
t Is a Structurally Novel Crohn's Disease-Associated Superantigen
T cell responses to enteric bacteria are important in inflammatory bowel disease. I2, encoded by the pfiT gene of Pseudomonas fluorescens, is a T-cell superantigen associated with human Crohn's disease. Here we report the crystal structure of pfiT at 1.7Å resolution and provide a functional analysis of the interaction of pfiT and its homolog, PA2885, with human class II MHC. Both pfiT and PA2885 bound to mammalian cells and stimulated the proliferation of human lymphocytes. This binding was greatly inhibited by anti-class II MHC HLA-DR antibodies, and to a lesser extent, by anti HLA-DQ and DP antibodies, indicating that the binding was class II MHC-specific. GST-pfiT efficiently precipitated both endogenous and in vitro purified recombinant HLA-DR1 molecules, indicating that pfiT directly interacted with HLA-DR1. Competition studies revealed that pfiT and the superantigen Mycoplasma arthritidis mitogen (MAM) competed for binding to HLA-DR, indicating that their binding sites overlap. Structural analyses established that pfiT belongs to the TetR-family of DNA-binding transcription regulators. The distinct structure of pfiT indicates that it represents a new family of T cell superantigens.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003837
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003837
Summary
T cell responses to enteric bacteria are important in inflammatory bowel disease. I2, encoded by the pfiT gene of Pseudomonas fluorescens, is a T-cell superantigen associated with human Crohn's disease. Here we report the crystal structure of pfiT at 1.7Å resolution and provide a functional analysis of the interaction of pfiT and its homolog, PA2885, with human class II MHC. Both pfiT and PA2885 bound to mammalian cells and stimulated the proliferation of human lymphocytes. This binding was greatly inhibited by anti-class II MHC HLA-DR antibodies, and to a lesser extent, by anti HLA-DQ and DP antibodies, indicating that the binding was class II MHC-specific. GST-pfiT efficiently precipitated both endogenous and in vitro purified recombinant HLA-DR1 molecules, indicating that pfiT directly interacted with HLA-DR1. Competition studies revealed that pfiT and the superantigen Mycoplasma arthritidis mitogen (MAM) competed for binding to HLA-DR, indicating that their binding sites overlap. Structural analyses established that pfiT belongs to the TetR-family of DNA-binding transcription regulators. The distinct structure of pfiT indicates that it represents a new family of T cell superantigens.
Introduction
T-cell receptors (TCRs) typically recognize antigens in the form of peptides or peptide-lipids bound to major histocompatibility complex (MHC) molecules. TCRs and class II MHC proteins may directly interact with viral or bacterial proteins known as superantigens (SAgs) to activate T cells. Strong primary T cell responses are elicited by microbial toxin superantigens via their interaction with TCR Vβ [1], [2], [3], [4], [5] elements, and in some cases to TCR Vα chains [6], [7], [8].
A novel enteric T cell superantigen, I2, and its full-length gene product pfiT, are encoded by a microbial gene associated with Crohn's disease (CD) [9], [10], [11]. Several lines of evidence has implicated I2 and pfiT in the pathogenesis of CD [10], [12], [13], [14], [15], [16], [17], [18]. The I2 sequence was selectively detected in active CD lesional colonic tissue, but not in healthy controls or inactive CD ileum. Serum antibody studies revealed that IgA seroreactivity to a recombinant I2 protein was CD specific. Such IgA antibodies specific for I2 were detected in 50–60% of patients (both children and adults) with CD, 10% of patients with ulcerative colitis, and 4% of healthy controls. Patients expressing I2 were distinguished by greater disease severity and progression, including a greater frequency of strictures, internal perforations, and small bowel surgery (72% in antibody-positive patients, versus 23% in antibody-negative patients) [19].
Through genetic analysis and genomic cloning, a full-length I2 gene (pfiT) was identified as a genomic element of Pseudomonas fluorescens, a normal environmental bacterium detectable in the human gastrointestinal tract [11]. Commensal bacteria have emerged as an important factor in the pathogenesis of CD [20], although P. fluorescens has had limited testing as a potential pathobiont in IBD [21].
Several lines of evidence suggest that the I2 protein pfiT is a novel T-cell SAg [9], [11]. I2 protein induced proliferation and IL-10 responses by normal CD4+ T cells [9]. The I2 response was dependent on MHC class II-mediated recognition, but the antigen did not require antigen processing, a feature typical of SAgs, and antibody blockade of I-Ab specifically blocked the T-cell proliferative response [9]. Selective activation was observed for the murine TCR-Vβ5 subpopulation, and pfiT also exhibited T-cell SAg bioactivity [11]. PA2885, the apparent Pseudomonas aeruginosa homolog of pfiT (78% sequence identity) also stimulated murine CD4+ T cell proliferation [11]. Accordingly, colonization by the I2 and pfiT-expressing microorganism, P. fluorescens, in IBD susceptible hosts, was speculated to provide a superantigenic stimulus pertinent to the pathogenesis of Crohn's disease.
In this study, we describe structural and functional studies of the CD-associated superantigen pfiT. We show that pfiT can stimulate the activation of both mouse splenocytes and human peripheral blood mononuclear cells (PBMCs). We show that pfiT specifically and directly interacts with class II MHC HLA-DR, and to a lesser extent HLA-DP or HLA-DQ, and show that pfiT shares an overlapping binding site on HLA-DR1 with other superantigens. Crystal structure of pfiT at high resolution revealed that pfiT belongs to the bacterial transcription factor family of tetracycline repressor (TetR). The distinct structure of pfiT relative to other SAgs suggests that it represents a novel family of SAg.
Results
Expression and purification of full-length pfiT and PA2885
Attempts to express and purify soluble His-tag versions of I2, pfiT, and PA2885 failed. Therefore, we grafted the inserts into the pGEX-6P-1 vector (GE HealthCare) and expressed the proteins as GST-fusion proteins. Although GST-I2 remained insoluble, GST-pfiT and GST-PA2885 could be directly affinity-purified from cell lysates under native conditions (Fig. 1A). To obtain tag-less proteins, the GST-fusion proteins were digested by the PreScission protease (GE HealthCare). All proteins, tagged or untagged, were further purified to homogeneity using size-exclusion chromatography (Fig. 1A).

pfiT stimulates the activation of both mouse splenocytes and human PBMC
Previously, I2 and pfiT were found to stimulate the activation of murine T cells with features most representative of a T cell superantigen [9], [11]. However, the proteins used in previous studies were purified from bacterial inclusion bodies and were possibly non-native. Therefore, we evaluated whether the proteins purified from soluble fractions were also immunostimulatory. Using pfiT purified directly from soluble fractions, activation of murine T cells was detected in a dose-dependent manner, although the magnitude of stimulation by pfiT was not as strong as that induced by the most potent known superantigen Staphylococcal enterotoxin (SE) B (SEB) (Fig. 1B).
Since the I2 gene was isolated from human CD patients, we anticipated that pfiT, the full-length protein encoded by I2, could act on human T lymphocytes. Therefore, we purified human T cells from peripheral blood mononuclear cells (PBMCs), and assayed T-cell proliferation in response to pfiT. As shown in Fig. 1C, both recombinant pfiT and its GST-fusion protein activated human T lymphocyte proliferation. To verify that pfiT and PA2885 could both activate human T cells, we labeled purified human T lymphocytes with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE) that permits visualization of proliferation by dye dilution during successive rounds of cell division. CFSE-labeled human T lymphocytes were then cultured with pfiT or PA2885; SEC and SEA were used as positive control SAgs, and media alone was used as a negative control. FACS analysis confirmed that CFSE-labeled human T-cells did not respond to the media control (Fig. 1D), but proliferated robustly to the SAg SEC (up to 5 generational CFSE-decremented populations, Fig. 1D). Although not as robust as SEC, pfiT and PA2885 triggered substantial T cell proliferation, comparable to levels induced by another positive control SAg, SEA (Fig. 1F–H). Overall, our results indicated that recombinant soluble pfiT and PA2885 were biologically active and could induce proliferation of both murine and human T cells.
Soluble pfiT binds to the cell surface of lymphocytes
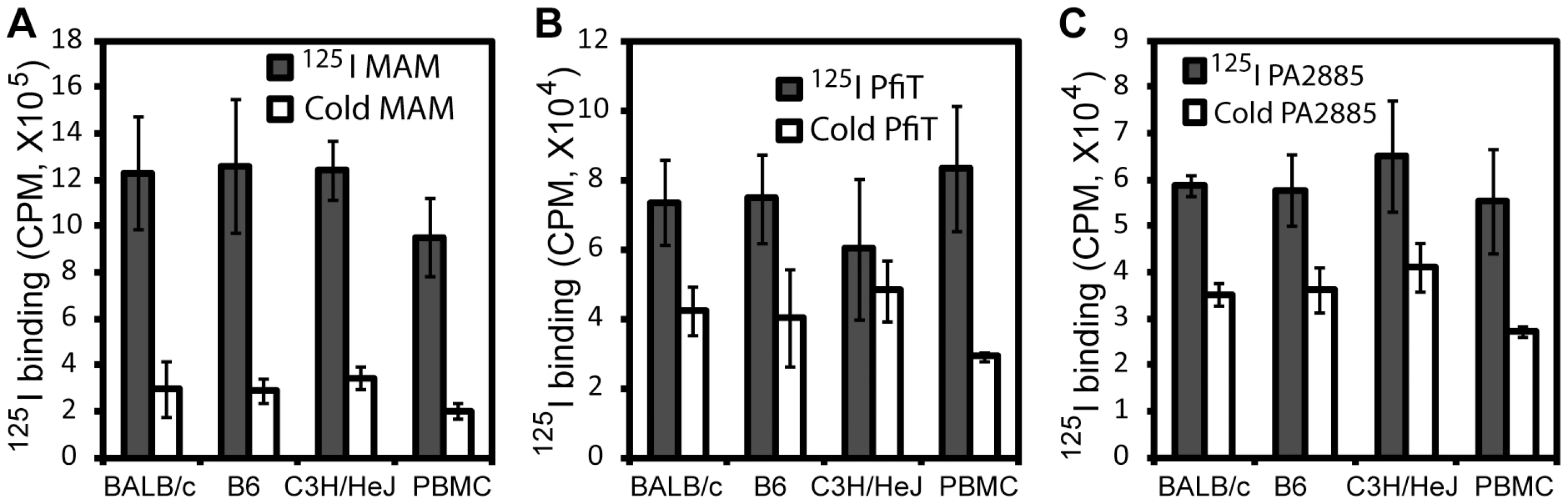
In order to identify human receptors responsible for presentation of pfiT and PA2885, we first performed an 125I-protein competition assay to investigate whether recombinant pfiT and PA2885 could bind lymphocytes. As test proteins, we labeled pfiT and PA2885 with 125I; as a positive control SAg, the Mycoplasma arthritidis-derived mitogen (MAM) was also produced and labeled, as described previously [7], [22]. Unlabeled MAM, pfiT, and PA2885 were used as cold competitors in a competition binding assay, as described previously [22]. As expected, 10-fold molar excess of MAM greatly (>75%) diminished binding of 125I-labeled MAM to the cell surface of both murine splenocytes and human PBMC (Fig. 2A). Similarly, excessive quantities of pfiT and PA2885 reduced about 40% of the binding of the proteins to murine splenocytes. Specific binding was much lower in C3H/HeJ mice (only 20% competition), perhaps reflecting a lower avidity for the class II MHC proteins of this haplotype. These results indicated that pfiT and PA2885 specifically bind to mouse and human cells.
Class II MHC is responsible for the binding of pfiT
We anticipated that the binding of pfiT and PA2885 to PBMCs would be dependent on human class II MHC. To test this prediction, and specify the class II MHC proteins responsible for binding, we labeled pfiT, PA2885, and a control SAg MAM with fluorescein isothiocyanate (FITC). We then used flow cytometry to analyze the binding of FITC-labeled proteins to the surface of human PBMC in the presence of various blocking antibodies against class II HLA molecules. In control studies, antibodies targeting class II HLA-DR reduced the binding of FITC-labeled MAM to the surface of PBMC by 70% (Fig. 3A), whereas anti-HLA-DQ and anti-HLA-DP antibodies did not significantly affect the binding. This is consistent with the fact that MAM is presented primarily by HLA-DR [7], [23], [24]. When the binding of FITC-labeled pfiT to human PBMC was tested (Fig. 3B), we observed a significantly reduction (30% and 25% compared to buffer control) with an anti-HLA-DR antibody, or when using a polyclonal antibody recognizing all three class II alleles (DR, DQ, and DP). Anti-HLA-DQ and anti-HLA-DP antibodies also moderately decreased the binding of FITC-labeled pfiT to human PBMCs. For PA2885, all anti-HLA antibodies reduced binding by more than 45%, where the greatest inhibition was obtained using anti-HLA-DR (75%), or anti-DR, DQ, DP antibodies (Fig. 3C). Our results indicate that pfiT and PA2885 specifically bind to class II MHC, and primarily to HLA-DR.

To confirm these observations, we independently evaluated HLA binding using a GST immunoprecipitation assay (Fig. 4). GST, GST-pfiT and GST-MAM were expressed and purified to homogeneity, and were immobilized to glutathione affinity beads. Using a cell lysate of Raji cells as a source of endogenous HLA-DR molecules, GST-MAM and GST-pfiT fusion proteins, but not the free GST, precipitated HLA-DR molecules (Fig. 4A). These data provided additional evidence that GST-pfiT, like GST-MAM, interacted with HLA-DR molecules. Next, using a bacterial expression system, we produced, refolded, and purified recombinant HLA-DR1 protein bound to a hemagglutinin (HA) peptide, as described previously [24]. Using this recombinant HLA-DR1/HA complex, we performed the GST immunoprecipitation assay. Again, free GST failed to precipitate the recombinant complex, whereas both GST-pfiT and GST-MAM efficiently precipitated the HLA-DR1/HA complex. Since in the latter case, all the materials were highly purified recombinant proteins, the results indicated that pfiT directly interacts with the HLA-DR/HA complex. Moreover, data from both studies suggested that the binding of HLA-DR to GST-pfiT was less than to GST-MAM, indicating that pfiT binds HLA-DR with apparent lower affinity than MAM.

To determine the binding affinity of pfiT with HLA-DR, we used the GST immunoprecipitation assay to titrate the HLA DR/HA complex. As shown in Fig. 4B and 4C, binding of the recombinant HLA-DR/HA complex to GST-pfiT was dose-dependent. Non-linear regression of the titration curve revealed that the dissociation constant (KD) for pfiT-HLA-DR1/HA interaction was about 13.5 µM. By comparison, the KD value for MAM binding to HLA-DR1/HA was in the sub-nanomolar range [22], [25]. This result supports our observation that pfiT binds HLA-DR more weakly than does MAM.
pfiT forms a dimer and binds HLA-DR1 in solution
To further investigate the interactions between pfiT and HLA-DR1, we performed analytical ultracentrifugation (AUC) sedimentation experiments. As it was known from our previous study [25] that HLA-DR1 exists as a monomer, we only performed sedimentation velocity (SV) experiment for pfiT to determine its oligomeration state. As shown in Fig. 5A, pfiT at a concentration of 3.6 µM showed a single peak with a sedimentation coefficient (S(20, w)) of 3.1 S. Transformation of C(S) distribution to c(M) size distribution [26] indicated a peak molecular weight (MW) of about 44 KDa, which is in a perfect agreement with a dimer form of pfiT with a MW of 45 KDa. Thus, our data demonstrate that pfiT forms a dimer in solution.

To determine the dissociation constant (KD) of pfiT dimer formation, we performed AUC sedimentation equilibrium experiments. By using SEDPHAT [27], we could readily model the sedimentation equilibrium data of pfiT with multiple concentrations and multiple speeds, using a single-species model. An apparent molecular mass of 48 KDa was obtained with a local root-mean-square (rms) deviation of 0.007 OD and a global χ2 of 1.5, with molecular mass being the only fitting parameter. This value is in good agreement with the theoretically calculated mass of 45 KDa for a pfiT dimer, suggesting again that pfiT exists as a dimer in solution. The somewhat high χ2 value indicated that a single-species model does not represent the experimental data very well. Indeed, when a monomer-dimer model was used in SEDPHAT [27], a much better fit was obtained, with a local rms deviation of 0.004 OD and a global χ2 of 0.7. A KD of 0.15 µM for the pfiT monomer-dimer equilibrium was obtained (Fig. 5B).
We next performed sedimentation equilibrium study of the pfiT/HLA-DR1 complex. pfiT was mixed with purified recombinant HLA-DR1/HA complex at three different molar ratios, and the sedimentation equilibrium was performed at three rotor speeds (Fig. 5C). Given that pfiT is a dimer and HLA-DR1 is a monomer, we chose a reversible (A+A)+B+BA+AB+B(AA)B+B(AA)BB complex model, with A referring to pfiT, (AA) representing pfiT dimer, and B representing HLA-DR1. By using SEDPHAT [27], a global analysis of the equilibrium data gave an excellent fit, with a global reduced χ2 of 0.5 and local rms error of 0.004 OD (Fig. 5C). From this analysis, we estimated the binding affinity (KD(AB)) as 17 µM for pfiT binding to the HLA-DR1/HA complex. The binding affinity (17 µM) determined using the AUC sedimentation equilibrium is in agreement with the value of 13.5 µM determined using the GST pull-down assay, and is much lower than the affinity (0.1 µM) for MAM-binding to recombinant HLA-DR1 using the same AUC method [25]. Overall, our results indicate that recombinant proteins pfiT and HLA-DR1 bind each other in solution.
The HLA-DR binding sites for pfiT and MAM overlap
In order to identify the site on HLA-DR to bind pfiT, we performed a competition GST immunoprecipitation assay, using tag-removed MAM or pfiT as a “cold” competitive binder. As shown in Fig. 6A and 6C, tag-less pfiT inhibited the binding of HLA-DR to immobilized GST-MAM in a dose-dependent manner; alternatively, free MAM decreased the binding of HLA-DR to GST-pfiT in a dose-responsive manner (Fig. 6B,C). Competition curve fitting of HLA-DR binding revealed that MAM inhibition of pfiT was more effective than pfiT inhibition of MAM. The IC50 values for MAM inhibition of pfiT binding and pfiT inhibition of MAM binding to HLA-DR were 5.4 µM and 47 µM, respectively (IC50: inhibitor concentration required to inhibit ligand binding by 50%). These results were consistent with the GST immunoprecipitation assay (Fig. 4A), where a similar quantity of GST-pfiT precipitated less HLA-DR than did GST-MAM. Overall, these results indicate that the HLA-DR binding sites for pfiT and MAM overlap.

Crystal structure of pfiT
In order to better understand the biological function of this Crohn's disease-related SAg, we next determined the crystal structure of pfiT at 1.7 Å resolution (Fig. 7, Table 1). Native crystals were first grown with a space group C2 and one molecule per asymmetric unit. Since a sequence search indicated that pfiT belongs to the TetR-family transcription regulator [11], we attempted to solve the structure using molecular replacement (MR) method with various structures of the TetR members. However, none of the MR calculations gave clear solutions. We therefore generated seleno-methionine (Se-Met)-substituted pfiT protein. The Se-Met pfiT was crystallized in the P21 space group with two molecules per asymmetric unit, which is different from that of the native crystal. After screening many crystals and diffraction data, we finally obtained clear structural solution at 2.4Å resolution from a set of diffraction data of the Se-Met pfiT crystal, using the single anomalous dispersion (SAD) phasing method. The structure was completed by iterative refinement and graphic modeling, using a set of diffraction data of the Se-Met crystal at 1.95Å resolution (Table 1). Structure of native crystals at 1.7Å resolution was determined using the MR method with the Se-Met pfiT structure as a search model (Table 1).


As shown in Fig. 7A & 7B, pfiT displays a completely α-helical structure that consists of ten α-helices. The structure of native pfiT in the C2 crystal form comprises residues 11–198. The N-terminal 10 residues are presumably disordered. The missing N-terminal residues can be traced without ambiguity for one monomer within the dimer of the Se-Met pfiT in the P21 crystal form (Fig. 7B). The N-terminus forms a short α-helix (α1) together with five residues GPLGS resulted from the GST-tag vector (Fig. 7B).
The Se-Met pfiT was crystallized as a dimer, which is consistent with results from our AUC solution studies indicating that pfiT exits as a dimer in solution (Fig. 5A & 5B). Although there is only one molecule in the asymmetric unit for the C2 crystal form of native pfiT, a dimer very similar to that in the P21 crystal form can be readily formed through crystallographic symmetry (Fig. 7C). Therefore, results from both crystal structure and solution studies indicated that pfiT exits as a dimer.
VAST [28] search indicated that pfiT belongs to the TetR-family transcription regulators [29], [30]. The structure of pfiT is most close to that of a putative TetR family protein from Vibrio parahaemolyticus (PDB code: 3HE0) (Cuff ME, Hendricks R, Moy S, Joachimiak A, unpublished data) with a root-mean-square-deviation of 1.72 Å (Fig. 7D). At a structural level, the TetR proteins all possess a helix-turn-helix DNA-binding domain (DBD) at their N-terminal ends, and have highly divergent C-termini postulated to be involved in the binding of inducing compounds [29], [30]. It is known that the TetR repressor function requires a TetR dimer to bind the DNA elements [31], [32], [33]. Similarly, the pfiT dimer is arranged in this manner of other TetR, as the dimers are all formed through the C-terminal helices [31], [32], [33], [34], [35], [36], [37] (Fig. 7E). However, the DBDs of the pfiT dimer are about 50Å apart, contrasting to an average 34 Å distance for most other TetR repressors in their active forms [31], [32], [33] (Fig. 7E). Similar widely-opened DBDs were also observed in the dimer of MexZ, a key repressor responsible for antibiotic resistance in P. aeruginosa [36]. In this regard, it has been proposed that the widely separated DBD dimer represents the released form of TetR [36].
Discussion
Superantigens (SAgs) activate large fractions of T cells expressing particular T-cell receptor (TCR) β chains, and are the pathogenic factors of many human diseases such as toxic shock syndrome and autoimmune diseases [4]. Although much is known about the structures and functions of the pyrogenic SAgs encoded by staphylococcal and streptococcal bacteria, far less is known about the non-pyrogenic SAgs, such as the Crohn's disease (CD)-associated protein I2 (pfiT). I2 and its full-length protein pfiT encoded by P. fluorescens are novel enteric SAgs; they are important pathogenic factors and serological markers of CD, an inflammatory bowel disease. The discovery of pathogenic factor and serological marker I2 (pfiT) suggest that these proteins play a role in the pathogenesis of Crohn's disease. However, how pfiT performs its biological function and its role in Crohn's disease are not known.
In this study, we determined the structure and identified an immunologic function of pfiT. Based upon the high resolution crystal structure of pfiT, we found that pfiT belongs to the TetR transcription regulator protein family. The TetR family members are key players in multidrug resistance, virulence, and pathogenicity processes caused by bacterial pathogens. They act as repressors by binding to consecutive DNA major grooves through the N-terminal DBDs [31], [32], [33], [34], [35], [36]. TetR proteins control the expression of genes involved in multidrug resistance, enzymes implicated in catabolic pathways, biosynthesis of antibiotics, osmotic stress, and pathogenicity of gram-negative and gram-positive bacteria [29], [30]. TetR proteins function as a dimer to bind their DNA elements [31], [32], [33]. Our results demonstrated that pfiT exists as a dimer in solution; and crystal structure also confirmed that pfiT is a dimer at structural level. Structure-based alignment of sequences of pfiT with those of TetR members with known structures of the protein-DNA complexes indicated that most of residues important for TetR members to bind their DNA elements are conserved in the pfiT sequence (Fig. 7F & 7H). Similar to QacR and TetR, pfiT also displays a positively charged surface at the putative DNA binding site (Fig. 7G). Although it is currently unknown whether pfiT binds DNA, these structural evidences suggest that pfiT might also function as a transcription regulator, like other TetR members. However, further investigation will be required to determine whether pfiT acts as both as a SAg and as a DNA-binding protein.
Pseudomonas is a large genus of Gram-negative aerobic γ proteobacteria, belonging to the family Pseudomonadaceae containing 191 species [38]. The genomes of many of these species have been sequenced [39], [40], [41], [42], [43]. BLAST search [44] indicated that many putative TetR-family members of the Pseudomonas bacteria share high sequence homology with pfiT (Fig. S1A). In addition, putative TetR members of a number of other bacterial species also display significant sequence homology with pfiT (Fig. S1B). It is currently unknown whether these other TetR members possess SAg activity. Nonetheless, our study suggests a new function for this group of proteins, at least for pfiT and PA2885.
We further investigated the function of the CD-associated enteric SAg pfiT. Our data revealed that pfiT can stimulate the activation of not only murine splenocytes, as previously reported [9], [11], but also human peripheral blood T cells. We demonstrated that the class II MHC molecule is the human receptor for the SAg pfiT. Addition of antibodies against all three class II HLA alleles greatly reduced the binding of pfiT to human PBMC. Addition of anti-HLA-DR antibody alone also greatly reduced pfiT binding to human PBMC, whereas anti-DQ and anti-DP antibodies only moderately reduced the binding. The results indicated that HLA-DR, and to a lesser extent, HLA-DQ and HLA-DP, is responsible for pfiT binding. Moreover, using purified recombinant proteins, we demonstrated that pfiT and HLA-DR interacted directly. Competition binding data suggested that the pfiT-binding site on HLA-DR overlaps with that for MAM, a phylogenetically and structurally distinct SAg.
Both pfiT and MAM are more selective for HLA-DR [45]. Using a genetic analysis, MAM was found to be immunologically active (T cell activation) in the context of murine H-2E (the murine homologue of HLA-DR), but also certain HLA-DQ and homologue H-2A alleles [45]. Using an antibody-blocking analysis, pfiT also showed immunologic activation with H-2Ab, but not H-2E or other HLA-D molecules ([9], [11], and present study). What might be the basis for this overlapping but distinct MHC-dependence of pfiT and MAM? Whereas staphylococcal and streptococcal SAgs share similar three dimensional structures, SAgs from other sources display various structural folds, ranging from completely β-sheets to completely α-helices [4], [24], [46]. Our structural study indicated that the structures of both pfiT and MAM are completely helical, although pfiT displays a different folding pattern [24]. Our competition binding measurements indicated that MAM can compete binding of pfiT to its human receptor HLA-DR. We speculate that the distinct folding patterns might yield differences in molecular contacts that could account for the divergent MHC fine specificities of these two SAgs. Alternatively, these differences may reflect the outcome of genetic versus antibody analysis. These questions await integrated assessment of these two SAgs using the same immunologic assay context, and direct structural analysis of binding with HLA-DR and H-2A.
In summary, the structural and functional studies of the pfiT-MHC interactions provide detailed insights into how a novel enteric SAg is recognized by its human receptor. Immune activity to I2, the peptide antigen encoded by pfiT, is a distinguishing biomarker for disease state and clinical phenotype of Crohn's disease [47], [48]. There is an intensifying focus on the interplay of environment and microbiota in Crohn's disease, both as a source of antigens in adaptive immune activation, and as a source of microbial products targeting innate immune and epithelial function [20], [49], [50]. This structural and functional study provides new insights into the SAg properties of pfiT, and accordingly, how P. fluorescens, a ubiquitous environmental microbial commensal, has emerged as an immunologic feature of Crohn's disease.
Materials and Methods
Ethics statement
Animals were housed and cared in accordance with standards of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) in AAALAC accredited facilities, and all animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committees of the Wadsworth Center Institutional Animal Care and Use Committee. Our institutional program that oversees the review & approval of animal use protocols uses the standards delineated in the Eighth edition of the “Guide For the Care and Use of Laboratory Animals” (the Guide), the “PHS Policy on Humane Care and Use of Laboratory Animals”, and the Animal Welfare Act Regulations (AWARs) for covered species. The Wadsworth Center is accredited by AAALAC International, and is an OLAW-Assured research institution (PHS Animal Welfare Assurance Number A3183-01).
The studies using anonymized human blood samples purchased from Research Blood Component was exempt and approved by the Institutional Review Board Committee of the Wadsworth Center.
Antibodies
Primary antibodies used in this study were anti-HLA-DR G-7 (Santa Cruz Biotechnology) and L243 (ATCC), anti-HLA-DQ (IVD12) (ATCC), anti-HLA-DP (BD Biosciences), anti-HLA-DR, DQ, DP (Tu39, BD Pharmingen), anti-CD19-APC (LT19, Abcam), anti-CD45-APC-H7 (2D1, BD biosciences), anti-CD3 (HIT3a, BD Biosciences), and anti-CD3-PerCP (MEM-57, Abcam), anti-GST (GE HealthCare). Control IgG 2H11 was a gift from Dr. Ellis Reinherz of Dana-Farber Cancer Institute.
Cloning, expression, and purification of pfiT, PA2885, and MAM
Full-length I2 encoded by P. fluorescens (pfiT) and its close homolog PA2885 from P. aeruginosa were cloned into pGEX-6P-1 expression vector (GE HealthCare) by grafting the inserts from the pQE30 vector (Qiagen). The His-tag pfiT and PA2885, prepared using the pQE-30 vector, were expressed and purified as described previously [51]. Expression and purification of pfiT, PA2885, MAM, and the selenomethionine-substituted (Se-Met) pfiT as GST-fusion proteins were carried out as described previously [24]. The GST-removed proteins were obtained by digestion of the GST-fusion proteins with the PreScission protease (GE HealthCare).
Preparation of mouse spleen lymphocytes
Spleens from naive mice were harvested by grinding them with frosted slides. Harvested cells were washed with Hank's balanced salt solution (HBSS) and then taken in 0.5 ml of hypotonic ammonium chloride solution to lyse the red blood cells (RBCs). Spleen lymphocytes free of RBC were then washed twice with HBSS by centrifugation, counted with a hemocytometer, and kept in complete tumor medium at 4°C until use.
Preparation of human PBMC
Buffy coats of human blood samples were purchased from Research Blood Component (Brighton, MA). 20 ml of Histopaque-1077 (Sigma) was pipetted into 50 ml conical centrifuge tubes and allowed sufficient time to reach the room temperature, after which 20 ml of fresh blood buffy coat was layered slowly on top of the Histopaque layer and centrifuged at 400 g for 30 minutes at room temperature. After centrifugation, the upper fluid layer was slowly removed, and the interfacial cellular layer was collected and transferred to another fresh 50 ml conical centrifuge tube. Cells were washed with phosphate buffered saline (PBS) once at room temperature and once at 4°C by centrifugation. Human T lymphocytes were further isolated from purified PBMC using the Pan T cell isolation kit II (Miltenyi Biotec Inc.) according to the manufacturer's instructions. The non-T cell components were also collected to serve as the antigen presenting cells in T-cell proliferation assay. Cells were counted by a hemocytometer, and either placed in RPMI1640 medium with 10% FBS on ice for culturing, or frozen at −80°C for future use.
For CFSE labeling experiment, purified T lymphocytes was labeled with 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE) (Sigma), according to the manufacturer's instructions.
Lymphocyte proliferation assay
1×106 lymphocytes isolated either from mouse spleen or from human PBMC were cultured with stimulating agents at a volume of 200 µl/well in CTM or RPMI1640 medium with 10% FBS in a 96 well culture plate. After 48 hour, 0.5 µCi of [3H] thymidine (New England Nuclear, Boston, MA) was added to the cultures and 16 h later the cells were harvested and [3H]thymidine incorporation was measured using a 1205 beta plate counter (Wallac, Gaithersburg, MD).
For CSFE experiments, CFSE-labeled human T cells (2×106 cells per well) were cultured with 1×105 antigen-presenting cells in the presence of various SAgs or controls in a 24-well flat-bottomed plate. After 96 hours, cells in each well were washed and resuspended with phosphate buffered saline (PBS). Anti-CD3-PerCP antibody was added and incubated at room temperature in dark for 20 minutes, and then washed by 1× PBS. Cells were finally resuspended in 500 µl 1× PBS for FACS analysis using a Becton Dickinson FACSCalibur (BD Biosciences).
125I binding assay
Purified pfiT, PA2885, and MAM were labeled with 125I as described previously [22]. Cells (5×105 cells/sample) were incubated with 125I-labeled proteins (25 nM, in 300 ml of PBS containing 1% BSA, 1 µM ZnCl2, and 0.02% NaN3). The reaction mixture was incubated at room temperature for two hours, washed with 1 ml PBS. Cells bound with labeled SAg were recovered after centrifugation over an oil cushion, and counted in a gamma counter.
FITC-protein binding assay
Purified pfiT, PA2885, and MAM were labeled with FITC according to the manufacturer's instructions. Human PBMCs (5×105 cells/sample) were incubated in a 50 µl volume with buffer control or various antibodies (100 nM) at room temperature for 20 minutes, then 50 µl of FITC-labeled proteins at 100 nM concentration was added along with anti-CD19-APC, and incubated in dark for 20 minutes. Cells were washed with 1× PBS, spun at 400 g for 5 minutes, and resuspended in 500 µl PBS for FACS analysis using a Becton Dickinson FACSCalibur (BD Biosciences).
GST pull down assay
GST, GST-MAM and GST-pfiT (200 µg) were immobilized on glutathione-sepharose 4B beads (GE HealthCare) and aliquoted at 10 µg/tube). Raji cell lysate (15 µl) or purified recombinant HLA-DR1/HA complex was applied to the aliquoted beads, and incubated at 4°C for 1 hour. The incubated beads were washed four times with a buffer containing 20 mM Tris, pH 7.4, 0.1 mM EDTA, and 100 mM NaCl), and subjected to western blot analysis using anti-HLA-DRα (G-7, Santa Cruz) or anti-GST antibody (GE HealthCare). Binding affinity (KD) or IC50 was evaluated by non-linear regression using the Prism software (GraphPad Software).
Analytical ultracentrifugation (AUC) sedimentation velocity
Sedimentation-velocity (SV) experiments were conducted at 20°C in a Beckman Optima XL-I analytical ultracentrifuge at a rotor speed of 50,000 rpm. Double-sector cells were loaded with 400 µl of pfiT at 3.6 µM and with 410 µl of reference solutions, respectively. Unless otherwise specified, the reference solution is Hepes-buffered saline (HBS) containing 10 mM Hepes buffer (pH 7.4), 150 mM NaCl, 2 mM DTT. Data were recorded with absorbance detection at wavelengths of 280 nm. Absorbance profiles were analyzed with the software SEDFIT (http://www.analyticalultracentrifugation.com) [27], using a model for continuous sedimentation coefficient distributions c(s) [26]. Distributions were calculated with maximum entropy regularization at a predetermined confidence level of 1 standard deviation.
AUC sedimentation equilibrium
Sedimentation equilibrium studies were conducted at a temperature of 20°C and at three rotor speeds for each protein or mixture. The individual protein or protein mixture (100 µl) at various concentrations in HBS was loaded into an Epon double-sector centerpiece. Reference cells were loaded with 110 µl of reference solution. For pfiT alone, sedimentation equilibrium of pfiT was analyzed at concentrations of 9 µM and 18 µM, with three rotor speeds (20,000, 25,000, and 30,000 rpm). For the pfiT/HLA-DR1/HA complex, mixtures of pfiT and DR1 at various molar ratios and concentrations, (66∶6.5, 44∶13, and 8∶24 in a real micromolar ratio of concentrations for pfiT:HLA-DR1), were used for sedimentation equilibrium analysis with three rotor speeds (20,000, 25,000, and 30,000 rpm).
Equilibrium absorbance profiles were acquired at either 280-nm (for pfiT alone) or 300-nm (for the pfiT-DR1 complex) wavelength. The equilibrium sedimentation data were analyzed using the software SEDPHAT (http://www.analyticalultracentrifugation.com) [27]. Data analysis was performed by global least-squares analysis of data from multiple concentrations and multiple rotor speeds, using conservation of mass constraints [27].
Crystallization, X-ray data collection, structure determination and refinement
Purified pfiT was concentrated to about 10–15 mg/ml with a buffer of 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 2 mM DTT. Crystals of pfiT were grown at room temperature in hanging drops, by mixing 2 µl of protein solution with an equal volume of reservoir solution containing 4–8% PEG 3350, 0.1 M Tris, pH 8.0, 0.2 M ammonium acetate, 5 mM MgCl2, 5 mM DTT, 5% isopropanol, and 5% glycerol. Microseeding was used to produce large crystals.
Native pfiT crystals belong to space group C2 (Table 1). The Se-Met substituted pfiT crystals belong to space group P21 (Table 1). Prior to data collection, all crystals were transferred to a reservoir solution containing 25% glycerol, and then flashed-cooled under a nitrogen stream at 100K, and stored in liquid nitrogen. Diffraction data of native crystal were collected to 1.7 Å resolution at 100K at beamline X4A of the National Synchrotron Light Source (NSLS) (Brookhaven National Laboratory). In order to solve the structure, diffraction data of the Se-Met crystals were collected to various resolutions (2.4 Å to 1.95 Å). All of the data were processed and scaled using HKL2000 [52] (Table 1).
The crystal structure of pfiT was first determined at 2.4 Å resolution using the single anomalous diffraction (SAD) phasing method with a SAD data collected at the Se peak wavelength, using the program SOLVE [53]. The electron density map was improved using the program RESOLVE [54]. Fragments containing about 55% of the pfiT residues could be automatically traced by the program RESOLVE. The model of a pfiT monomer was finally completed by manually fitting the electron density map with the pfiT sequence using the program TURBO FRODO [55]. The second pfiT molecule of the dimer in the asymmetric unit was then generated using the NCS symmetry. The structure was then fully refined, using a diffraction data at higher resolution (1.95 Å) with the PHENIX program suite [56]. The crystal structure of native pfiT was determined using the molecular replacement methods using PHENIX [56]. Structural refinement was carried out using PHENIX [56] (Table 1).
The final refinement statistics are summarized in Table 1. Atomic coordinates have been deposited in the Protein Data Bank as entries 4MO7 and 4MXM.
Supporting Information
Zdroje
1. KapplerJW, PullenA, CallahanJ, ChoiY, HermanA, et al. (1989) Consequences of self and foreign superantigen interaction with specific V beta elements of the murine TCR alpha beta. Cold Spring Harb Symp Quant Biol 54 Pt 1 : 401–407.
2. MarrackP, KapplerJ (1990) The staphylococcal enterotoxins and their relatives. Science 248 : 1066.
3. KotzinBL, LeungDY, KapplerJ, MarrackP (1993) Superantigens and their potential role in human disease. Adv Immunol 54 : 99–166.
4. LiH, LleraA, MalchiodiEL, MariuzzaRA (1999) The structural basis of T cell activation by superantigens. Annu Rev Immunol 17 : 435–466.
5. McCormickJK, YarwoodJM, SchlievertPM (2001) Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol 55 : 77–104.
6. PeterssonK, PetterssonH, SkartvedNJ, WalseB, ForsbergG (2003) Staphylococcal enterotoxin h induces valpha-specific expansion of T cells. J Immunol 170 : 4148–4154.
7. WangLM, ZhaoYW, LiZ, GuoY, LindsayJL, et al. (2007) Crystal Structure of a Complete Ternary Complex Between T-Cell Receptor, Superantigen, and peptide-MHC Class II Molecule. Nat Struct & Mol Biol 14 : 169–171.
8. SalineM, RodstromKE, FischerG, OrekhovVY, KarlssonBG, et al. (2010) The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nature communications 1 : 119.
9. DalwadiH, WeiB, KronenbergM, SuttonCL, BraunJ (2001) The Crohn's disease-associated bacterial protein I2 is a novel enteric t cell superantigen. Immunity 15 : 149–158.
10. SuttonCL, KimJ, YamaneA, DalwadiH, WeiB, et al. (2000) Identification of a novel bacterial sequence associated with Crohn's disease. Gastroenterology 119 : 23–31.
11. WeiB, HuangT, DalwadiH, SuttonCL, BrucknerD, et al. (2002) Pseudomonas fluorescens encodes the Crohn's disease-associated I2 sequence and T-cell superantigen. Infect Immun 70 : 6567–6575.
12. LandersCJ, CohavyO, MisraR, YangH, LinYC, et al. (2002) Selected loss of tolerance evidenced by Crohn's disease-associated immune responses to auto - and microbial antigens. Gastroenterology 123 : 689–699.
13. ArnottID, LandersCJ, NimmoEJ, DrummondHE, SmithBK, et al. (2004) Sero-reactivity to microbial components in Crohn's disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am J Gastroenterol 99 : 2376–2384.
14. IltanenS, TervoL, HalttunenT, WeiB, BraunJ, et al. (2006) Elevated serum anti-I2 and anti-OmpW antibody levels in children with IBD. Inflamm Bowel Dis 12 : 389–394.
15. SpivakJ, LandersCJ, VasiliauskasEA, AbreuMT, DubinskyMC, et al. (2006) Antibodies to I2 predict clinical response to fecal diversion in Crohn's disease. Inflamm Bowel Dis 12 : 1122–1130.
16. MundwilerML, MeiL, LandersCJ, ReveilleJD, TarganS, et al. (2009) Inflammatory bowel disease serologies in ankylosing spondylitis patients: a pilot study. Arthritis research & therapy 11: R177.
17. AshornS, ValinevaT, KaukinenK, AshornM, BraunJ, et al. (2009) Serological responses to microbial antigens in celiac disease patients during a gluten-free diet. Journal of clinical immunology 29 : 190–195.
18. SuzukiH, FukudaY, KoizukaH, TomitaT, HoriK, et al. (2008) Dietary antigens in Crohn's disease: antibodies to porcine pancreatic amylase. The American journal of gastroenterology 103 : 656–664.
19. MowWS, VasiliauskasEA, LinYC, FleshnerPR, PapadakisKA, et al. (2004) Association of antibody responses to microbial antigens and complications of small bowel Crohn's disease. Gastroenterology 126 : 414–424.
20. MorganXC, TickleTL, SokolH, GeversD, DevaneyKL, et al. (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome biology 13: R79.
21. KimSC, TonkonogySL, AlbrightCA, TsangJ, BalishEJ, et al. (2005) Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128 : 891–906.
22. Etongue-MayerP, LangloisMA, OuelletteM, LiH, YounesS, et al. (2002) Involvement of zinc in the binding of mycoplasma arthritidis-derived mitogen to the proximity of the HLA-DR binding groove regardless of histidine 81 of the beta chain. Eur J Immunol 32 : 50–58.
23. BaccalaR, SmithLR, VestbergM, PetersonPA, ColeBC, et al. (1992) Mycoplasma arthritidis mitogen. V beta engaged in mice, rats, and humans, and requirement of HLA-DR alpha for presentation. Arthritis Rheum 35 : 434–442.
24. ZhaoY, LiZ, DrozdS, GuoY, MouradW, et al. (2004) Crystal structure of Mycoplasma arthritidis mitogen complexed with HLA-DR1 reveals a novel superantigen fold and a dimerized superantigen-MHC complex. Structure 12 : 277–288.
25. LiHM, ZhaoY, GuoY, LiZ, EiseleL, et al. (2007) Zinc induces dimerization of the class II MHC molecule that leads to cooperative binding to a superantigen. J Biol Chem 282 : 5991–6000.
26. SchuckP, PeruginiMA, GonzalesNR, HowlettGJ, SchubertD (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 82 : 1096–1111.
27. VisticaJ, DamJ, BalboA, YikilmazE, MariuzzaRA, et al. (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem 326 : 234–256.
28. GibratJF, MadejT, BryantSH (1996) Surprising similarities in structure comparison. Curr Opin Struct Biol 6 : 377–385.
29. RamosJL, Martinez-BuenoM, Molina-HenaresAJ, TeranW, WatanabeK, et al. (2005) The TetR family of transcriptional repressors. Microbiology and molecular biology reviews : MMBR 69 : 326–356.
30. Martinez-BuenoM, Molina-HenaresAJ, ParejaE, RamosJL, TobesR (2004) BacTregulators: a database of transcriptional regulators in bacteria and archaea. Bioinformatics 20 : 2787–2791.
31. HinrichsW, KiskerC, DuvelM, MullerA, TovarK, et al. (1994) Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science 264 : 418–420.
32. OrthP, SchnappingerD, HillenW, SaengerW, HinrichsW (2000) Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat Struct Biol 7 : 215–219.
33. MillerDJ, ZhangYM, SubramanianC, RockCO, WhiteSW (2010) Structural basis for the transcriptional regulation of membrane lipid homeostasis. Nature structural & molecular biology 17 : 971–975.
34. SchumacherMA, MillerMC, GrkovicS, BrownMH, SkurrayRA, et al. (2001) Structural mechanisms of QacR induction and multidrug recognition. Science 294 : 2158–2163.
35. GuR, SuCC, ShiF, LiM, McDermottG, et al. (2007) Crystal structure of the transcriptional regulator CmeR from Campylobacter jejuni. Journal of Molecular Biology 372 : 583–593.
36. AlguelY, LuD, QuadeN, SauterS, ZhangX (2010) Crystal structure of MexZ, a key repressor responsible for antibiotic resistance in Pseudomonas aeruginosa. Journal of structural biology 172 : 305–310.
37. SchumacherMA, MillerMC, GrkovicS, BrownMH, SkurrayRA, et al. (2002) Structural basis for cooperative DNA binding by two dimers of the multidrug-binding protein QacR. Embo J 21 : 1210–1218.
38. EuzebyJP (1997) List of Bacterial Names with Standing in Nomenclature: a folder available on the Internet. International journal of systematic bacteriology 47 : 590–592.
39. LoperJE, HassanKA, MavrodiDV, DavisEW2nd, LimCK, et al. (2012) Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS genetics 8: e1002784.
40. SilbyMW, Cerdeno-TarragaAM, VernikosGS, GiddensSR, JacksonRW, et al. (2009) Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome biology 10: R51.
41. KimbrelJA, GivanSA, HalgrenAB, CreasonAL, MillsDI, et al. (2010) An improved, high-quality draft genome sequence of the Germination-Arrest Factor-producing Pseudomonas fluorescens WH6. BMC genomics 11 : 522.
42. OrtetP, BarakatM, LalaounaD, FochesatoS, BarbeV, et al. (2011) Complete genome sequence of a beneficial plant root-associated bacterium, Pseudomonas brassicacearum. Journal of bacteriology 193 : 3146.
43. PaulsenIT, PressCM, RavelJ, KobayashiDY, MyersGS, et al. (2005) Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nature biotechnology 23 : 873–878.
44. MorgulisA, CoulourisG, RaytselisY, MaddenTL, AgarwalaR, et al. (2008) Database indexing for production MegaBLAST searches. Bioinformatics 24 : 1757–1764.
45. ColeBC, SawitzkeAD, AhmedEA, AtkinCL, DavidCS (1997) Allelic polymorphisms at the H-2A and HLA-DQ loci influence the response of murine lymphocytes to the Mycoplasma arthritidis superantigen MAM. Infect Immun 65 : 4190–4198.
46. DonadiniR, LiewCW, KwanAH, MackayJP, FieldsBA (2004) Crystal and solution structures of a superantigen from Yersinia pseudotuberculosis reveal a jelly-roll fold. Structure 12 : 145–156.
47. MendozaJL, AbreuMT (2009) Biological markers in inflammatory bowel disease: practical consideration for clinicians. Gastroenterol Clin Biol 33 Suppl 3: S158–173.
48. PrideauxL, De CruzP, NgSC, KammMA (2012) Serological antibodies in inflammatory bowel disease: a systematic review. Inflamm Bowel Dis 18 : 1340–1355.
49. HouJK, AbrahamB, El-SeragH (2011) Dietary intake and risk of developing inflammatory bowel disease: a systematic review of the literature. Am J Gastroenterol 106 : 563–573.
50. SmithPM, HowittMR, PanikovN, MichaudM, GalliniCA, et al. (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341 : 569–573.
51. RayD, ShahA, TilgnerM, GuoY, ZhaoY, et al. (2006) West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. Journal of virology 80 : 8362–8370.
52. OtwinowskiZ, MinorW (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology 276 : 307–326.
53. TerwilligerTC, BerendzenJ (1999) Automated structure solution for MIR and MAD. Acta Crystallogr D Biol Crystallogr D55 : 849–861.
54. TerwilligerTC (2000) Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr 56(Pt 8): 965–972.
55. Roussel A, Cambillau C (1989) TURBO FRODO. Silicon Graphics Geometry Partners Directory. Mountain View, CA: Silicon Graphics. pp. 77–78.
56. AdamsPD, AfoninePV, BunkocziG, ChenVB, DavisIW, et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66 : 213–221.
57. NichollsA, SharpKA, HonigB (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 : 281–296.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells