1038/nature13595 25056061
55. C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC), Danesh J, Hingorani A, Wensley F, Casas JP, Smeeth L, Samani NJ, et. al. Collaborative pooled analysis of data on C-reactive protein gene variants and coronary disease: judging causality by Mendelian randomisation. Eur J Epidemiol. 2008;23 : 531–540. doi: 10.1007/s10654-008-9249-z 18425592
56. Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302 : 37–48. doi: 10.1001/jama.2009.954 19567438
57. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37 : 658–665. doi: 10.1002/gepi.21758 24114802
58. Dastani Z, Hivert M-F, Timpson N, Perry JRB, Yuan X, Scott RA, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607. doi: 10.1371/journal.pgen.1002607 22479202
59. Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359 : 1897–1908. doi: 10.1056/NEJMoa0707402 18971492
60. Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet. 2012;380 : 572–580. doi: 10.1016/S0140-6736(12)60312-2 22607825
61. Cole TJ. Sympercents: symmetric percentage differences on the 100 log(e) scale simplify the presentation of log transformed data. Stat Med. 2000;19 : 3109–3125. 11113946
62. Rice JA. Mathematical statistics and data analysis. 2nd ed. Belmont (California): Duxbury Press; 1995.
63. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81 : 559–575. 17701901
64. Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381 : 1371–1379. doi: 10.1016/S0140-6736(12)62129-1 23453885
65. Power RA, Steinberg S, Bjornsdottir G, Rietveld CA, Abdellaoui A, Nivard MM, et al. Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat Neurosci. 2015;18 : 953–955. doi: 10.1038/nn.4040 26053403
66. Zakharyan R, Chavushyan A, Khoyetsyan A, Stahelova A, Arakelyan A, Boyajyan A, et al. Genetic variants of the inflammatory C-reactive protein and schizophrenia in Armenian population: a pilot study. Int J Immunogenet. 2010;37 : 407–410. doi: 10.1111/j.1744-313X.2010.00942.x 21182750
67. Singh B, Chaudhuri TK. Role of C-reactive protein in schizophrenia: an overview. Psychiatry Res. 2014;216 : 277–285. doi: 10.1016/j.psychres.2014.02.004 24565000
68. Wium-Andersen MK, Ørsted DD, Nordestgaard BG. Elevated C-reactive protein associated with late - and very-late-onset schizophrenia in the general population: a prospective study. Schizophr Bull. 2014;40 : 1117–1127. doi: 10.1093/schbul/sbt120 23996346
69. Miller BJ, Culpepper N, Rapaport MH. C-reactive protein levels in schizophrenia: a review and meta-analysis. Clin Schizophr Relat Psychoses. 2014;7 : 223–230. doi: 10.3371/CSRP.MICU.020813 23428789
70. Dickerson F, Stallings C, Origoni A, Vaughan C, Khushalani S, Yang S, et al. C-reactive protein is elevated in schizophrenia. Schizophr Res. 2013;143 : 198–202. doi: 10.1016/j.schres.2012.10.041 23218564
71. Fernandes BS, Steiner J, Bernstein H-G, Dodd S, Pasco JA, Dean OM, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry. 2016;21 : 554–564. doi: 10.1038/mp.2015.87 26169974
72. Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet. 2003;73 : 34–48. doi: 10.1086/376549 12802786
73. International Schizophrenia Consortium, Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460 : 748–752. doi: 10.1038/nature08185 19571811
74. Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460 : 753–757. doi: 10.1038/nature08192 19571809
75. Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460 : 744–747. doi: 10.1038/nature08186 19571808
76. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43 : 969–976. doi: 10.1038/ng.940 21926974
77. Al-Asmari SM, Kadasah S, Arfin M, Tariq M, Al-Asmari A. Genetic variants of interleukin-10 gene promoter are associated with schizophrenia in Saudi patients: A case-control study. N Am J Med Sci. 2014;6 : 558–565. doi: 10.4103/1947-2714.145466 25535603
78. Saviouk V, Chow EWC, Bassett AS, Brzustowicz LM. Tumor necrosis factor promoter haplotype associated with schizophrenia reveals a linked locus on 1q44. Mol Psychiatry. 2005;10 : 375–383. doi: 10.1038/sj.mp.4001582 15340354
79. Hänninen K, Katila H, Saarela M, Rontu R, Mattila KM, Fan M, et al. Interleukin-1 beta gene polymorphism and its interactions with neuregulin-1 gene polymorphism are associated with schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2008;258 : 10–15. doi: 10.1007/s00406-007-0756-9 17901998
80. Sekar A. A natural allelic series of complex structural variants and its influence on the risk of lupus and schizophrenia. 2014 [cited 19 May 2016]. Digital Access to Scholarship at Harvard. Available: http://dash.harvard.edu/handle/1/13070061.
81. Vaez A, Jansen R, Prins BP, Hottenga J-J, de Geus EJC, Boomsma DI, et al. In silico post genome-wide association studies analysis of C-reactive protein loci suggests an important role for interferons. Circ Cardiovasc Genet. 2015;8 : 487–497. doi: 10.1161/CIRCGENETICS.114.000714 25752597
82. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, et al. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92 : 997–1009. doi: 10.1111/j.1471-4159.2004.02954.x 15715651
83. Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. 2008;180 : 3866–3873. 18322194
84. Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47 : 4–16. 20686195
85. Marsman A, van den Heuvel MP, Klomp DWJ, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull. 2013;39 : 120–129. doi: 10.1093/schbul/sbr069 21746807
86. Chung K-H, Huang S-H, Wu J-Y, Chen P-H, Hsu J-L, Tsai S-Y. The link between high-sensitivity C-reactive protein and orbitofrontal cortex in euthymic bipolar disorder. Neuropsychobiology. 2013;68 : 168–173. doi: 10.1159/000353613 24051690
87. Hong EP, Kim DH, Suh JG, Park JW. Genetic risk assessment for cardiovascular disease with seven genes associated with plasma C-reactive protein concentrations in Asian populations. Hypertens Res. 2014;37 : 692–698. doi: 10.1038/hr.2014.56 24671014
88. Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, et al. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation. 2007;115 : 1020–1028. doi: 10.1161/CIRCULATIONAHA.106.664854 17283257
89. Zhou H, Li Y, Huang G, Gu X, Zeng J, Li Y, et al. Interleukin 6 augments mechanical strain-induced C-reactive protein synthesis via the stretch-activated channel-nuclear factor κ B signal pathway. Heart. 2013;99 : 570–576. doi: 10.1136/heartjnl-2012-303355 23257175
90. Timpson NJ, Lawlor DA, Harbord RM, Gaunt TR, Day INM, Palmer LJ, et al. C-reactive protein and its role in metabolic syndrome: Mendelian randomisation study. Lancet. 2005;366 : 1954–1959. doi: 10.1016/S0140-6736(05)67786-0 16325697
91. Davey Smith G, Lawlor DA, Harbord R, Timpson N, Rumley A, Lowe GDO, et al. Association of C-reactive protein with blood pressure and hypertension: life course confounding and Mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol. 2005;25 : 1051–1056. doi: 10.1161/01.ATV.0000160351.95181.d0 15731495
92. Spector TD, Hart DJ, Nandra D, Doyle DV, Mackillop N, Gallimore JR, et al. Low-level increases in serum C-reactive protein are present in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum. 1997;40 : 723–727. 9125256
93. Bos SD, Suchiman HED, Kloppenburg M, Houwing-Duistermaat JJ, le Graverand MPH, Seymour AB, et al. Allelic variation at the C-reactive protein gene associates to both hand osteoarthritis severity and serum high sensitive C-reactive protein levels in the GARP study. Ann Rheum Dis. 2008;67 : 877–879. doi: 10.1136/ard.2007.079228 18055473
94. Vlad SC, Neogi T, Aliabadi P, Fontes JDT, Felson DT. No association between markers of inflammation and osteoarthritis of the hands and knees. J Rheumatol. 2011;38 : 1665–1670. doi: 10.3899/jrheum.100971 21572158
95. Kerkhof HJM, Bierma-Zeinstra SMA, Castano-Betancourt MC, de Maat MP, Hofman A, Pols HAP, et al. Serum C reactive protein levels and genetic variation in the CRP gene are not associated with the prevalence, incidence or progression of osteoarthritis independent of body mass index. Ann Rheum Dis. 2010;69 : 1976–1982. doi: 10.1136/ard.2009.125260 20511616
96. Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379 : 1214–1224. doi: 10.1016/S0140-6736(12)60110-X 22421340
97. Anand SS, Yusuf S. C-reactive protein is a bystander of cardiovascular disease. Eur Heart J. 2010;31 : 2092–2096. doi: 10.1093/eurheartj/ehq242 20675658
98. Brunner EJ, Kivimäki M, Witte DR, Lawlor DA, Davey Smith G, Cooper JA, et al. Inflammation, insulin resistance, and diabetes—Mendelian randomization using CRP haplotypes points upstream. PLoS Med. 2008;5:e155. doi: 10.1371/journal.pmed.0050155 18700811
99. Timpson NJ, Nordestgaard BG, Harbord RM, Zacho J, Frayling TM, Tybjærg-Hansen A, et al. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. Int J Obes 2005. 2011;35 : 300–308. doi: 10.1038/ijo.2010.137
100. Wium-Andersen MK, Orsted DD, Nordestgaard BG. Elevated C-reactive protein, depression, somatic diseases, and all-cause mortality: a Mendelian randomization study. Biol Psychiatry. 2014;76 : 249–257. doi: 10.1016/j.biopsych.2013.10.009 24246360
101. Ryberg H, An J, Darko S, Lustgarten JL, Jaffa M, Gopalakrishnan V, et al. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve. 2010;42 : 104–111. doi: 10.1002/mus.21683 20583124
102. Kok EH, Alanne-Kinnunen M, Isotalo K, Luoto T, Haikonen S, Goebeler S, et al. CRP gene variation affects early development of Alzheimer’s disease-related plaques. J Neuroinflammation. 2011;8 : 96. doi: 10.1186/1742-2094-8-96 21831326
103. Song I-U, Chung S-W, Kim J-S, Lee K-S. Association between high-sensitivity C-reactive protein and risk of early idiopathic Parkinson’s disease. Neurol Sci. 2011;32 : 31–34. doi: 10.1007/s10072-010-0335-0 20532580
104. Pasco JA, Nicholson GC, Williams LJ, Jacka FN, Henry MJ, Kotowicz MA, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197 : 372–377. doi: 10.1192/bjp.bp.109.076430 21037214
105. Prasad K. C-reactive protein (CRP)-lowering agents. Cardiovasc Drug Rev. 2006;24 : 33–50. doi: 10.1111/j.1527-3466.2006.00033.x 16939632
106. Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet. 2013;14 : 507–515. doi: 10.1038/nrg3457 23774735
107. Vimaleswaran KS, Berry DJ, Lu C, Tikkanen E, Pilz S, Hiraki LT, et al. Causal relationship between obesity and vitamin D status: bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013;10:e1001383. doi: 10.1371/journal.pmed.1001383 23393431
108. Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33 : 30–42. doi: 10.1093/ije/dyh132 15075143
109. Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J. 2014;35 : 1917–1924. doi: 10.1093/eurheartj/ehu208 24917639
110. Hwang Y, Kim J, Shin JY, Kim JI, Seo JS, Webster MJ, et al. Gene expression profiling by mRNA sequencing reveals increased expression of immune/inflammation-related genes in the hippocampus of individuals with schizophrenia. Transl Psychiatry. 2013;3:e321. doi: 10.1038/tp.2013.94 24169640
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2016 Číslo 6
- Alternativní léčebné možnosti u hypercholesterolemie při intoleranci statinů
- Vliv kombinace nutraceutik na remodelaci levé komory srdeční u osob s metabolickým syndromem
- Nutraceutika a jejich ovlivnění mírného kardiometabolického rizika
- Princip účinku medu v léčbě chronických i infikovaných ran
- Superoxidovaný roztok a jeho využití v léčbě ran
Nejčtenější v tomto čísle
- Why Most Clinical Research Is Not Useful
- Agreements between Industry and Academia on Publication Rights: A Retrospective Study of Protocols and Publications of Randomized Clinical Trials
- Inter-pregnancy Weight Change and Risks of Severe Birth-Asphyxia-Related Outcomes in Singleton Infants Born at Term: A Nationwide Swedish Cohort Study
- Strengthening the Reporting of Observational Studies in Epidemiology—Nutritional Epidemiology (STROBE-nut): An Extension of the STROBE Statement