
The Cyst Wall Protein CST1 Is Critical for Cyst Wall Integrity and Promotes Bradyzoite Persistence
Toxoplasma gondii infects up to one third of the world's population. A key to the success of T. gondii as a parasite is its ability to persist for the life of its host as bradyzoites within tissue cysts. The glycosylated cyst wall is the key structural feature that facilitates persistence and oral transmission of this parasite. Because most of the antibodies and reagents that recognize the cyst wall recognize carbohydrates, identification of the components of the cyst wall has been technically challenging. We have identified CST1 (TGME49_064660) as a 250 kDa SRS (SAG1 related sequence) domain protein with a large mucin-like domain. CST1 is responsible for the Dolichos biflorus Agglutinin (DBA) lectin binding characteristic of T. gondii cysts. Deletion of CST1 results in reduced cyst number and a fragile brain cyst phenotype characterized by a thinning and disruption of the underlying region of the cyst wall. These defects are reversed by complementation of CST1. Additional complementation experiments demonstrate that the CST1-mucin domain is necessary for the formation of a normal cyst wall structure, the ability of the cyst to resist mechanical stress, and binding of DBA to the cyst wall. RNA-seq transcriptome analysis demonstrated dysregulation of bradyzoite genes within the various cst1 mutants. These results indicate that CST1 functions as a key structural component that confers essential sturdiness to the T. gondii tissue cyst critical for persistence of bradyzoite forms.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003823
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003823
Summary
Toxoplasma gondii infects up to one third of the world's population. A key to the success of T. gondii as a parasite is its ability to persist for the life of its host as bradyzoites within tissue cysts. The glycosylated cyst wall is the key structural feature that facilitates persistence and oral transmission of this parasite. Because most of the antibodies and reagents that recognize the cyst wall recognize carbohydrates, identification of the components of the cyst wall has been technically challenging. We have identified CST1 (TGME49_064660) as a 250 kDa SRS (SAG1 related sequence) domain protein with a large mucin-like domain. CST1 is responsible for the Dolichos biflorus Agglutinin (DBA) lectin binding characteristic of T. gondii cysts. Deletion of CST1 results in reduced cyst number and a fragile brain cyst phenotype characterized by a thinning and disruption of the underlying region of the cyst wall. These defects are reversed by complementation of CST1. Additional complementation experiments demonstrate that the CST1-mucin domain is necessary for the formation of a normal cyst wall structure, the ability of the cyst to resist mechanical stress, and binding of DBA to the cyst wall. RNA-seq transcriptome analysis demonstrated dysregulation of bradyzoite genes within the various cst1 mutants. These results indicate that CST1 functions as a key structural component that confers essential sturdiness to the T. gondii tissue cyst critical for persistence of bradyzoite forms.
Introduction
Toxoplasma gondii, an Apicomplexan, is an obligate intracellular protozoan parasite that can cause severe human disease. It is estimated that a third of the human population is chronically infected with T. gondii [1], with prevalence rates ranging from a few percent to nearly 80% depending on the population [2]. This parasite can cause lethal encephalitis in immune compromised individuals such as those with AIDS or organ transplant recipients on immune suppressive medications. It is also the cause of a devastating congenital disease, which may result in blindness and mental retardation if infection occurs in a T. gondii seronegative pregnant woman. During acute infection, the parasites proliferate as the fast-growing tachyzoite life cycle form, which causes a disseminated systemic infection. This disseminated acute infection is controlled by interferon-γ and T cell responses. In response to stress signals during acute infection, such as the immune response or programmed spontaneous differentiation responses, tachyzoites differentiate into the slow-growing bradyzoite life cycle stage that remains latent in the host. Bradyzoites can form tissue cysts in brain, muscles and visceral organs and when tissue cysts are orally ingested the released bradyzoites differentiate into tachyzoites, causing an acute infection in a new host. Bradyzoite differentiation processes and the development and maintenance of tissue cysts are critical for transmission of T. gondii infection. Evidence suggests that the latent tissue cysts evade the immune response [3] and can persist for the host life span [4]. It is likely tissue cysts occasionally rupture and any released parasites [5] are cleared by immune system. In the absence of an effective immune response these released organisms can differentiate into tachyzoites causing an acute infection. Thus, tissue cysts serve as reservoir for the reactivation of the toxoplasmosis when the host becomes immune compromised with conditions such as AIDS or organ transplantation.
Tissue cysts can range from 5 to 100 µm in size containing just a few to thousands of encysted bradyzoites. Tissue cysts can be found in any organ, but are especially prevalent in the central nervous system. The bradyzoites within the tissue cyst are covered by a prominent translucent 0.25 to 0.75 µm thick cyst wall structure (cyst wall) which can be visualized using electron microscopy [6]. The cyst wall forms beneath a modified parasitophorous vacuole membrane containing bradyzoites. The cyst wall is highly glycosylated and stains easily with periodic acid-Schiff, Dolichos biflorus lectin (DBA), and succinylated wheat germ agglutinin [6]–[8]. These carbohydrate modifications of the cyst wall are hypothesized to mask cyst wall proteins from host immune responses and to provide structural and chemical resistance against environmental stress, facilitating transmission of this pathogen [9].
The biogenesis, composition, and functions of the cyst wall are not yet well defined. A cyst wall glycoprotein CST1 was discovered more than a decade ago [10]. This protein, CST1, binds to DBA lectin, suggesting that it is a glycoprotein that contains N-acetyl-galactosamine. CST1 localized to the in vivo and in vitro cyst wall, but was not found associated with the tachyzoite parasitophorous vacuole. The corresponding gene, CST1, has not previously been identified, as the available monoclonal antibody 73.18 [11] to CST1 recognizes a glycoepitope and attempts to identify the glycoprotein recognized by this monoclonal antibody were unsuccessful (Weiss LM, unpublished). While some progress has occurred, cyst wall biology is still poorly understood despite the clinical and biological importance of this structure for transmission and latency in this important protozoan infection [12]. We produced a new monoclonal antibody library to T. gondii tissue cysts and used a combination of microscopic, genetic and proteomic approaches to identify cyst wall components. Using this approach we identified CST1, the gene corresponding to CST1, and characterized the effect of a knockout of this gene on T. gondii.
Results
Monoclonal antibody SalmonE binds to the cyst wall
To identify cyst wall proteins, a hybridoma library was created from mice immunized with a lysate of T. gondii ME49 cysts purified from the brains of mice with chronic T. gondii infection. From this library, we screened monoclonal antibodies by immunofluorescence against ME49 T. gondii in vitro cysts (bradyzoite-) and tachyzoite-containing vacuoles. Among the 189 cyst-wall positive hybridomas, we identified an mAb clone SalmonE that reacted with bradyzoite-containing parasitophorous vacuoles and uniformly stained the limiting parasitophorous vacuole membrane of BAG1-positive parasites (bradyzoites) but did not stain vacuoles containing BAG1 negative parasites (Figure 1A). BAG1 negative vacuoles were positive for SAG1, a tachyzoite specific marker (data not shown). This candidate cyst wall reactive monoclonal antibody was used to further characterize the cyst wall.

Since in vitro cysts do not completely differentiate, we examined the localization of SalmonE in cysts isolated from mice with chronic T. gondii infection. Within these more mature cysts there is a more organized cyst wall structure, and the bradyzoites within these cysts enter Go and arrest in the cell cycle [12], [13]. T. gondii (ME49 strain) in vivo brain cysts harvested from infected mice were labeled with SalmonE and analyzed using immuno-electron microscopy (Figure 1B). SalmonE recognizes the diffuse thick layer of the cyst wall beneath the limiting membrane of the cyst wall in a distribution similar to the reactivity of the mAb specific for CST1 as well as the DBA lectin [10].
CST1, the mAb SalmonE reactive molecule, is a SRS glycoprotein with an extended mucin domain
To determine the target of monoclonal antibody SalmonE, antigens from ME49 in vitro derived bradyzoite lysates were immunoprecipitated with mAb SalmonE, separated with SDS-PAGE and the two major candidate protein bands (a low signal intensity 150 kDa band and a high signal intensity high molecular weight band in the stacking gel) were excised and analyzed by MALDI-TOF mass spectrometry. The high molecular band had two peptides that matched the predicted gene product of TGME49_064660 (peptides: RGGGFLTTYTLNVPRL and KEFLRPLADLVPGASLKL, MASCOT, p<10−7, Figure 2A), which had been annotated as SRS44 in a published analysis of SRS domain-containing proteins [14]. The low molecular weight band had two peptides that matched SRS13 (peptides: KLPEKPAAAVAR and LTLDAGPPQATTLCYK), a glycoprotein we also subsequently characterized (Tomita and Weiss, in preparation). Polyclonal murine antiserum raised to the first 200 amino acids of SRS13 did not react with the cyst wall (data not shown). There was no other protein identified from SalmonE immunoprecipitated bands. To verify that the TGME49_064660 gene product is responsible for the cyst wall staining of monoclonal antibody SalmonE, mouse antiserum was raised against recombinant TGME49_064660 protein consisting of the first 200 amino acid of the predicted gene (Figure 2A, rTGME49_064660). Probing in vitro cysts with the anti-rTGME49_064660 serum revealed a similar pattern of staining as seen with the monoclonal antibody SalmonE (Figure 1A and 2B). This verified that TGME49_064660 is indeed a cyst wall gene. After completion of molecular verification (see below for details) TGME49_064660 was identified as CST1, the gene corresponding to the previously identified protein CST1 [10].

The cyst wall localization as well as its identification as an SRS protein suggested that CST1 should be a secreted protein; however, the current annotated TGME49_064660 gene product in ToxoDB.org does not contain a potential signal peptide sequence (SignalP 4.0 prediction [15]). Examination of the upstream sequences (Figure S1A), suggested that the gene model was incorrect. Use of 5′ rapid amplification of cDNA ends (RACE) demonstrated additional coding sequence, a revised 5′ UTR (Figure S1A) and also confirmed that CST1 (TGME49_064660) does not extend into the predicted upstream gene TGME49_064670. Sequence analysis of the 5′ RACE product revealed an in-frame methionine codon located 43 residues upstream of the annotated predicted start site (Figure S1A). The protein predicted using this upstream methionine codon encodes a high probability signal peptide with a cleavage site; therefore, it is likely that CST1 translation begins at this methionine, 43 residues upstream of the annotated initiator methionine codon.
CST1 contains thirteen SRS domains (Figure 2A) and is unique among the SRS family proteins in having such a large number of SRS domains. Another striking feature of the predicted protein is the presence of a 263 amino acid stretch with multiple threonine-rich tandem repeats of T5–11[R/I]K2P; this region has homology to the mucin-like domains in a major glycoprotein of Cryptosporidium parvum (GP900, CMU_014140) (Figure 2A). Since mucin domains are typically extensively O-glycosylated on Ser or Thr residues, the probability of O-glycosylation at this mucin domain was assessed using neural network model NetOGlyc 3.1 [16]. Of the 157 threonines in the mucin domain, 95% were predicted to be O-glycosylated using NetOGlyc 3.1(Figure 3A).
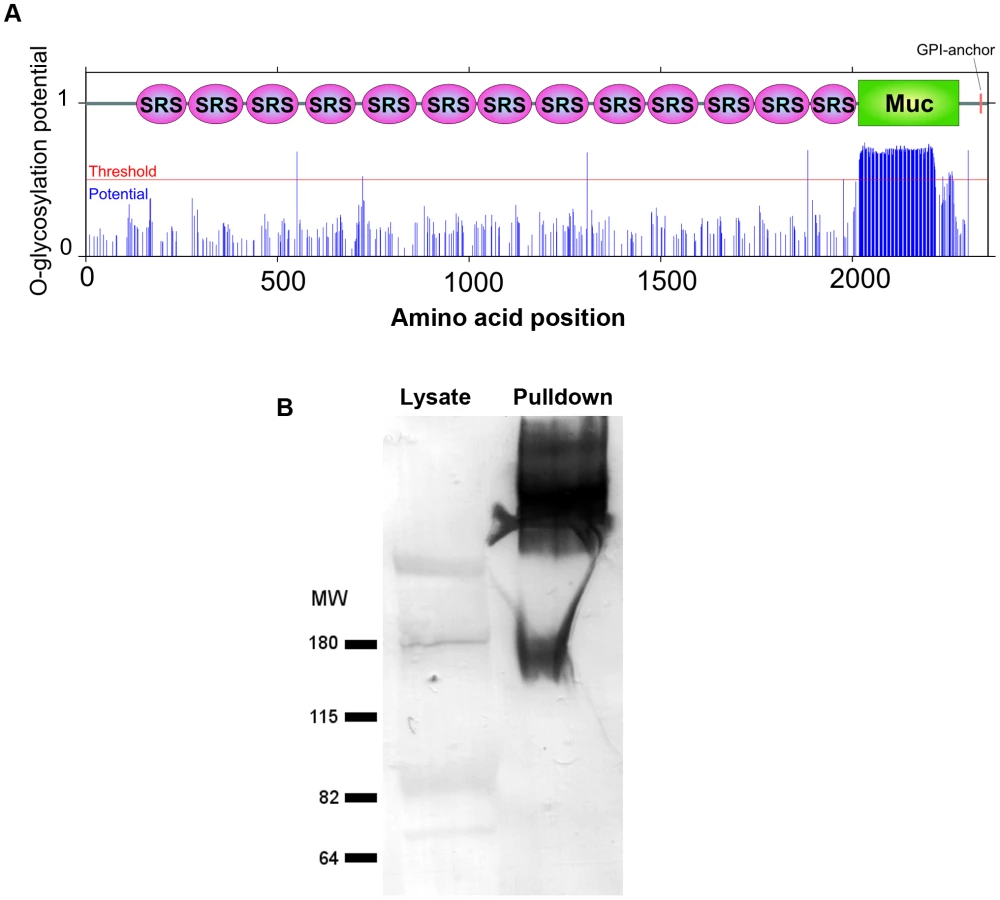
To investigate whether the CST1 is O-glycosylated, SalmonE-immunoprecipitates were probed with Dolichos biflorus lectin (DBA), a marker for the cyst wall that recognizes GalNAc [17] (Figure 3B). DBA lectin overlays verified that CST1, the TGME49_064660 gene product, is a glycoprotein. CST1 mRNA is expressed in type I (RH), type II (P/ME49) and type III (CTG) strains as evidenced by expression data from www.ToxoDB.org and our RNA-seq data (Figure S1).
CST1 is not required for cyst formation
To understand the function of CST1 we deleted the entire CST1 gene (Figure 4A) in the PruΔku80 background [18]. This strain has a high frequency of homologous recombination that facilitates the development of knock-outs and also contains GFP under the control of bradyzoite specific LDH2 promoter so that the brain cysts containing bradyzoites can readily be identified by fluorescence microscopy [18]. The deletion of the CST1 (Δcst1 T. gondii strain) was verified by PCR (Figure S1B) as well as by RNA-seq (Figure S1C).

T. gondii lysates of parasites grown in vitro at pH 8.1 (bradyzoites) and pH 7 (tachyzoites) were probed with mAb SalmonE by immunoblot (Figure 4B). The mAb SalmonE reactive band at pH 8.1 is seen in the stacking gel, suggesting that this is a high molecular mass antigen with extensive post translational glycosylation that may prevent entry into the resolving gel. While the mAb SalmonE reactivity is virtually absent in parasites grown at pH 7, a strong signal was observed in lysates from wild-type parasites grown at pH 8.1. These CST1 bands also bind to DBA lectin consistent with the presence of glycosylation in this protein (Figure S2).
Since CST1 was originally defined by reactivity to DBA and recognition by mAb 73.18 [10], [11], the CST1 deficient strain (Δcst1) should not be recognized by mAb 73.18. Immunopurified CST1, and cell lysates from pH 8.1 treated T. gondii Pru wild type or Δcst1 cultures were probed with the previously described CST1 specific mAb 73.18 and mAb SalmonE (Figure 5A). Monoclonal antibody SalmonE and mAb 73.18 had similar patterns of reactivity on immunoblot, and, as expected, the Δcst1 did not have the major immunoreactive band and had lost the characteristic cyst wall labeling seen with DBA, mAb SalmonE (Figure 4C), or mAb 73.18 (Figure 5B). These cysts that did not stain with DBA, mAb SalmonE or 73.18 were still positive with BAG1 antibody (Figure 6). Examination of brains of mice infected with Δcst1 T. gondii demonstrated that cyst formation could still occur in this knockout strain (Fig. 7).



Collectively, these results indicate CST1 is not required for bradyzoite or cyst formation and that CST1 is the cyst wall protein recognized by mAb SalmonE, DBA, and mAb 73.18. Both DBA and mAb 73.18 recognize glycoepitopes, and we noted that while cyst wall reactivity was lost in Δcst1, there was some residual reactivity seen within the parasites by mAb SalmonE, DBA and mAb 73.18, suggesting that other less abundant glycoepitopes that react with mAb SalmonE, DBA, and mAb 73.18 are present in bradyzoites.
Complementation of Δcst1 strain reveals that the mucin domain is required for DBA lectin reactivity
To examine the role of glycosylation in CST1 function, we complemented the Δcst1 strain with two variants of CST1 (Figure 4A). One Δcst1 line was complemented with a full-length cDNA (Δcst1::cst1) and the other with a CST1 lacking the 789 bp region coding for the mucin domain (Δcst1::cst1Δmuc). Following transfection and selection, the presence of those complemented genes were verified by PCR (Figure S1B) and RNA-seq (Figure S1C). Stage specific expression of CST1 was equivalent to that seen in the wild type parasite in the complemented Δcst1::cst1 T. gondii strain as verified by immunoblot with mAb SalmonE (Figure 4B). IFA of pH 8.1 treated Δcst1::cst1 in vitro cysts demonstrated that the strain complemented with full-length cDNA of CST1 has the correct localization of CST1 to the cyst wall, as well as the restoration of DBA staining (Figure 4C) and mAb 73.18 staining (Figure 5B) of the cyst wall. These cysts remained IFA positive for BAG1 (Figure 6).
In contrast, the Δcst1::cst1Δmuc parasites lack the major mAb SalmonE and DBA reactive band in immunoblot (Figure 4B and S2), and lack cyst wall staining with DBA, mAb SalmonE, and mAb 73.18 in IFA (Figure 4C and 5B). The expression and localization of CST1Δmuc protein was verified with a polyclonal antibody produced against recombinant CST1 (rTGME49_064660 AA1-200) (Figure S3).
Δcst1 parasites form fewer brain cysts and exhibit a fragile cyst wall phenotype
To determine the effect of CST1 deletion or mucin domain deletion in vivo, C57BL/6 mice were infected with wild type, Δcst1, Δcst1::cst1, and Δcst1::cst1Δmuc parasites at 200 parasites per mouse. The mouse survival rates (Figure 7A) during acute infection with each parasite line were not statistically different (n = 20, Log-rank test). The number of brain cysts per mouse at 4 weeks after infection (Figure 7B) was reduced by 41% in the Δcst1 strain (p<0.05, Mann-Whitney U test). Complementation with full length (Δcst1::cst1), but not mucin-null (Δcst1::cst1Δmuc), restored the cyst number level back to the wild type levels. Histological analysis of the brains suggested that inflammation was less severe in Δ cst1 than the wild type (Figure S4); however this did not achieve statistical significance. Cysts were produced in vivo by all mutants (Figure 7C) and there was no difference in the size of cysts produced by these mutants. Brains from mice infected with the wild type, Δ cst1, Δ cst1::cst1Δmuc, and Δcst1::cst1 parasites were fed to Balb/cDM1 and all were capable of transmitting infection.
During the brain cyst isolation procedure, cysts are subjected to mechanical stress to release them from brain tissue to purify them by isopycnic centrifugation [19]. The wild type cysts stayed intact during this procedure, but Δcst1 brain cysts were much more fragile and broke apart when homogenized using a pestle tissue homogenizer to purify cysts from brain samples [19] (Figure 8A). Despite many attempts, we were unable to develop a reliable procedure to purify intact Δcst1 cysts from mouse brains or a method to standardize cyst inocula to compare the transmissibility of cysts from our mutant strains.

To further investigate the fragile phenotype, we examined the ultrastructure of the brain cysts by electron microscopy. Figure 8B demonstrates a wild type brain cyst, which has the classic organized cyst wall with an underlying amorphous granular layer. In contrast, the Δcst1 brain cysts lack this organization and displayed a disrupted layer. Independently isolated Δcst1 clones had the same fragile phenotype. Full length cDNA complementation of Δcst1 (Δcst1::cst1) rescued the fragile brain cyst phenotype as well as restoring the cyst wall layer as seen by TEM (Figure 8). In contrast, complementation of Δcst1 parasite with cst1Δmuc gene did not rescue the fragile cyst wall phenotype or correct the disruption of cyst wall layer seen by TEM (Figure 8). Measurements of the cyst wall confirmed a significant decrease in the cyst wall thickness with disruption of cst1: WT 153±28 nm, Δcst1 24±10 nm, Δcst1::cst1 284±65 nm, and Δ cst1::cst1Δmuc, 34±14 nm (p<0.05 WT vs Δcst1, WT vs Δ cst1::cst1Δmuc) as well as an increase in cyst wall thickness in the Δcst1::cst1 strain (p<0.05 WT vs Δ cst1::cst1).
To determine if cyst wall thickness affected cyst fragility, we compared cyst fragility in individual brains using a relatively vigorous disruption method with a small sintered glass pestle tissue homogenizer (size A: 0.1–0.15 mm clearance, frosted inner glass surface) that disrupts a significant fraction of wild-type cysts in brain homogenate. For this experiment, brains (n = 4 per group) were cut in half, the cysts in the right half were homogenized unfixed, and the left half was fixed with 4% paraformaldehyde overnight at 4°C prior to homogenization with the pestle tissue homogenizer. Using this procedure 100±0% of Δcst1 and 100±0% Δcst1::cst1Δmuc cysts were broken. Interestingly, fewer Δcst1::cst1 (15±3%) cysts were broken than wild type cysts (61±8%), suggesting that the increased thickness of the cyst wall seen on TEM (Figure 8) with Δcst1::cst1 parasites does protect the cysts/bradyzoites from mechanical stress.
Δcst1 parasites replicate slower during in vitro bradyzoite development
To examine any growth defects of the Δcst1 parasite, parasite growth was measured with incorporation of 3H-uracil in pH 7.1 (tachyzoite stage) and pH 8.1 (bradyzoite differentiation) medium. The deletion of CST1 resulted in a reduction in the growth rate at pH 8.1 of the Δcst1 parasite compared to the growth rate of the wild type (WT) parasites (Figure 9A, p<0.005), this reduction in growth was not seen at pH 7 (Figure S5). This slower growth phenotype seen with bradyzoite inducing condition in Δcst1 parasites was rescued by full length CST1 complementation, but only partially by the mucin-null CST1 (Figure 9B).

The Δcst1 strain has dysregulated expression of bradyzoite and pH-induced genes
Since there was reduction in growth rate at pH 8.1 in vitro for the Δcst1 parasite and fewer brain cysts in vivo, we investigated whether the cst1 mutants might have global changes in gene expression under the bradyzoite-inducing conditions. The transcriptome of parasites cultured at pH 7 (tachyzoite) was compared with the transcriptome of parasites cultured at pH 8.1 (bradyzoite) for 3 days using RNA-seq. Figure 10A shows the heat map of top 50 upregulated genes at bradyzoite conditions in wild type (WT) T. gondii (see Table S1 for a complete list of these genes). Expression of 49 of 50 genes was less efficiently induced by pH shock in the Δcst1 strain compared to control WT parasites. The complementation of full-length CST1 (Δcst1::cst1) restored the majority of these genes back to their wild type level. However, the mucin-null complement (Δcst1::cst1Δmuc) was not able to restore expression of these genes to wild type levels. Figure 10B shows the fold change in gene expression for several known bradyzoite specific genes. These bradyzoite genes also follow a similar pattern of reduced gene upregulation with Δcst1, restoration with Δcst1::cst1 and only a partial restoration with Δcst1::cst1Δmuc. The altered gene expression pattern was not evident for housekeeping or tachyzoite specific genes. This RNA-seq data suggests that lack of CST1 disrupts bradyzoite differentiation, but not enough to prevent in vitro and in vivo cyst formation.

Discussion
This study identifies the gene encoding the major cyst wall DBA-binding protein CST1. CST1 is a SRS containing protein with an extended mucin domain. The mucin domain of CST1 is necessary for DBA binding and is a major domain for glycosylation of this protein. Δcst1 parasites can differentiate and form mouse brain cysts without CST1; however, CST1 and CST1 glycosylation is required for formation of an organized cyst wall layer that confers structural rigidity to the cyst wall. This is the first cyst wall protein that has been shown to be essential to establish the physical integrity of in vivo brain cysts. Complementation demonstrates that the mucin domain of CST1 is necessary for the cyst wall organization and rigidity. In addition to its role in structural stability of the cyst wall, a lack of CST1 also reduces in vitro growth rate, mouse brain cyst number, and pH 8-induced bradyzoite specific gene upregulation in T. gondii. These results suggest that expression of CST1 or glycosylation of CST1 in early cyst development influences the expression pattern of genes during bradyzoite differentiation.
Previous work has suggested that the cyst wall contains several glycoproteins including CST1 [10], a proteophosphoglycan [11] and other unknown glycoproteins reacting with s-WGA [8]. CST1 is highly unusual SRS protein in that it has a large mucin domain and thirteen SRS domains. The glycoprotein gp900, from another Apicomplexan parasite Cryptosporidium parvum, has a large mucin-like domain that has 68% sequence similarity with the mucin domain of CST1. Other than the presence of mucin-like domain, there is no sequence similarity between these two glycoproteins. The C. parvum gp900 has a transmembrane domain, is expressed on the plasma membrane, and is shed into the environment [20]. The gp900 protein is localized to the tethers on the inner surface of oocyst walls [20], [21]. This suggests that gp900, a large mucin-like protein, is important for making a structurally rigid enclosure for this parasite. Other smaller glycoproteins (gp40 and gp15) are present in the oocyst wall tethers of C. parvum [20], [21]. Our studies with CST1 in T. gondii tissue cysts suggest phylogenetic conservation of the functions of these secreted structural glycoproteins in the Apicomplexa.
The protozoan parasite Trypanosoma cruzi has up to 850 highly glycosylated and GPI-anchored surface mucin genes that form a stage specific mosaic coat on their cell surface. It is suggested that these glycoproteins have a protective role against the proteases in the intestine of the insect vector [22], function in attachment to the host cell, and as an immune evasion mechanism [23]. CST1, which has large mucin domain and a predicted GPI-anchor (as with other SRS domain containing proteins), may have comparable functions in terms of protecting bradyzoites in the cyst from the proteases present in the gastrointestinal tract during the oral infection or in the surrounding necrotic tissues when the host dies [22]. The ability of mucins to retain large amount of water probably protects parasites by preventing dehydration, facilitating parasite transmission to the next host.
Of the 189 cyst-wall positive hybridomas we identified, 34 had an identical immunoblot pattern as mAb SalmonE. This suggests that CST1 may be highly immunogenic toward the Th2 pathway. Cysts are present not only in the central nervous system, but also in the visceral organs and muscles. CST1 from ruptured cysts can induce a strong antibody response and this may facilitate the clearance of cysts by the immune system when their host cells are dead. Cloned hybridomas specific to CST1 included other classes of antibodies (e.g. IgG2b and IgM), therefore, CST1 does not only elicit the production of IgE antibody.
In a previous study, CST1 was detected by its reactivity to the mAb 73.18 in the R5 strain of T. gondii, an atovoquone resistant mutant that spontaneously formed cysts more readily [24]. In this strain CST1 was reported as a 116 kDa band on 2D SDS-PAGE [10]. In our current study, both mAb SalmonE and mAb 73.18 detected two distinct bands (Figures 5A, S6), one migrating at 150 kDa (SRS13, Weiss, unpublished data) and one band (CST1) in the stacking gel (>210 kDa). Similar immunoblot patterns were seen using Pru, ME49 or the ME49 mutant R5 (Figure S6). Both bands also react with DBA lectin indicating that the SRS13 has similar glycoepitope as CST1. This SRS13 reactivity is still present in the Δcst1 strain and does not localize to cyst wall (Tomita and Weiss, unpublished data) and mAb SalmonE staining of the 150 kDa band is absent in a Δsrs13 strain (Figure S6). Monoclonal antibody SalmonE, mAb 73.18, and DBA all display no cyst wall staining in the Δcst1 strain, consistent with SRS44 being CST1, a major cyst wall glycoprotein identified by our laboratory over 15 years ago [10], [11]. During previous studies we were not able to detect the band in the stacking gel since the stacking gel was separated from the resolving gel before the transfer to a membrane. The discrepancy in the molecular-weight of 116 kDa and 150 kDa may be due to the difference in gel conditions or molecular markers.
CST1 is one of several cyst wall proteins that are induced during bradyzoite development. Recently, a screening of insertional mutants for a reduction of in vitro cyst development led to the identification of another T. gondii cyst wall protein, proteophosphoglycan (TgPPG) [25]. TgPPG is expressed in the cyst wall and is probably highly glycosylated, as evidenced by its retention in the stacking gel on SDS-PAGE. Disruption of TgPPG gene results in the delay in cyst wall formation and bradyzoite conversion; however, complementation only rescued cyst wall formation measured by the DBA staining, but not bradyzoite differentiation as measured by BAG1 expression. In another recent study, transcriptomic analysis of brain cysts yielded two distinct cyst wall proteins Bradyzoite Pseudokinase 1 (BPK1) and Microneme Adhesive Repeat domain-containing protein 4 (MCP4) [13]. Subsequent study demonstrated that BPK1 plays a role in effective oral transmission [26]. Finally, several GRA proteins have been demonstrated to localize the cyst wall in bradyzoite parasitophorous vacuoles, as well as to the dense granules in both tachyzoites and bradyzoties [27]. Deletion of cyst wall associated GRA6 was shown to dramatically decrease tissue cyst burdens in mice [18]. The biological functions of these cyst wall proteins await further study.
Toxoplasma gondii is one of the most successful parasites partly because it forms persistent latent cysts that last for the life of its hosts, and the cyst wall is a critical biological structure for this persistence. CST1 functions as a key structural component reinforcing the cyst wall structure and conferring resistance to physical stress to the T. gondii cyst. The fragile cyst phenotype in the Δcst1 strain suggests that transmission and persistence could be affected by this gene deletion, but this hypothesis could not be fully tested, as intact viable cysts cannot be purified from mouse brains to perform quantitative oral challenge experiments [10], [11].
There are 5 putative UDP:GalNAc:polypeptide N-acetylgalactosaminyltransferases (ppGalNAc-T) in the T. gondii genome (Toxo DB). Two of these, T1 and T3, were expressed in insect cells and their glycosyl transferase activity was confirmed [28]. Both transferases turned out to be “follow-up” transferases, only transfering to the pre-O-glycosylated peptide. At this point, it is not known which glycosyl transferasase is responsible for the heavy glycosylation of CST1 mucin domain. A recent study showed that the nucleotide sugar transporter (TgNST1) is necessary for the glycosylation of cyst wall proteins such as CST1 [29]. Deletion of TgNST1 resulted in fewer brain cysts and the authors concluded that glycosylation of the cyst wall is required for persistence of bradyzoites, but did not identify which glycoproteins were most critical. Our studies show a similar phenotype for parasites lacking CST1, consistent with CST1 being the major cyst wall glycoprotein involved in the maintenance of brain cyst burden. Furthermore, parasites expressing a mutant CST1 lacking the mucin domain have a similar defect, demonstrating that post translational glycosylation of the CST1 mucin domain is critical for CST1 biological function. Therefore enzymes involved in glycosylation of cyst wall proteins including candidate enzymes such as T. gondii ppGalNAc-Ts have potential as therapeutic agents to prevent T. gondii bradyzoite persistence.
Materials and Methods
T.gondii cell culture and strains
Human foreskin fibroblasts (HFF) were maintained in 10% fetal bovine serum pH 7 DMEM with penicillin-streptomycin at 5% CO2. Confluent monolayers were infected with Type II strains ME49, the reference genome strain, or PruΔku80 strain of T. gondii, which is widely used for genetic studies [18]. For in vitro bradyzoite differentiation, parasite strains were grown in differentiation medium (DMEM medium adjusted to pH 8.1 with 10 mM HEPES and 1% fetal bovine serum with penicillin-streptomycin) for 3 days at 0.5% CO2.
Ethics statement
All animal experiments were conducted according to the U.S.A. Public Health Service Policy on Humane Care and Use of Laboratory Animals. Animals were maintained in an AAALAC-approved facility and all protocols were approved by the Institutional Care Committee of the Albert Einstein College of Medicine, Bronx, New York(Animal Protocols 20121104, 20121109 and 20121110; Animal Welfare Assurance number A3312-01).
No human samples were used in these experiments. Human foreskin fibroblasts were obtained from ATCC.
Monoclonal antibody production
BALB/cdm1 mice, which have a deletion in the Ld gene at the HLA-2L locus and produce more brain cysts than wild type BALB/c [30], were infected with ME49 strain of T. gondii and treated with sulfamerazine at 30 mg/L in drinking water to minimize death from the acute infection. Four weeks after the infection, brain cysts were isolated using previously described isopycnic centrifugation [19]. Briefly, brains were isolated and homogenized in PBS with a pestle tissue homogenizer with clearance of 0.15–0.23 mm (Thomas Scientific) for 10 times. Percoll was added to 40% of the total volume and centrifuged at 27,000×g for 20 minutes. The middle layer was recovered and centrifuged with equal volume of PBS at 100×g for 10 minutes. Cysts were then subjected to ten freeze-thaw cycles, (3 minutes each: 100% ethanol-dry ice bath followed by room temperature water bath) and emulsified with an equal volume of Freund's complete adjuvant. The emulsion was injected into BALB/c mice subcutaneously. Two months later, spleens were isolated from the immunized mice and fused with myeloma cell line to create hybridoma libraries. Using the IFA, ELISA and immunoblot, those hybridoma supernatants were screened against parasites that were cultured in pH 8.1 medium. IFA patterns were similar for two Type II strains ME49 and Pru. Subcloned hybridoma cells were cultured in CELLine bioreactor (Integra) for large-scale production of monoclonal antibodies.
Transmission electron microscopy
The BALB/cdm1 mice were infected with Pru strain of T. gondii for 4 weeks in the presence of sulfamerazine at 30 mg/L in drinking water. After they were harvested, whole brains were fixed in 4% paraformaldehyde in PBS for overnight, followed by homogenization and isopycnic centrifugation as described above. While wild-type cysts did not require fixation prior to processing, fixation of Δcst1 infected brains was necessary in order to prevent breakage of fragile brain cysts. The isolated cysts were fixed with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1M sodium cacodylate buffer, postfixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded series of ethanol and embedded in LX112 resin (LADD Research Industries, Burlington VT). Ultrathin sections were cut on a Reichert Ultracut UCT, stained with uranyl acetate followed by lead citrate and viewed on a JOEL 1200EX transmission electron microscope at 80 kv.
For immunoelectron microscopy, the cysts were fixed with 4% paraformaldehyde 0.05% glutaraldehyde in 0.1M sodium cacodylate buffer, dehydrated through a graded series of ethanol, with a progressive lowering of the temperature to −50°C in a Leica EMAFS, embedded in Lowicryl HM-20 monostep resin (Electron Microscopy Sciences), and polymerized using UV light. Ultrathin sections were cut on a Reichert Ultracut E, immunolabeled with SalmonE, and then stained with uranyl acetate followed by lead citrate. Stained sections were viewed on a JOEL 1200EX transmission electron microscope at 80 kv.
Identification of the SalmonE reactive glycoprotein by mass spectrometry
SalmonE was crosslinked to the Protein L agarose beads with disuccinimidyl suberate following the manufacturer's protocol (Thermo Scientific). Human foreskin fibroblasts were infected with ME49 strain of T. gondii and incubated for 3 days in pH 8.1 medium with 10% FBS at 0.5% CO2. Cells were lysed with 1% TritonX-100 in PBS with proteinase inhibitor cocktail and incubated with SalmonE-beads for 2 hours at 4°C. The beads were extensively washed with 1% TritonX-100 PBS and eluted with 0.1M glycine at pH 2.5. The eluate was neutralized and separated on SDS-PAGE. The gel was stained with Coomassie Brilliant Blue and a visible high molecular-weight band was excised. The protein in the band was reduced and alkylated using TCEP and iodoacetamide then digested with trypsin in 25 mM ammonium bicarbonate/0.01% ProteaseMax at 50°C for 1 hour. The resulting digest was cleaned with C18 ziptip and the peptides eluted onto a MALDI plate with a saturated solution of α-cyanohydroxycinnamic acid in 70% acetonitrile/0.1% trifluoroacetic acid. MS/MS analysis of the digested sample was carried out using the AB Sciex 4800 MALDI-TOF-TOF (Applied Biosystems), operated at 20 kV accelerating voltage in the reflector positive ion mode. The MS/MS data generated were converted to mgf files and searched against EPICDB [31] using the in-house Mascot Protein Search engine (Matrix Science) for protein identification.
Production of CST1 mouse antisera
RNA was isolated from ME49 strain of T. gondii and a cDNA library was created using SuperScript III First Strand Kit (Invitrogen). The first 200 peptides of the CST1 (TGME49_064660), as predicted in ToxoDB (i.e. this does not include the probable N-terminal extension of CST1 gene predicted using SignalP 4.0 [15]), was amplified by PCR, cloned into the pET32 vector and used to transform BL21 competent E. coli. Recombinant CST1 protein was expressed using Overnight Express Autoinduction System 1 (Novagen), purified with nickel column, and separated on SDS-PAGE. The band was cut out and emulsified with Freund's complete adjuvant and immunized into BALB/c mice intraperitoneally. Three months after the immunization, antisera were collected and probed against in vitro cysts using IFA.
CST1 (TGME49_064660; SRS44) knock-out
Type II Prugniaud strain with deletion of the Ku80 gene (PruΔku80) [18] was used as the background strain for the creation of Δcst1 strain. The construct for the knockout was built as previously described [18]. Briefly, 1 kb upstream and downstream genomic DNA sequence of TGME_064660 gene were amplified from the parental PruΔku80 strain. These fragments were concatenated into pRS416 yeast shuttle vector (ATCC) flanking the selectable marker hypoxanthine-xanthine-guanine phosphoribosyltransferase [HXGPRT] cassette using the yeast strain ATCC#90845. This construct deletes whole CST1 gene as well as 205 nucleotides 5′ upstream region from the predicted start site. This 5′ region includes the probable start site and signal peptides. All the primers used in the plasmid construct are listed in the Supplemental material (Table S2). The parasites were transfected with the linearized Δcst1 vector and subcloned in the presence of 25 µg/ml mycophenolic acid and 50 µg/ml xanthine. Integration of Δcst1 vector at the TGME49_064660 locus was verified by PCR. Lack of CST1 protein expression was confirmed with IFA and immunoblot.
Complementation of CST1
For CST1 complementation, DNA fragments were concatenated into pSMART-BAC plasmid (Lucigen) using In-Fusion system (Clontech) following the manufacture's protocol. The homologous sequences 1 kb upstream and downstream of UPRT coding region for the UPRT locus were isolated from genomic DNA. Flanking sequences 1.3 kb 5′ and 3′ of CST1 were isolated from Pru genomic DNA. TGME49_064660 cDNA was generated from RNA harvested from Pru strain cultured at pH 8.1. The fragments were concatenated in the following order, 5′ UPRT recombination sequence, TGME49_064660 upstream element, TGME49_064660 cDNA, TGME49_064660 3′ UTR, and 3′ UPRT recombination sequence.
For the mucin domain null mutant complementation, two fragments (base 1–6006 and 6766–7035 with appropriate adapters) were used instead of whole cDNA. This mucin domain null vector replaces the mucin domain (nt 6007–6765) with a 1xFLAG sequence. The parasites were transfected with complementation vectors and subcloned in the presence of 5 µM 5-fluorodeoxyuridine (FUDR). Integration of complementing vectors at the UPRT locus was verified with PCR.
Our attempts to use the HXGPRT selectable marker present in the Δcst1 parasite to complement the endogenous cst1 locus were not successful. This was probably due to the low level expression of HXGPRT at the cst1 locus that was not sufficient for negative selection using 6-thioxanthine, but was sufficient for positive selection in constructing the knockout using mycophenolic acid (MPA). We have experienced similar problems using the Δku80 system with the HGXPRT selectable marker and have found that removal of this marker by negative selection is not feasible at all loci and needs to be evaluated for each knock-out. Therefore complementation was performed at the UPRT locus for the cst1 gene since this target provides a direct selection and the loss of UPRT does not influence cyst development or cyst burdens in mice [18].
5′ RACE
RNA was isolated using RNeasy mini with DNase treatment from HFF infected with ME49 strain of T. gondii culture in pH 8.1 medium for 3 days. 5′ UTR was amplified with FirstChoice RLM-RACE kit (Ambion) with gene specific primer GGGCGGGTCGAAAATGTTG following the manufacturer's protocol. The DNA was sequenced with ABI 3730 DNA analyzer.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde in PBS for 30 minutes on ice and then permeablized with 0.2% TritonX100/0.1% glycine/0.2% bovine serum albumin (BSA) in PBS on ice for 20 minutes. The cells were washed with 0.2% BSA in PBS three times and blocked with 1% BSA in PBS at 4°C overnight. All incubation with primary and secondary antibodies was done at 37°C for 90 minutes in a moist chamber. Concentration of the antibodies and probes used were 20 µg/ml for FITC-conjugated DBA (Vector Lab), 1∶50 for antibody 73.18, 1∶100 for SalmonE, 1∶25 for mouse polyclonal anti-CST1(1–200), 1∶50 for rabbit anti-BAG, 1∶200 anti-FLAG M2 (Sigma) all in 1% BSA/PBS. Cells were washed three times with 0.2% BSA/PBS then incubated with appropriate secondary antibody at 1∶500. After incubation, the cells were washed three times with 0.2% BSA/PBS and mounted with ProLong Gold antifade (Invitrogen). Photomicrographs were taken either with a SP5 confocal microscope (Leica) or Microphoto-FXA epifluorescence microscope (Nikon).
Immunoblotting
HFF cells were infected with T. gondii in normal DMEM medium or differentiation medium and kept in 5% or 0.5% CO2 respectively. Cells were harvested at 3 dpi with cell scraper, centrifuged at 3000×g for 15 minutes and lysed with 1% TritonX100, 1% SDS, protease inhibitor cocktail in PBS. The samples were resolved in 10% SDS-PAGE at 100 V and transferred at 250 mA for 90 minutes on the PVDF membrane Immobilon-FL (Millipore). The membrane was blocked overnight with 5% non-fat dry milk (NFDM) in PBST, then probed with SalmonE at 1∶250 and anti-GRA1 at 1∶500 in 1% BSA/PBS at 4°C on shaker overnight. Following antibody incubation the membrane was washed three times with PBST and then incubated with donkey anti-mouse antibody conjugated with IRDye800 (Licor) at 1∶40,000 in 5% NFDM/PBST at room temperature for 90 minutes. The membrane was then scanned using an Odyssey (Licor) imaging system.
For the Immunoprecipitation-Immunoblot experiments, SDS was omitted from the lysis buffer to minimize the disruption of antigen-antibody interaction. Immunoprecipitation was performed as described in the mass spectrometry method section. The gel was transferred to nitrocellulose membrane and probed with DBA conjugated with alkaline phosphatase (EY Laboratories) at 1∶100 in PBST for 90 minutes at room temperature. The membrane was then washed three times with PBST and developed with BCIP/NBT color development substrate (Promega). The image of the membrane was scanned using a desktop scanner. For double labeling with SalmonE and 73.18 (CST1 reactive; [10], [11], samples were run in 6% SDS-PAGE gel with 3% stacking gel. The membrane was probed with 73.18 (1∶20) then with anti-mouse IRDye700 (1∶20000). After the membrane was scanned, it was re-probed with SalmonE (1∶100) then rat anti-mouse IgE antibody (1∶2000) then anti-rat IRDye800 (1∶20000).
Parasite growth assay
For WT and Δcst1 parasites, a growth assay was performed using a previously described 3H-uracil incorporation method [32]–[34]. Since mammalian host cells are not capable of uracil uptake but T. gondii is [33], [34], the incorporation of radio-labeled uracil is used to measure the growth of parasites. Briefly, HFF monolayers in 12-well tissue culture plates were infected (100 or 10,000 parasites/well) in either pH 7.1 or pH 8.1 medium and incubated in 5% or 0.5% CO2 respectively. Cells were incubated with 1 ml of 2 µCi/ml 3H-uracil per well for 24 hours before the each harvest time point. At 24, 48, 72, or 96 hours after infection, medium was removed and cells were lysed with 1% SDS and 100 µg/ml of unlabeled uracil in PBS on ice. Nucleic acids were precipitated with 10% trichloroacetic acid on ice for 2 hours. The contents of each well were filtered with the glass fiber filters. The radioactivity of 3H on the filter was measured with a scintillation counter.
For assessment of Δcst1::cst1 and Δcst1::cst1Δmuc parasite growth, microscopic observation of the growth of intracellular parasites was used, as the disruption of the UPRT gene locus results in parasites that no longer incorporate uracil. Parasites were grown as described above then fixed in 4% paraformaldehyde, permeabilized with 0.2% TritonX100 and probed with anti-GRA1 antibody to visualize the parasites. For pH 8 (bradyzoite permissive condition) a total of 600 host cells were examined for each time point/condition and the number of intracellular parasites per 600 cells determined. Bradyzoite vacuoles have been reported to be resistant to lysis so direct counting of vacuoles was used to evaluate growth. For pH 7 (tachyzoite permissive condition) parasites were grown as described above (10,000 parasite initial inoculum), cells were scraped from each well of a 24 well plate, centrifuged and then re-suspended in 100 µl of 0.5% saponin/PBS, pipetted 20 times to ensure host cell lysis and then counted in a Neubauer hemocytometer in triplicate.
Parasite challenge, cyst count, and histology
Female 4 to 8 week old C57/BL6 mice (Jackson Laboratory) were infected with 200 tachyzoites intraperitoneally. Any observed mortality was recorded until 28 days after infection when the mice were sacrificed and brains harvested. Brains were fixed in 4% PFA in PBS overnight. Right brain halves were partially homogenized with a syringe with PBS into 600 µl suspension then 120 µl were counted under fluorescent microscope. Left halves of each brain were fixed in 10% neutral buffered formalin for additional 72 hours and processed for paraffin embedding. Samples for histopathology were sectioned to a thickness of 5 µm and stained using hematoxylin and eosin (H&E) stains. Slides were analyzed for the presence of tissue cysts, inflammation, and gliosis by light microscopy and graded on a scale of 0–5 (where 0 = no lesions; 1 = minimal lesions; 2 = mild lesions; 3 = moderate lesions; 4 = marked lesions; and 5 = severe lesions). Sections were graded by the pathologist in a blinded fashion to avoid confirmation bias.
Deep RNA sequencing (RNA-seq)
HFF cells grown in seven 150 mm tissue culture plates were infected with parasites for each strain WT Pru, Δcst1, Δcst1::cst1 and Δcst1::cst1Δmuc in regular medium (pH 7 DMEM with 10% fetal bovine serum, incubated in 5% CO2). Eight hours later, free parasites were removed by washing with PBS and replaced with regular medium (pH 7, 5% CO2) or differentiation medium (pH 8.1 DMEM with 1% fetal bovine serum, 10 mM HEPES, incubated in 0.5% CO2). Three days after the infection, cells were harvested, passed through 27G needle twice to lyse HFF cells and filtered through 3 µm pore polycarbonate membrane to remove HFF cells. This separation process was performed on ice. Purified parasites were pelleted at 1000×g for 15 minutes 4°C.
RNA was extracted using TRIzol Reagent (Invitrogen), followed by genomic DNA removal and cleaning using RNase-Free DNase Set kit and Mini RNease kit (Qiagen). Integrity of the RNA samples was assessed using the Aligent 2100 Bioanalyzer. RNA samples having RNA Integrity Number between 9 and 10 were used in this work. MicroPoly(A)Purist Kit (Ambion) was used for enrichment of transcripts. The SOLiD Total RNA-Seq Kit was used to construct template cDNA for RNA-Seq following the protocol recommended by Applied Biosystems. Briefly, mRNA was fragmented using chemical hydrolysis followed by ligation with strand specific adapters and reverse transcription was used to generate cDNA. The cDNA fragments, 150 to 250 bp in size, were subsequently isolated by electrophoresis in 6% Urea-TBE acrylamide gel. The isolated cDNA was amplified through 15 amplification cycles to produce the required number of templates for the SOLiD EZ Bead system, which was used to generate the template bead library for ligation base sequencing by the either SOLiD4 or 5500xl SOLiD instrument (LifeTechnologies). The 50-base short read sequences produced by the SOLiD sequencer were mapped in color space using the Whole Transcriptome analysis pipeline in Life Technologies LifeScope software version 2.5 against the genome of T. gondii strain ME49 using the default mapping setting. Both Fasta and GFF files were obtained from ToxoDB website (www.toxoDB.org; Release 6.1). The output of the Whole Transcriptome analysis generated (1) a gene counts file, with the base counts summed to a single value across the entire gene length, and with a RPKM value also given for each gene; (2) a BAM file containing the sequence of every mapped read and its mapped location; (3) two pairs of *.wig files (one pair for the two strands on each chromosome) giving the mapped counts at each base position; and (4) a statistics summary on alignment and filtering report. Fold change in gene upregulation at bradyzoite induction was calculated by dividing the RPKM values of pH 8.1 by that of pH 7. The genes with low level expression (RPKM<5) in pH 8.1 WT parasites were removed. The top 50 genes that were upregulated in WT parasites were plotted with gplot heatmap.2 in R.
Supporting Information
Zdroje
1. TenterAM, HeckerothAR, WeissLM (2000) Toxoplasma gondii: from animals to humans. International Journal for Parasitology 30 : 1217–1258.
2. PappasG, RoussosN, FalagasME (2009) Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. International Journal for Parasitology 39 : 1385–1394.
3. FergusonDJ, HutchisonWM (1987) The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Archiv A, Pathological anatomy and histopathology 411 : 39–43.
4. HillD, DubeyJP (2002) Toxoplasma gondii: transmission, diagnosis and prevention. Clinical Microbiology and Infection 8 : 634–640.
5. FergusonDJ, HutchisonWM, PettersenE (1989) Tissue cyst rupture in mice chronically infected with Toxoplasma gondii. An immunocytochemical and ultrastructural study. Parasitology Research 75 : 599–603.
6. DubeyJP, LindsayDS, SpeerCA (1998) Structures of Toxoplasma gondii Tachyzoites, Bradyzoites, and Sporozoites and Biology and Development of Tissue Cysts Clinical Microbiology Reviews. 11 : 267–299.
7. SimsTA, HayJ, TalbotIC (1988) Host-parasite relationship in the brains of mice with congenital toxoplasmosis. The Journal of Pathology 156 : 255–261.
8. BoothroydJC, BlackM, BonnefoyS, HehlA, KnollLJ, et al. (1997) Genetic and biochemical analysis of development in Toxoplasma gondii. Philosophical transactions of the Royal Society of London Series B, Biological sciences 352 : 1347–1354.
9. WeissLM, KimK (2000) The development and biology of bradyzoites of Toxoplasma gondii. Frontiers in Bioscience 5: D391–405.
10. ZhangYW, HalonenSK, MaYF, WittnerM, WeissLM (2001) Initial characterization of CST1, a Toxoplasma gondii cyst wall glycoprotein. Infection and Immunity 69 : 501–507.
11. WeissLM, LaPlaceD, TanowitzHB, WittnerM (1992) Identification of Toxoplasma gondii bradyzoite-specific monoclonal antibodies. The Journal of Infectious Diseases 166 : 213–215.
12. Weiss LM, Kim K (2007) Bradyzoite development. In: Weiss LM, Kim K, editors. Toxoplasma gondii: The Model Apicomplexan. Elsevier Ltd. pp. 341–366.
13. BuchholzKR, FritzHM, ChenX, Durbin-JohnsonB, RockeDM, et al. (2011) Identification of tissue cyst wall components by transcriptome analysis of in vivo and in vitro Toxoplasma gondii bradyzoites. Eukaryotic Cell 10 : 1637–1647.
14. WasmuthJD, PszennyV, HaileS, JansenEM, GastAT, et al. (2012) Integrated Bioinformatic and Targeted Deletion Analyses of the SRS Gene Superfamily Identify SRS29C as a Negative Regulator of Toxoplasma Virulence. mBio 3: e00321–12.
15. PetersenTN, BrunakS, Von HeijneG, NielsenH (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods 8 : 785–786.
16. JuleniusK, GuptaR (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15 : 153–164.
17. EtzlerME, KabatEA (1970) Purification and characterization of a lectin (plant hemagglutinin) with blood group A specificity from Dolichos biflorus. Biochemistry 9 : 869–877.
18. FoxBA, FallaA, RommereimLM, TomitaT, GigleyJP, et al. (2011) Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryotic Cell 10 : 1193–1206.
19. CornelissenAWCA, OverdulveJP, HoenderboomJM (1981) Separation of Isospora (Toxoplasma) gondii cysts and cystozoites from mouse brain tissue by continuous density - gradient centrifugation. Parasitology 83 : 103.
20. BarnesDA, BonninA, HuangJX, GoussetL, WuJ, et al. (1998) A novel multi-domain mucin-like glycoprotein of Cryptosporidium parvum mediates invasion. Molecular and Biochemical Parasitology 96 : 93–110.
21. ChatterjeeA, BanerjeeS, SteffenM, O'ConnorRM, WardHD, et al. (2010) Evidence for mucin-like glycoproteins that tether sporozoites of Cryptosporidium parvum to the inner surface of the oocyst wall. Eukaryotic Cell 9 : 84–96.
22. MortaraRa, Da SilvaS, AraguthMF, BlancoSa, YoshidaN (1992) Polymorphism of the 35 - and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi metacyclic trypomastigotes. Infection and Immunity 60 : 4673–4678.
23. BuscagliaCa, CampoVa, FraschACC, Di NoiaJM (2006) Trypanosoma cruzi surface mucins: host-dependent coat diversity. Nature Reviews Microbiology 4 : 229–236.
24. McFaddenDC, TomavoS, BerryEA, BoothroydJC (2000) Characterization of cytochrome b from Toxoplasma gondii and Qo domain mutations as a mechanism of atovaquone-resistance. Molecular and Biochemical Parasitology 108 : 1–12.
25. CraverMPJ, RooneyPJ, KnollLJ (2010) Isolation of Toxoplasma gondii development mutants identifies a potential proteophosphogylcan that enhances cyst wall formation. Molecular and Biochemical Parasitology 169 : 120–123.
26. BuchholzKR, BowyerPW, BoothroydJC (2013) Bradyzoite Pseudokinase 1 Is Crucial for Efficient Oral Infectivity of the Toxoplasma gondii Tissue Cyst. Eukaryotic Cell 12 : 399–410.
27. FergusonDJJP (2004) Use of molecular and ultrastructural markers to evaluate stage conversion of Toxoplasma gondii in both the intermediate and definitive host. International Journal for Parasitology 34 : 347–360.
28. Stwora-WojczykMM, DzierszinskiF, RoosDS, SpitalnikSL, WojczykBS (2004) Functional characterization of a novel Toxoplasma gondii glycosyltransferase: UDP-N-acetyl-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase-T3. Archives of Biochemistry and Biophysics 426 : 231–240.
29. CaffaroCE, KoshyAa, LiuL, ZeinerGM, HirschbergCB, et al. (2013) A Nucleotide Sugar Transporter Involved in Glycosylation of the Toxoplasma Tissue Cyst Wall Is Required for Efficient Persistence of Bradyzoites. PLoS Pathogens 9: e1003331.
30. BrownC (1990) Class I MHC genes and CD8+ T cells determine cyst number in Toxoplasma gondii infection. The Journal of Immunology 145 : 3438–3441.
31. Madrid-AlisteCJ, DybasJM, AngelettiRH, WeissLM, KimK, et al. (2009) EPIC-DB: a proteomics database for studying Apicomplexan organisms. BMC Genomics 10 : 38.
32. ZhangYW, KimK, MaYF, WittnerM, TanowitzHB, et al. (1999) Disruption of the Toxoplasma gondii bradyzoite-specific gene BAG1 decreases in vivo cyst formation. Molecular Microbiology 31 : 691–701.
33. PfefferkornER, PfefferkornLC (1977) Toxoplasma gondii: specific labeling of nucleic acids of intracellular parasites in Lesch-Nyhan cells. Experimental Parasitology 41 : 95–104.
34. SchwartzmanJD, PfefferkornER (1981) Pyrimidine synthesis by intracellular Toxoplasma gondii. The Journal of Parasitology 67 : 150–158.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells