
The Host Protein Calprotectin Modulates the Type IV Secretion System via Zinc Sequestration
Helicobacter pylori is a bacterium that colonizes the stomach and causes gastric diseases. Some strains of H. pylori possess a secretion system that has the capacity to inject a cancer-causing protein into host cells. The activity of this secretion system contributes to the development of inflammation and is linked to the development of gastric cancer. Here, we show that the host protein calprotectin, which has the ability to bind and sequester nutrient metals from invading pathogens, can directly repress H. pylori secretory activity and the production of secretion-associated pili in a zinc-dependent manner. H. pylori-infected animals lacking calprotectin trend toward having more gastric inflammation and a significantly lower bacterial burden than infected animals that express calprotectin; these differences are not observed when animals are infected with a strain of H. pylori that lacks an active secretion system. Thus, a better understanding of how nutritional immunity modulates this secretion system could help us develop novel antimicrobial therapeutic strategies targeting secretory processes in H. pylori.
Published in the journal:
. PLoS Pathog 10(10): e32767. doi:10.1371/journal.ppat.1004450
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004450
Summary
Helicobacter pylori is a bacterium that colonizes the stomach and causes gastric diseases. Some strains of H. pylori possess a secretion system that has the capacity to inject a cancer-causing protein into host cells. The activity of this secretion system contributes to the development of inflammation and is linked to the development of gastric cancer. Here, we show that the host protein calprotectin, which has the ability to bind and sequester nutrient metals from invading pathogens, can directly repress H. pylori secretory activity and the production of secretion-associated pili in a zinc-dependent manner. H. pylori-infected animals lacking calprotectin trend toward having more gastric inflammation and a significantly lower bacterial burden than infected animals that express calprotectin; these differences are not observed when animals are infected with a strain of H. pylori that lacks an active secretion system. Thus, a better understanding of how nutritional immunity modulates this secretion system could help us develop novel antimicrobial therapeutic strategies targeting secretory processes in H. pylori.
Introduction
Helicobacter pylori is a Gram-negative bacterial pathogen that colonizes half of the world's population and contributes to a variety of disease outcomes, including peptic or duodenal ulcer disease, gastric adenocarcinoma, and mucosal-associated lymphoid tissue (MALT) lymphoma [1]. The most common manifestation of H. pylori-related disease is chronic gastric inflammation (non-atrophic chronic gastritis), which can potentially advance to multifocal atrophic gastritis, a precancerous lesion [2]. H. pylori-induced gastritis is characterized by neutrophil and mononuclear leukocyte infiltration to the lamina propria, involving cells in both the innate and adaptive arms of the immune response.
This gastric mucosal inflammatory response to H. pylori is enhanced if persons are infected with strains that possess a cag - type IV secretion system (cag T4SS). The cag T4SS is a macromolecular assembly that is responsible for translocating the oncogenic effector molecule, CagA and peptidoglycan, into host cells [3], [4]. These translocated effectors elicit a variety of host cell responses, including activation of nuclear factor κB (NFκB) and secretion of the proinflammatory cytokines, IL-1β and IL-8 [5]–[8]. The latter is associated with recruitment of innate immune cells, including neutrophils [9]. Neutrophil recruitment to the gastric mucosa is also enhanced by Th17 and Th1 responses.
Neutrophils are capable of controlling microbial infections via phagocytosis and subsequent production of reactive oxygen and reactive nitrogen intermediates, neutrophil extracellular trap (NET) formation, and production of antimicrobial factors [10], [11]. One such example is calprotectin (CP) [12]. CP, a heterodimer of S100A8 and S100A9 subunits (also known as Mrp8/14, calgranulin A/B, cystic fibrosis antigen). CP comprises about 50% of the neutrophil's cytoplasmic protein content and is a critical component of the host nutrient withholding process termed nutritional immunity [13], [14]. Humans and other mammals restrict access to essential metals through this nutritional immunity mechanism as a means to prevent infection with pathogenic organisms.
CP binds manganese and zinc with high affinity, effectively starving bacteria of these essential nutrients. There are two transition metal binding sites in CP; site 1 (S1; 6 His site) binds manganese and zinc, and site 2 (S2; 3 His Asp site) binds zinc only [15], [16]. Mutagenesis of CP's two metal binding sites has produced a site 1 mutant (ΔS1) that binds zinc only, a site 2 mutant (ΔS2) capable of binding both manganese and zinc, and a double site mutant (DS CP) incapable of binding manganese and zinc. Previous reports have indicated that CP exhibits antimicrobial activity against numerous microorganisms, including Salmonella enterica serovar Typhimurium, Staphylococcus aureus, Escherichia coli, Borrelia burgdorferi, Listeria monocytogenes, Candida albicans, Acinetobacter baumannii, Staphylococcus epidermidis, Staphylococcus lugdunensis, Enterococcus faecalis, Pseudomonas aeruginosa, and Shigella flexneri [12], [14], [15], [17]–[22]. CP has been demonstrated to enhance pathogenic Salmonella persistence in the inflamed gut [18] as well as increase neutrophil killing of S. aureus [14]. Concentrations of calprotectin in the tissue have been reported to be as high as 20 mg/ml in response to bacterial infections [23]. Expression of CP subunits S100A8 and S100A9 in inflamed gastric tissues of H. pylori-infected persons has been reported in the literature [24]. However, the interaction between CP and H. pylori has not been previously investigated.
H. pylori-associated inflammation is dynamic and recently published results indicate that H. pylori can modulate its cag T4SS activity in response to inflammation [25]. Here, we report a study to determine the role of CP in control of H. pylori colonization and pathogenesis. We find that gastric CP levels are elevated in H. pylori-infected humans and rodents, and that most of the CP localizes to neutrophils in H. pylori-infected tissues. We also demonstrate that H. pylori-infected CP-deficient mice (A9−/ − mice) have decreased bacterial burden and a trend towards increased gastric inflammation compared to infected WT mice, a phenotype which is not observed in mice infected with an H. pylori isogenic cagE mutant, which lacks cag T4SS activity. Finally, we show that CP represses cag T4SS activity and cag T4SS-associated pilus production via zinc sequestration.
Results
Calprotectin is elevated in H. pylori-infected mice and humans
In order to determine if CP is elevated in the context of H. pylori infection, real-time RT-PCR analysis of s100a8 and s100a9 transcripts in RNA isolated from either mouse or human gastric tissue was performed. For the first mouse study, RNA was isolated from gastric tissue of mice that had been infected with H. pylori PMSS1 or SS1 for 1, 2, and 3 months and s100a8 and s100a9 expression was compared to that of uninfected mice. CP subunit s100a8 was significantly increased in gastric tissue in response to H. pylori infection (Figure 1A). Transcript levels of CP subunit s100a9 did not significantly increase, but it has been demonstrated that the S100A9 protein is stabilized by increased S100A8 expression [18]. Corresponding inflammation scores are presented for these infections (Figure 1B). In a second study, human gastric biopsy samples were divided into H. pylori-negative samples and H. pylori-positive samples. Gene expression of both s100a8 and s100a9 subunits were elevated in H. pylori-infected biopsy samples compared to H. pylori-negative samples (Figure 1C).

CP expression in the stomach localizes to neutrophils
CP was elevated in response to H. pylori infection in both human and murine stomach tissues, a phenomenon we hypothesize is driven by the increased presence of neutrophils. An immunohistochemical staining approach was used to evaluate the localization of CP within H. pylori PMSS1 or SS1-infected murine stomach tissues. Microscopy analyses revealed that the majority of CP is localized in association with neutrophils within the gastric tissue (Figure 1D).
Since immunohistochemistry staining revealed that CP was mainly localized in proximity to host neutrophils (Figure 1D) and since CP comprises 40–60% of the total protein in the neutrophil cytoplasm [26], [27], we hypothesized that recruitment of neutrophils to the H. pylori-infected stomachs correlates with increases in CP expression. To test this hypothesis, IL-17 receptor A-deficient mice (IL-17RA-/-) were used. Previously published data indicate that these mice have a defect in IL-17 signaling, a prerequisite for the maintenance of neutrophil recruitment to the stomach during chronic H. pylori infection [28]. At 3 months post-infection, H. pylori infected IL-17RA-/ - mice exhibit significantly decreased PMN infiltration compared to H. pylori infected WT mice [28]. Thus, the 3 month time point was chosen for these analyses. Real-time RT-PCR analysis of CP subunit expression revealed that PMSS1-infected IL-17RA-/ - mice have diminished s100a8 and s100a9 expression compared to PMSS1-infected wild-type (WT) animals (Figure 1E), demonstrating that increased abundance of CP was correlated to the presence of a neutrophilic infiltrate.
CP affects growth and viability of H. pylori
CP has been demonstrated to inhibit bacterial growth via sequestration of nutrient manganese and zinc [16]–[18]. We hypothesized that CP is elevated in response to H. pylori infection as part of a host strategy to inhibit bacterial proliferation within the gastric niche. To test this proposal, in vitro growth assays were performed in modified bacteriological medium. Analysis of bacterial growth curves (OD600) and colony forming units (CFU/mL) revealed that wild-type CP (CP) at 300 µg/mL significantly inhibited H. pylori growth (Figure 2 and Figure S1). The addition of exogenous manganese and zinc (50 µM of zinc chloride and 50 µM manganese chloride) restored growth to control levels. In addition to investigating the ability of CP to inhibit growth of H. pylori, the effects of three previously generated mutants of CP's metal binding sites (DS CP, ΔS1, and ΔS2) were investigated [15]. The DS CP harboring inactivation of both S1 (manganese and zinc binding) and S2 (zinc binding alone) sites was unable to inhibit bacterial growth (Table S1). The ΔS1 mutant at 1200 µg/mL was able to inhibit bacterial growth, as was the ΔS2 mutant (Table S1). These results indicate that CP inhibited H. pylori growth in vitro at concentrations above 300 µg/mL, and that the antibacterial activity is dependent on CP's ability to sequester metal.

CP affects H. pylori colonization and H. pylori-induced inflammation in a mouse model
Because CP can inhibit the growth of H. pylori in vitro, we hypothesized that this host protein would also contribute to control of the bacterial burden in vivo. To test this hypothesis, both WT and CP-deficient (A9-/-) mice were orogastrically infected with H. pylori strain SS1 or PMSS1; the former strain lacks a functional cag T4SS, while the latter expresses a functional cag T4SS. In analysis of animals at 6 weeks post-infection, A9-/ - mice infected with the SS1 strain had significantly higher levels of H. pylori compared to WT mice infected with the SS1 strain (Figure 3A, p = 0.014), A9-/ - mice infected with PMSS1 had significantly fewer CFU per gram of tissue compared to PMSS1-infected WT mice (Figure 3C, p = 0.0325). Bacterial burden and inflammation have been shown to have a reciprocal relationship in H. pylori models of murine infection [29]–[31]. This suggests that CP-deficient mice may have an increase in the inflammatory response to H. pylori. To test this hypothesis, inflammation in WT and A9-/ - mice was evaluated via histological analysis and scoring. The A9-/ - mice infected with SS1 did not have significant differences in inflammation compared to WT mice (Figure 3B), but A9-/ - mice infected with PMSS1 had significantly higher inflammation scores than WT-infected animals (p = 0.04; Figure 3D). These results suggested that the absence of CP results in increased gastric inflammation during H. pylori infection with a strain expressing a functional cag T4SS (the SS1 strain lacks a functional cag T4SS), which may explain the decreased bacterial burden. We also investigated the effect of CP on chronic infection in the mice at later time points, up to 3 months post infection. At this timepoint, there were no significant differences in the colonization in the H. pylori-infected A9-/ - mice compared to WT mice, but there was a trend toward increased inflammation in H. pylori-infected A9-/ - mice compared to H. pylori-infected WT mice (SS1 infection p = 0.06; PMSS1 infection p = 0.13). We observed that the stomachs of A9-/ - mice became infected with fungus by 3 months, which complicates the interpretation of results at this timepoint.

We hypothesized that CP modulates the cag T4SS since the activity of this virulence factor is associated with H. pylori-induced inflammation. To test this hypothesis, WT and A9-/ - mice were infected with either PMSS1 or an isogenic H. pylori PMSS1 cagE mutant. Since CagE is the ATPase that powers cag T4SS assembly, H. pylori strains deficient for CagE do not have a functional cag T4SS and do not form cag T4SS pili [32]. Again, when infected with the PMSS1 strain, the bacterial burden was significantly lower (Figure 3E) and there was a trend toward higher inflammation in the A9-/ - infected mice compared to the WT mice (Figure S2A). When infected with the PMSS1 cagE mutant, there was no significant difference in bacterial burden (Figure 3E) or inflammation (Figure S2) in A9-/ - mice compared to WT mice at 6 weeks post-infection. To further quantify the gastritis in these mice, flow cytometry was performed at 6 weeks post-infection. There was a trend toward greater numbers of gastric PMNs and monocytes in PMSS1-infected A9-/ - mice compared to PMSS1-infected WT mice (p = 0.057 and p = 0.097, respectively; Figure S2B, C). Assessment of CP levels (transcript levels of s100a8 and s100a9) by realtime rtPCR showed no difference in expression when comparing H. pylori infected WT mice infected with PMSS1 or the PMSS1 cagE mutant (Table S2). Thus differences in bacterial burden and inflammation between PMSS1-infected WT and the A9-/ - mice are likely cag T4SS-dependent (Figure 3E and Figure S2). These data suggest that CP may repress the activity of the cag T4SS in vivo.
CP inhibits activity of the H. pylori cag T4SS
A functional cag T4SS translocates the effector molecule, CagA, into host cells, where it is then phosphorylated [25]. Moreover, a functional cag T4SS is necessary for activation of NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) in human AGS gastric epithelial cells, which is a result of both CagA translocation and peptidoglycan recognition by NOD1 [33]. As a result of these cellular signaling events, IL-8 is produced and secreted by the AGS gastric epithelial cells. Therefore, the functional activity of the cag T4SS was measured with three assays; CagA translocation and phosphorylation (Figure 4), NFκB activation (Figure 5A), and IL-8 secretion by gastric epithelial cells (Figure 5B).
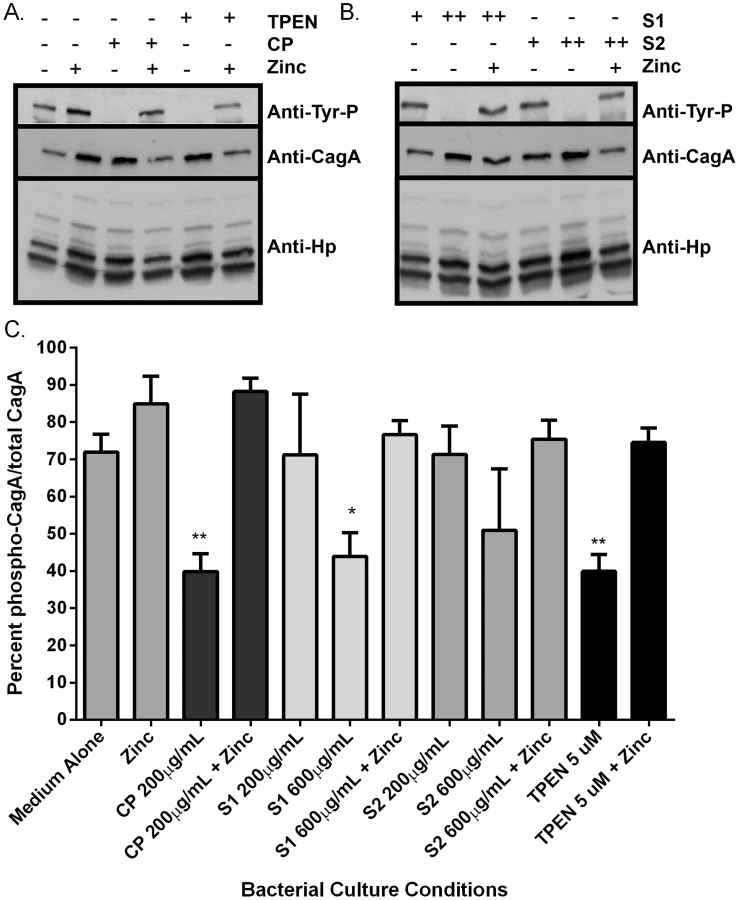

As an initial assessment of cag T4SS function, an assay for CagA translocation into AGS cells based on detecting phosphorylated CagA was performed. H. pylori were cultured in the presence or absence of CP for 4–6 hours prior to a 4 hour co-culture with AGS cells. Following washes to remove unbound bacteria, lysates of H. pylori-bound AGS cells were generated and separated by SDS-PAGE. The levels of phospho-CagA and total CagA in the extracts were determined by immunoblot analyses [25]. As shown in Figure 4A and C, reduced levels of phosphorylated CagA in H. pylori-AGS co-cultures were observed when H. pylori was pre-treated with sub-inhibitory (growth) doses of CP.
As mentioned earlier, activity of the cag T4SS leads to NFĸB activation and IL-8 secretion by gastric epithelial cells. To assess the ability of CP to inhibit cellular activation, H. pylori were cultured in the presence or absence of CP for 4–6 hours prior to a 1 hour co-culture with an AGS-NFĸB luciferase reporter cell line. As show in Figure 5, CP inhibits the ability of the cag T4SS to activate NFĸB (Figure 5A). To determine if CP also inhibits IL-8 secretion in this co-culture system, H. pylori were cultured in the presence or absence of CP for 4–6 hours prior to a 4 hour co-culture with AGS. IL-8 secretion was significantly lower in supernatants from AGS co-cultures with H. pylori pre-treated with CP compared to AGS co-cultures with untreated H. pylori (Figure 5B). To investigate whether these results were an effect of reduced adherence, a bacterial adherence assay was performed. There were no significant changes in H. pylori adherence as a consequence of treatment with either CP or TPEN alone or in the presence of an exogenous source of nutrient zinc (Figure S3).
To investigate which metal binding sites are critical for this phenotype and to determine if this phenotype is due to manganese sequestration or zinc sequestration, the same experiments were performed with CP ΔS1, ΔS2, and DS mutants. Mutagenesis of either the S1 site (manganese and zinc binding) or the S2 site (zinc binding alone) decreased the efficiency of CP's ability to repress the CagA translocation (Figure 4) and cag T4SS activity (Figure 5A and B). While 200 µg/ml of CP was sufficient to repress CagA translocation, NFkB activation and IL-8 production, due to a reduced ability to bind metal, higher levels (600 µg/ml) of ΔS1 and ΔS2 CP were needed to observe the same effects. The DS mutant, which does not bind any metals, was unable to ablate the cag T4SS-dependent NFĸB activation and had no effect on IL-8 induction (Figure 5A and B). Since the S1 mutant, which can only bind zinc, is still functional, nutrient zinc sequestration is responsible for the cag T4SS repression phenotype. The observation that CP is acting on the cag T4SS through a zinc sequestration-dependent pathway was consistent with experiments showing that the inhibitory effects of CP on both CagA phosphorylation and the activity of the cag T4SS were reversible with the addition of an exogenous zinc source (Figure 4 and 5).
To confirm that the effect of CP on the repression of cag T4SS was due to zinc chelation, a synthetic metal chelator which preferentially binds zinc, N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN), was tested for its ability to repress the cag T4SS. Bacteria pre-exposed to TPEN at concentrations below that necessary to inhibit growth (5 µM, Figures 2 and S1B) translocated less CagA than did untreated bacteria, as determined by immunoblot analysis for phospho-CagA (Figure 4A). Bacteria pre-exposed to TPEN caused less NFkB activation within the host cell (Figure 5A) and showed a diminished capacity to induce IL-8 secretion (Figure 5B). The addition of exogenous zinc to the H. pylori cultures reversed the phenotype, and the activity of the cag T4SS was restored (Figure 4 and 5). These data suggest that the activity of the cag T4SS is reduced by CP through a zinc-sequestration-dependent process.
Metal sequestration by CP inhibits cag T4SS pilus biogenesis
Our co-culture experiments demonstrated that metal sequestration by CP leads to abrogated phosphorylation of CagA and inhibition of downstream cellular activation in gastric epithelial cells. We next tested the hypothesis that the observed inhibition of the cag T4SS-dependent phenotype was attributable to inhibition of cag T4SS pilus production. To test this, field emission gun-scanning electron microscopy (FEG-SEM) analysis of the bacterial-gastric epithelial cell (H. pylori-AGS cell) co-cultures was performed to visualize the cag T4SS pili, as previously described [25], [32], [34]. Briefly, bacteria were cultured for 4–6 hours prior to co-culture with AGS cells in the presence or absence of WT or mutant forms of CP or the synthetic zinc chelator, TPEN, alone or in the presence of an exogenous source of nutrient zinc. Bacteria were co-cultured with gastric cells in the absence of additives for 4 hours before samples were processed and analyzed by high resolution FEG-SEM. Pre-treatment of H. pylori with CP or TPEN reduced the number of cag T4SS pili visible at the host pathogen interface (Figure 6). The addition of exogenous zinc restored WT pili formation, suggesting that the zinc-chelation is responsible for this reduced pilus formation. Similarly, the ΔS1 and ΔS2 mutant CP proteins were unable to repress pilus formation at the same concentration as WT (200 µg/mL). However, when increasing the concentration of the ΔS1 and ΔS2 mutants to high concentrations (600 µg/ml), cag T4SS pilus formation was repressed. The higher concentration of both ΔS1 and ΔS2 mutant CP proteins was consistent with earlier results, where 600 µg/ml of these mutant proteins was necessary to observe decreased NFkB and IL-8 expression. Conversely, the DS mutant CP did not repress cag T4SS pilus formation, even at very high concentrations (1200 µg/ml). These data indicate that CP inhibits the production of cag T4SS-associated pili through zinc sequestration.

Discussion
Results from these studies indicate that zinc homeostasis plays an important role in regulating the cag T4SS in H. pylori. Previous reports have shown that when H. pylori interacts with gastric epithelial cells, the cag T4SS translocates CagA into host cells leading to activation of c-Src and changes in the cytoskeleton [34]–[36]. A functional cag T4SS also increases the activation of NOD and NFkB [33], which ultimately results in the production of IL-8 and the recruitment of neutrophils [34]–[36]. Based on our results, we propose a model which is presented in Figure 7. In response to H. pylori infection, neutrophils are recruited to the site of infection. CP is deposited, and nutrient manganese and zinc are sequestered. Sequestration of zinc by CP represses cag T4SS pilus formation and CagA translocation, and results in diminished NFκB activation and IL-8 secretion. With reduced IL-8, less inflammation develops and the end result of CP-dependent zinc-sequestration is increased bacterial persistence.

H. pylori infection elicits a robust neutrophil response, resulting in increased expression of CP in the infected tissue, a result that agrees with previously published observations [24], [37]. In other models of infection, CP levels increase in response to bacterial infection [14], [18], [38], and levels vary from tissue to tissue [39]. It is likely that CP levels are dynamic within host gastric tissue during H. pylori infection because inflammation varies in a time - and location-dependent manner and is shaped by host genetics [2], [40], [41]. We hypothesized that CP is an important mediator of host-H. pylori interactions. Our results suggest that CP's activity against H. pylori may be dose-dependent. CP inhibits growth of H. pylori by sequestration of nutrient metals, an observation that agrees with the results seen with numerous other pathogens and supports the proposal that CP is part of the host response designed to restrict H. pylori burden in vivo. Yet when CP is absent, the bacterial burden is reduced by the increased inflammatory response. These data suggest that whether CP is present or absent, there is cross regulation between the bacteria and the host immune response, leading to a level of inflammation which controls bacterial burden but does not necessarily induce enough inflammation to completely clear the infection, and therefore, the bacteria persist.
In addition to indicating that CP restricts the growth of H. pylori, our work reveals that CP represses the activity of the cag T4SS, an important virulence factor that has been associated with carcinogenesis [42]. By utilizing CP mutants harboring inactivation of the metal binding sites within CP, our work demonstrated that both the site 1 and the site 2 mutant proteins have the capacity to inhibit the activity and biogenesis of pili associated with the cag T4SS. Since the CP mutant that can only bind zinc is still capable of inhibiting cag T4SS function and pilus biogenesis, this result proves that nutrient zinc sequestration is responsible for the cag T4SS repression phenotype, a result that is supported by restoration of cag T4SS activity and pilus biogenesis when an exogenous source of nutrient zinc is provided. At present, the components of the cag T4SS are not well defined and controversial [43], therefore it is not possible to determine whether pilus production is blocked at the level of pilus assembly, at the level of translation of pilus components or through some other mechanism. Interestingly, the cag T4SS has been reported to be induced by chelation of nutrient iron, a result that is reciprocal to the regulation imposed by sequestration of nutrient zinc, but still supports the contribution of micronutrients to the regulation of this important bacterial organelle [44]. There are other examples of reciprocal regulation of virulence factors by nutrient iron and zinc among bacterial pathogens. For example, in the gastrointestinal pathogen, enteropathogenic E. coli (EPEC), the expression and secretion of EPEC-associated secreted proteins (Esp) have been shown to be repressed by nutrient zinc and induced by nutrient iron [45].
Furthermore, CP-mediated metal sequestration has been associated with changes in bacterial virulence in other pathogens. For example, in the presence of CP, S. aureus superoxide defense is repressed, leading to diminished resistance to innate immune cells [16]. These previously published results reveal that CP can, in addition to repressing bacterial growth, also repress bacterial virulence to promote host defense strategies. In our murine model of infection, CP represses a major inflammation-promoting virulence factor, the cag T4SS. This activity may contribute to bacterial evasion of the immune system.
CP has been shown to be induced in the context of bacterial infection, and the utility of CP-deficient mice (A9 -/ - mice) in these studies has proven to be important [14], [17], [18]. In a model of S. aureus infection, CP-deficient mice had an approximately 1-log increase in bacterial burden in their livers compared to WT mice [16]. Similarly, in a model of A. baumannii infection, CP-deficient mice exhibited a significant increase in bacterial burden relative to WT mice at 36 hours post-infection, but not at 72 hour post-infection, indicating that CP is important for controlling early infection, but other components of the host system can clear bacteria independently of CP [17]. Conversely, in a model of S. Typhimurium, Salmonella overcomes zinc sequestration by calprotectin to colonize the gut through production of the CznABC transporter. CznABC was also required to promote the growth of S. Typhimurium over that of competing commensal bacteria in the inflamed gut [18]. H. pylori bacterial burden is decreased in CP-deficient mice compared to WT mice, indicating that the activity of CP is not restricted to antimicrobial activity against H. pylori, but can also regulate bacterial virulence which may promote chronic colonization within the host gastric niche. In other words, CP repression of the cag T4SS could promote chronic colonization by reducing inflammation and increasing bacterial burdens.
In addition to repressing bacterial virulence, CP has other effects on bacterial pathogens. S. aureus upregulate two metal uptake systems, MntABC and MntH, to promote manganese acquisition in the presence of CP [39]. The metal-acquisition properties are important for staphylococcal resistance to CP as well, and mutants in mntABC or mntH have decreased growth compared to WT strains in increasing concentrations of CP [39]. Similarly, both A. baumannii and S. Typhimurium upregulate zinc acquisition machinery to resist the nutrient sequestration imposed by CP in inflamed tissues. Mutants in zinc acquisition in both of these pathogens have diminished capacity to compete with WT bacteria in rodent models of infection [17], [18]. Taken together, these data support a model in which zinc acquisition provides a selective advantage for invading pathogens in the presence of inflammation-associated antimicrobial peptides such as CP. It also supports the diverse roles that both manganese and zinc availability play in microbial responses within the vertebrate host.
The trace element zinc is essential for cell function, and zinc co-factors are prevalent in the bacterial proteome [46]. Less than 0.1% of zinc is found in serum plasma [47], indicating the vast majority of zinc is stored in the intracellular space, and thereby, unavailable to extracellular pathogens. Additionally, the immune system produces zinc-chelating molecules such as S100A8 and S100A9 that have the capacity to bind and sequester nutrient zinc from invaders [48]. Thus, zinc homeostasis is emerging as an important feature of the host-pathogen interaction. Zinc exposure has been associated with decreased adherence, biofilm formation and virulence factor expression in enteroaggregative Escherichia coli (EAEC) [49]. Additionally, macrophages have been shown to utilize intraphagosomal zinc accumulation as a strategy to poison bacterial pathogens such as Mycobacterium tuberculosis [50]. To combat this, M. tuberculosis elaborates heavy metal efflux P-type ATPases, metallothioneins, and a zinc exporter [50]. This strategy is not uncommon, as pathogens such as Streptococcus pyogenes and Pseudomonas aeruginosa, have been shown to express zinc efflux systems that are crucial for colonization of an animal host [51], [52]. Similarly, H. pylori encodes a novel metal efflux pump, CznABC, which is required for resistance to cadmium, nickel and zinc intoxication, as well as colonization of a vertebrate host [53]. These results implicate zinc as an important micronutrient signal at the host-pathogen interface that bacteria sense and respond to accordingly. H. pylori has likely evolved to sense the presence of zinc as well as the deprivation of zinc by CP to induce the appropriate cellular responses. Thus, CP is an important inflammation-associated environmental signal to the bacterial cell.
CP also has the ability to act as a damage-associated molecular pattern molecule (DAMP) within the host [54]. It has been demonstrated that CP activates toll-like receptor 4 (TLR4) and signaling through nuclear factor ĸB, which ultimately leads to increased inflammatory responses [54], [55]. Additionally, CP is hypothesized to interact with the receptor for advanced glycation end products (RAGE) to promote chronic inflammation [56]. In the context of our murine model of infection, CP itself could potentially contribute to inflammatory processes. However, in the presence of an H. pylori infection, the presence of CP is associated with diminished inflammation, which can be attributed to the zinc-dependent regulation of the proinflammatory cag T4SS.
In conclusion, we propose a model (Figure 7) in which nutrient zinc acts as a signal to induce the cag T4SS and promote H. pylori-dependent inflammation. After a neutrophil response develops through cag T4SS-driven IL-8 production or through induction of the Th17 response, deposition of CP sequesters zinc and manganese. The result of this sequestration is that the cag T4SS is switched off. This tightly controlled regulation may contribute to bacterial immune evasion and promote chronicity.
Materials and Methods
Ethics statement
All animal experiments were done in concordance with the Animal Welfare Act, U.S. federal law, and NIH guidelines. All experiments were carried out under an ACORP protocol approved by Vanderbilt University Institutional Animal Care and Use Committee (IACUC; V/10/410 and V/13/240) and the Department of Veteran's Affairs, a body that has been accredited by the Association of Assessment and Accreditation of Laboratory Animal Care (AAALAC). The human study protocol was approved by the Vanderbilt University and the Nashville Department of Veterans Affairs Institutional Review Board (#5190). Human subjects gave informed written consent.
Bacterial strains, cell lines and culturing
H. pylori strains SS1 (a mouse-adapted clinical isolate), PMSS1 (the clinical isolate from which SS1 was derived), and the PMSS1 cagE isogenic mutant (PMSS1 cagE::aphA, a gift from M. Amieva) were used for these studies. PMSS1 was selected because it has a functional cag PAI and has the ability to colonize mice, and SS1 was selected due to its ability to colonize mice, although it lacks a functional cag PAI [57]. For infection assays, H. pylori strains were cultured on tryptic soy agar plates supplemented with 5% sheep blood or in Brucella broth supplemented with 10% fetal bovine serum at 37°C in room air supplemented with 5% CO2. For bacterial growth assays, H. pylori were grown in 60% Brucella broth plus 40% calprotectin (CP) buffer [16] supplemented with 10% fetal bovine serum (FBS) alone or supplemented with 50 µM zinc chloride and 50 µM manganese chloride, with increasing concentrations of CP at 37°C in room air supplemented with 5% CO2. Bacterial growth was quantified at 4, 12 and 24 hours by spectrophotometric reading of OD600, and at both 4 and 24 hours bacteria were subjected to serial dilution and plating onto tryptic soy agar plates supplemented with 5% sheep blood for enumeration of viable bacterial cells (CFU/mL).
For H. pylori-AGS cell co-culture assays, bacteria were grown in 60% Brucella broth plus 40% CP buffer supplemented with 10% FBS alone or supplemented with 100 µM zinc chloride, in the presence of 200 µg/mL WT CP, or 200–1200 µg/ml of ΔS1, ΔS2, double site mutant (DS CP) CP mutants. H. pylori were also cultured in the presence of the synthetic zinc chelator, N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) (Sigma Aldrich) at a concentration of 5 µM. Bacteria for AGS co-cultures were enumerated by using the OD600 and bacterial coefficient established for this H. pylori strain using our spectrophotometer (this coefficient was determined by plating to enumerated viable bacteria). Purification of WT and mutant CP proteins was performed as previously described [16]. Adherence assay methods are in the Materials and Methods S1 file.
AGS human gastric cells (ATCC) were cultured to 70% confluency in RPMI medium supplemented with 10% FBS, 2 mM L-glutamine, and 10 mM HEPES buffer at 37°C in room air plus 5% CO2.
IL-8 ELISA
H. pylori strains grown in various conditions of zinc availability were co-cultured with AGS cells at a multiplicity of infection of 100∶1 (as determined by spectrophotometric reading at OD600) for 4 hours. Cellular supernatants were collected and IL-8 was measured using an anti-human IL-8 ELISA (R&D), as previously described [32]. Bacteria grown in the presence of 5 µM TPEN or CP protein (WT, ΔS1 CP mutant, ΔS2 CP mutant, DS CP mutant) at either 200 µg/mL or 600 µg/mL were compared to those grown in the medium alone or in the presence of chelator plus 100 µM zinc chloride.
NF-kB reporter assay
Human gastric AGS cells were stably transfected with an NF-kB-luciferase reporter, as previously described [25]. Briefly, eukaryotic cells were grown to 70% confluency, bacteria were added at a multiplicity of infection of 20∶1 (as determined by spectrophotometric reading at OD600), and co-cultured for 4 hours. Supernatants were collected, cells were lysed and luciferase activity was measured with the Promega E4030 luciferase assay system (Promega, Madison, WI). As a positive control, H. pylori PMSS1 grown in medium alone was used and normalized for 100% luciferase activity.
Immunoblotting for phospho-CagA and total CagA
Phospho-CagA was detected by immunoblot, as previously described [25]. For detection of phospho-CagA, AGS cells were co-cultured with bacteria, as described for the IL-8 ELISA assays. AGS cells were washed twice with RPMI medium containing 1 mM sodium orthovanadate and pelleted by centrifugation (15,000 RPM for 3 minutes). Pellets were lysed in NP-40 lysis buffer containing Completer Mini EDTA-free protease inhibitor (Roche, Indianapolis, IN) and 2 mM sodium orthovanadate. Proteins were separated by soluble fractionation using 7.5% SDS-PAGE and immunoblotting with an anti-phosphotyrosine antibody (anti-PY99, Santa Cruz) or anti-H. pylori antibody, a polyclonal rabbit anti-H. pylori antibody described in [58]). Immunoreactive bands were visualized by ECL following incubation of the blot with HRP-conjugated secondary antibody. Detection of CagA and H. pylori soluble proteins was performed by stripping the blot with Restore Buffer (Pierce), and reprobing with either anti-CagA or anti-H. pylori primary antibodies.
Animals and experimental H. pylori infection
Permission to use male and female IL-17RA-/ - mice for the establishment of a breeding colony was obtained from Amgen (Seattle, WA). S100A9-/ - mice were a gift from Wolfgang Nacken (Institute of Experimental Dermatology, University of Münster, 48149 Münster, Germany) to E.P.S. The generation of these mice was previously described [59]. CP-deficient mice lack the S100A9 component of the heterodimer and exhibit destabilization of S100A8 protein as well [60], resulting in deficiencies in metal sequestration [14]. Amgen's IL-17RA-/ - mouse breeding colony is maintained at Taconic Farms. Helicobacter-free male IL-17RA-/-, S100A9-/ - and WT mice (all C57BL/6 background; 8–10 weeks old, were used in all experiments). Mice were orogastrically infected with 5×108 CFU H. pylori in 0.5 mL of Brucella broth twice over a 2 day period.
Processing of mouse stomachs
At different time points post-infection mice were sacrificed by carbon dioxide inhalation and the glandular stomach was removed for analyses. The stomach was removed from each mouse by excising between the esophagus and the duodenum. The forestomach (nonglandular portion) was removed from the glandular stomach and discarded. The stomach was rinsed gently with PBS to remove food and cut into three longitudinal strips spanning both the antrum and corpus, which were used for quantitative bacterial culture, RNA extraction/real-time RT-PCR analyses, and histological examination. For culturing of H. pylori from the stomach, gastric tissue was placed into Brucella broth-10% FBS for immediate processing. For RNA extraction, the stomach was placed in RNAlater solution (Ambion) before being processed. A longitudinal strip from the greater curvature of the stomach was excised and placed in 10% normal buffered formalin for 24 hours, embedded in paraffin and processed routinely for hematoxylin and eosin (H&E) staining. Indices of inflammation and injury were scored by a single pathologist (M.B.P.) who was blinded to the identity of the mice. Acute and chronic inflammation in the gastric antrum and corpus were graded on a 0–3 scale. Acute inflammation was graded based on density of neutrophils and chronic inflammation was graded based on the density of lamina propria mononuclear cell infiltration independent of lymphoid follicles. Total inflammation was calculated as a sum of acute and chronic inflammation in the corpus and the antrum allowing for quantification of total inflammation on a scale of 0–12 [61]. Immunohistochemistry was performed using commercially available polyclonal rabbit anti-S100A9 antibody (Cat # NB110-89726, Novus Biologicals, Littleton, CO).
For quantitative culture, the gastric tissue was homogenized in Brucella broth using a tissue tearor (BioSpec Products, Inc. Bartlesville, OK). Serial dilutions of the homogenate were plated on trypticase soy agar plates containing 5% sheep blood, 10 µg/ml nalidixic acid, 100 µg/ml vancomycin, 2 µg/ml amphotericin, and 200 µg/ml bacitracin. After 5 to 7 days of culture under microaerobic conditions generated by CampyPak Plus Gas Pak system, H. pylori colonies were counted. Flow cytometry methods are in the Materials and Methods S1 file.
Because our study comparing PMSS1 and PMSS1 cagE mutant was designed to elucidate the regulation of the cag T4SS in response to a neutrophil-associated antimicrobial protein, the six week post-infection time point was an appropriate timepoint when neutrophils would be present [29] and the cag T4SS would still be functional [25], [57], [62].
RNA isolation and real-time RTPCR
RNA was isolated from the stomach using the TRIZOL isolation protocol (Invitrogen, Carlsbad, CA) with slight modifications, as previously described [61]. RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). For real time RT-PCR, we used the relative gene expression method [63]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the normalizer, and tissue from a 5 uninfected WT mouse stomachs (or 5 uninfected human biospies) served as the pooled calibrator sample. All real time RT-PCR was performed using an Applied Biosystems StepOne Plus real time PCR instrument. Levels of cytokine expression are indicated as “relative units”, based on comparison of tissue from H. pylori-infected mice with tissue from uninfected mice (calibrator tissue) [63]. Primer and probe sets were purchased as Taqman Gene Expression Assays from Applied Biosystems (as pre-designed assays the annealing temperatures and amplicon length are available on their website). Primer and probe sets for eukaryotic genes were purchased as Taqman Gene Expression Assays from Applied Biosystems [S100A8 (Mm01220132_g1), S100A9 (Mm00656925_m1), GAPDH (Mm99999915_g1), human S100A8 (Hs00374264_g1), human S100A9 (Hs00610058_m1), and human GAPDH (Hs99999905_m1)].
Field emission gun-scanning electron microscopy of cag T4SS pili
H. pylori cag T4SS pili were imaged by field emission gun-scanning electron microscopy (FEG-SEM) analysis using methods previously described [32]. Briefly, H. pylori cells grown under various conditions of zinc availability were co-cultured at a multiplicity of infection of 50∶1 with AGS human gastric epithelial cells on poly-L-lysine-treated coverslips (BD Biosciences) for 4 h at 37°C in the presence of 5% CO2. Cells were fixed with 2.0% paraformaldehyde, 2.5% glutaraldehyde in 0.05 M sodium cacodylic acid buffer for 1 h at room temperature. Samples were washed three times with cacodylic acid buffer before secondary fixation with 1% osmium tetroxide. Cells were subjected to sequential dehydration with increasing concentrations of ethanol before being dried at the critical point, mounted onto SEM stubs, painted with colloidal silver at the sample edge, and sputtered with 20 nm of gold-palladium coating. Samples were visualized with an FEI Quanta 250 FEG-SEM at high vacuum and micrographs were analyzed with Image J software.
Statistical analysis
Statistical analysis of bacterial burden was performed after log transformation using unpaired two-tailed Student's t-test. Statistical analyses of IL-8 secretion, pilus quantification, expression data and luciferase activity were performed using paired two-tailed Student's t-test. Statistical analyses of histological inflammation scores were performed using Mann-Whitney U analysis. All data were derived from at least three separate biological replicates unless specified otherwise. Statistical analyses were performed using GraphPad Prism Software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. StolteM (1992) Helicobacter pylori gastritis and gastric MALT-lymphoma. Lancet 339 : 745–746.
2. CorreaP, PiazueloMB (2012) Evolutionary History of the Helicobacter pylori Genome: Implications for Gastric Carcinogenesis. Gut Liver 6 : 21–28.
3. SteinM, RappuoliR, CovacciA (2000) Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A 97 : 1263–1268.
4. AllisonCC, KuferTA, KremmerE, KaparakisM, FerreroRL (2009) Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 183 : 8099–8109.
5. MaedaS, AmarsanaaJ, MitsunoY, HirataY, AkanumaM, et al. (2002) Relationship between nuclear factor-kappaB activation and virulence factors of Helicobacter pylori in Japanese clinical isolates. J Gastroenterol Hepatol 17 : 556–562.
6. SharmaSA, TummuruMK, BlaserMJ, KerrLD (1998) Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol 160 : 2401–2407.
7. MaedaS, AkanumaM, MitsunoY, HirataY, OguraK, et al. (2001) Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem 276 : 44856–44864.
8. FoxJG, WangTC, RogersAB, PoutahidisT, GeZ, et al. (2003) Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 124 : 1879–1890.
9. OrrTS, ElliottEV, AltounyanRE, SternMA (1980) Modulation of release of neutrophil chemotactic factor (NCF). Clin Allergy 10 Suppl: 491–496.
10. KaplanMJ, RadicM (2012) Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189 : 2689–2695.
11. RadicM, KaplanMJ (2012) Jumbled NETs promote vasculitis. Arthritis Rheum 64 : 3498–3501.
12. SohnlePG, HunterMJ, HahnB, ChazinWJ (2000) Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14). J Infect Dis 182 : 1272–1275.
13. YuiS, NakataniY, MikamiM (2003) Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull 26 : 753–760.
14. CorbinBD, SeeleyEH, RaabA, FeldmannJ, MillerMR, et al. (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319 : 962–965.
15. DamoSM, Kehl-FieTE, SugitaniN, HoltME, RathiS, et al. (2013) Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc Natl Acad Sci U S A 110 : 3841–3846.
16. Kehl-FieTE, ChitayatS, HoodMI, DamoS, RestrepoN, et al. (2011) Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10 : 158–164.
17. HoodMI, MortensenBL, MooreJL, ZhangY, Kehl-FieTE, et al. (2012) Identification of an Acinetobacter baumannii Zinc Acquisition System that Facilitates Resistance to Calprotectin-mediated Zinc Sequestration. PLoS Pathog 8: e1003068.
18. LiuJZ, JellbauerS, PoeAJ, TonV, PesciaroliM, et al. (2012) Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11 : 227–239.
19. LoomansHJ, HahnBL, LiQQ, PhadnisSH, SohnlePG (1998) Histidine-based zinc-binding sequences and the antimicrobial activity of calprotectin. J Infect Dis 177 : 812–814.
20. LusitaniD, MalawistaSE, MontgomeryRR (2003) Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect Immun 71 : 4711–4716.
21. SteinbakkM, Naess-AndresenCF, LingaasE, DaleI, BrandtzaegP, et al. (1990) Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336 : 763–765.
22. ZaiaAA, SappingtonKJ, NisapakultornK, ChazinWJ, DietrichEA, et al. (2009) Subversion of antimicrobial calprotectin (S100A8/S100A9 complex) in the cytoplasm of TR146 epithelial cells after invasion by Listeria monocytogenes. Mucosal Immunol 2 : 43–53.
23. ClohessyPA, GoldenBE (1995) Calprotectin-mediated zinc chelation as a biostatic mechanism in host defence. Scand J Immunol 42 : 551–556.
24. LeachST, MitchellHM, GeczyCL, ShermanPM, DayAS (2008) S100 calgranulin proteins S100A8, S100A9 and S100A12 are expressed in the inflamed gastric mucosa of Helicobacter pylori-infected children. Can J Gastroenterol 22 : 461–464.
25. BarrozoRM, CookeCL, HansenLM, LamAM, GaddyJA, et al. (2013) Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9: e1003189.
26. GebhardtC, NemethJ, AngelP, HessJ (2006) S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol 72 : 1622–1631.
27. UrbanCF, ErmertD, SchmidM, Abu-AbedU, GoosmannC, et al. (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5: e1000639.
28. AlgoodHM, AllenSS, WashingtonMK, PeekRMJr, MillerGG, et al. (2009) Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J Immunol 183 : 5837–5846.
29. AlgoodHM, Gallo-RomeroJ, WilsonKT, PeekRMJr, CoverTL (2007) Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol Med Microbiol 51 : 577–586.
30. EatonKA (2006) Response to “eradication of Helicobacter pylori and resolution of gastritis in the gastric mucosa of IL-10-deficient mice”. Helicobacter 11 : 135–136.
31. LeeCW, RaoVP, RogersAB, GeZ, ErdmanSE, et al. (2007) Wild-type and interleukin-10-deficient regulatory T cells reduce effector T-cell-mediated gastroduodenitis in Rag2-/ - mice, but only wild-type regulatory T cells suppress Helicobacter pylori gastritis. Infect Immun 75 : 2699–2707.
32. ShafferCL, GaddyJA, LohJT, JohnsonEM, HillS, et al. (2011) Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog 7: e1002237.
33. HuttonML, Kaparakis-LiaskosM, TurnerL, CardonaA, KwokT, et al. (2010) Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun 78 : 4523–4531.
34. JohnsonEM, GaddyJA, CoverTL (2012) Alterations in Helicobacter pylori triggered by contact with gastric epithelial cells. Front Cell Infect Microbiol 2 : 17.
35. NotoJM, PeekRMJr (2012) The Helicobacter pylori cag Pathogenicity Island. Methods Mol Biol 921 : 41–50.
36. Sanchez-ZaucoNA, TorresJ, Perez-FigueroaGE, Alvarez-ArellanoL, Camorlinga-PonceM, et al. (2013) Impact of cagPAI and T4SS on the inflammatory response of human neutrophils to Helicobacter pylori infection. PLoS One 8: e64623.
37. MuellerA, O'RourkeJ, GrimmJ, GuilleminK, DixonMF, et al. (2003) Distinct gene expression profiles characterize the histopathological stages of disease in Helicobacter-induced mucosa-associated lymphoid tissue lymphoma. Proc Natl Acad Sci U S A 100 : 1292–1297.
38. MooreJL, BeckerKW, NicklayJJ, BoydKL, SkaarEP, et al. (2014) Imaging mass spectrometry for assessing temporal proteomics: Analysis of calprotectin in Acinetobacter baumannii pulmonary infection. Proteomics 14 : 820–828.
39. Kehl-FieTE, ZhangY, MooreJL, FarrandAJ, HoodMI, et al. (2013) MntABC and MntH contribute to systemic Staphylococcus aureus infection by competing with calprotectin for nutrient manganese. Infect Immun 81 : 3395–3405.
40. KodamanN, PazosA, SchneiderBG, PiazueloMB, MeraR, et al. (2014) Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A 111 : 1455–1460.
41. RoligAS, ShanksJ, CarterJE, OttemannKM (2012) Helicobacter pylori requires TlpD-driven chemotaxis to proliferate in the antrum. Infect Immun 80 : 3713–3720.
42. FrancoAT, IsraelDA, WashingtonMK, KrishnaU, FoxJG, et al. (2005) Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A 102 : 10646–10651.
43. JohnsonEM, GaddyJA, VossBJ, HennigEE, CoverTL (2014) Genes Required for Assembly of Pili Associated with the Helicobacter pylori cag Type IV Secretion System. Infect Immun 82 : 3457–3470.
44. NotoJM, GaddyJA, LeeJY, PiazueloMB, FriedmanDB, et al. (2013) Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J Clin Invest 123 : 479–492.
45. CraneJK, NaeherTM, ShulginaI, ZhuC, BoedekerEC (2007) Effect of zinc in enteropathogenic Escherichia coli infection. Infect Immun 75 : 5974–5984.
46. AndreiniC, BertiniI, CavallaroG, HollidayGL, ThorntonJM (2008) Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem 13 : 1205–1218.
47. Zalups RK, Koropatnick J (2010) Systematic Zinc Homeostasis. Cellular and molecular biology of metals. Boca Raton, FL: CRC Press: Taylor & Francis Group. pp. 201–202.
48. Diaz-OchoaVE, JellbauerS, KlausS, RaffatelluM (2014) Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front Cell Infect Microbiol 4 : 2.
49. MedeirosP, BolickDT, RocheJK, NoronhaF, PinheiroC, et al. (2013) The micronutrient zinc inhibits EAEC strain 042 adherence, biofilm formation, virulence gene expression, and epithelial cytokine responses benefiting the infected host. Virulence 4 : 624–633.
50. BotellaH, PeyronP, LevillainF, PoinclouxR, PoquetY, et al. (2011) Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe 10 : 248–259.
51. OngCL, GillenCM, BarnettTC, WalkerMJ, McEwanAG (2014) An Antimicrobial Role for Zinc in Innate Immune Defense Against Group A Streptococcus. J Infect Dis
52. DieppoisG, DucretV, CailleO, PerronK (2012) The transcriptional regulator CzcR modulates antibiotic resistance and quorum sensing in Pseudomonas aeruginosa. PLoS One 7: e38148.
53. StahlerFN, OdenbreitS, HaasR, WilrichJ, Van VlietAH, et al. (2006) The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect Immun 74 : 3845–3852.
54. LoserK, VoglT, VoskortM, LuekenA, KupasV, et al. (2010) The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat Med 16 : 713–717.
55. RivaM, KallbergE, BjorkP, HanczD, VoglT, et al. (2012) Induction of nuclear factor-kappaB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology 137 : 172–182.
56. IbrahimZA, ArmourCL, PhippsS, SukkarMB (2013) RAGE and TLRs: relatives, friends or neighbours? Mol Immunol 56 : 739–744.
57. ArnoldIC, LeeJY, AmievaMR, RoersA, FlavellRA, et al. (2011) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140 : 199–209.
58. LohJT, FriedmanDB, PiazueloMB, BravoLE, WilsonKT, et al. (2012) Analysis of Helicobacter pylori cagA promoter elements required for salt-induced upregulation of CagA expression. Infect Immun 80 : 3094–3106.
59. YeP, RodriguezFH, KanalyS, StockingKL, SchurrJ, et al. (2001) Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194 : 519–527.
60. ManitzMP, HorstB, SeeligerS, StreyA, SkryabinBV, et al. (2003) Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol 23 : 1034–1043.
61. HorvathDJJr, WashingtonMK, CopeVA, AlgoodHM (2012) IL-23 Contributes to Control of Chronic Helicobacter pylori Infection and the Development of T Helper Responses in a Mouse Model. Front Immunol 3 : 56.
62. SierraJC, HobbsS, ChaturvediR, YanF, WilsonKT, et al. (2013) Induction of COX-2 expression by Helicobacter pylori is mediated by activation of epidermal growth factor receptor in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol 305: G196–203.
63. GiuliettiA, OverberghL, ValckxD, DecallonneB, BouillonR, et al. (2001) An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods 25 : 386–401.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 10
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Novel Cyclic di-GMP Effectors of the YajQ Protein Family Control Bacterial Virulence
- MicroRNAs Suppress NB Domain Genes in Tomato That Confer Resistance to
- The ESAT-6 Protein of Interacts with Beta-2-Microglobulin (β2M) Affecting Antigen Presentation Function of Macrophage
- Characterization of Uncultivable Bat Influenza Virus Using a Replicative Synthetic Virus