
Squalene Synthase As a Target for Chagas Disease Therapeutics
Chagas disease is caused by the protozoan parasite Trypanosoma cruzi and affects eight million individuals, primarily in Latin America. Currently there is no cure for chronic T. cruzi infections. Unlike humans, this parasite use a variety of sterols (e.g. ergosterol, 24-ethyl-cholesta-5,7,22-trien-3 beta ol, and its 22-dihydro analogs), rather than cholesterol in their cell membranes, so inhibiting endogenous sterol biosynthesis is an important therapeutic target. Here, we report the first structure of the parasite's squalene synthase, which catalyzes the first committed step in sterol biosynthesis, as well as the structures of a broad range of squalene synthase inhibitors active against the clinically relevant intracellular stages, opening the way to new approaches to treating this neglected tropical disease.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004114
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004114
Summary
Chagas disease is caused by the protozoan parasite Trypanosoma cruzi and affects eight million individuals, primarily in Latin America. Currently there is no cure for chronic T. cruzi infections. Unlike humans, this parasite use a variety of sterols (e.g. ergosterol, 24-ethyl-cholesta-5,7,22-trien-3 beta ol, and its 22-dihydro analogs), rather than cholesterol in their cell membranes, so inhibiting endogenous sterol biosynthesis is an important therapeutic target. Here, we report the first structure of the parasite's squalene synthase, which catalyzes the first committed step in sterol biosynthesis, as well as the structures of a broad range of squalene synthase inhibitors active against the clinically relevant intracellular stages, opening the way to new approaches to treating this neglected tropical disease.
Introduction
Many millions of individuals are infected with the so-called “World's most neglected diseases”. These include the leishmaniases, with ∼12 million individuals affected [1], and in Latin America, Chagas disease. The latter affects ∼8 million individuals [2] including ∼300,000 in the United States, according to the US Centers for Disease Control and Prevention [3]). The global burden of Chagas disease is estimated to be ∼$7 billion a year [4]. There are no cures available for the chronic form of the disease which can involve cardiac myopathy, mega-oesophagus and mega-colon, although clinical trials with the azole drug posaconazole and a ravuconazole prodrug are in progress [5], [6]. Both of these compounds function by blocking the ergosterol biosynthesis pathway [7], [8] shown in Figure 1A, as described in a recent review [9].

Ergosterol is an essential membrane sterol in many trypanosomatid parasites and plays the same structural role as does cholesterol in humans. It is synthesized in a lengthy series of reactions beginning with the condensation of dimethylallyl diphosphate (DMAPP, Figure 1A) with two molecules of iso-pentenyl diphosphate (IPP) to form farnesyl diphosphate (FPP) in a reaction catalyzed by farnesyl diphosphate synthase (FPPS), a reaction that is inhibited by bisphosphonate drugs [10]. Two FPP molecules then condense in a “head-to-head” fashion to form presqualene diphosphate (the “first-half” reaction) which then undergoes loss of diphosphate, rearrangement, and reduction by NADPH to form squalene (the “second-half” reaction), Figure 1A [11], both reactions being catalyzed by squalene synthase (SQS). Squalene is epoxidized (by O2/squalene epoxidase) to form oxidosqualene, which is then electro-cyclized by oxidosqualene cyclase (OSC) to form lanosterol. Lanosterol is demethylated by the 14-α demethylase/P450 system (CYP51), the target of the azole drugs, and after several more steps, ergosterol, 24-ethyl-cholesta-5,7,22-trien-3-β-ol, and its 22-dihydro analogs are produced. Yeasts and fungi also produce ergosterol and the azole drugs were originally developed as anti-fungals [12] but were later found to have potent activity against T. cruzi [7], [13]. More recently, SQS inhibitors, quinuclidines (Figure 1B), originally developed as cholesterol-lowering drug leads [14], have also been found to kill T. cruzi, in vitro and in vivo [15]. However, more selective SQS inhibitors are of interest since they would reduce potential side-effects on steroidogenesis [16]. To begin to contemplate how to design such selective quinuclidine species, it is desirable to first learn more about how these compounds inhibit both human and trypanosomatid SQS, but to date, no such structures have been reported.
There is also interest in the development of SQS inhibitors with completely different structures and properties, including compounds that might have multiple sites of action in the ergosterol biosynthesis pathway (polypharmacology), as well as different tissue distributions. Other SQS inhibitors that have been discovered include the thiocyanate WC-9 [17] as well as the bisphosphonates ibandronate and incadronate. These bisphosphonates inhibit both human SQS (HsSQS) and HsFPPS [18]–[20] and block cholesterol biosynthesis [19]. Ibandronate is used clinically to treat osteoporosis and functions by inhibiting FPP biosynthesis in osteoclasts. Unfortunately, ibandronate binds tightly to human bone mineral [21] so this so-called nitrogen-containing bisphosphonate would not be a good anti-infective lead since it is rapidly removed from the circulation, but more lipophilic bisphosphonates [22]–[24] have poorer bone-binding capacity and have been shown to kill parasitic protozoa, such as malaria parasites (Plasmodium spp.), both in vitro and in vivo [23], [24]. In malaria parasites, unlike the situation with T. cruzi, there is no squalene synthase, and cell growth inhibition by lipophilic bisphosphonates is primarily at the level of FPPS/GGPPS (geranylgeranyl diphosphate synthase) inhibition [23].
The structure of human SQS has been reported [25] but gave little insight into the SQS mechanism of action. More recently, we reported [26] the structures of a bacterial SQS homolog, dehydrosqualene synthase (CrtM) from Staphylococcus aureus which carries out the same first-half reaction as does SQS, formation of presqualene diphosphate (PSPP, Figure 1A) from FPP. With CrtM, PSPP then loses diphosphate, and the resulting carbocation rearranges and loses a proton to form dehydrosqualene, and we obtained a quinuclidine inhibitor-bound structure, proposed to mimic one of the carbocation intermediates in catalysis [27]. Based on these results and those of others [28], [29] the SQS mechanism of action shown in Figure S1 is suggested. There have, however, been no structures of any trypanosomatid SQS enzyme. Here, we report the structures of human SQS and T. cruzi SQS bound to a substrate-like inhibitor (S-thiolo-farnesyldiphosphate, FSPP), as well as the structures of both enzymes bound to two potent quinuclidine inhibitors (E5700 and ER119884, Figure 1B) which suggest routes to selective inhibitor development. We also report six x-ray structures of lipophilic bisphosphonate inhibitors bound to TcSQS and/or HsSQS, as well as the activity of a series of lipophilic bisphosphonates against T. cruzi FPPS, TcSQS, and solanesyl diphosphate synthase (TcSPPS, involved in ubiquinone-9 biosynthesis, Figure 1A), and against T. cruzi amastigotes, plus, we demonstrate synergistic effects of E5700 and posaconazole against amastigotes.
Results and Discussion
Structures of T. cruzi and human squalene synthase bound to FSPP
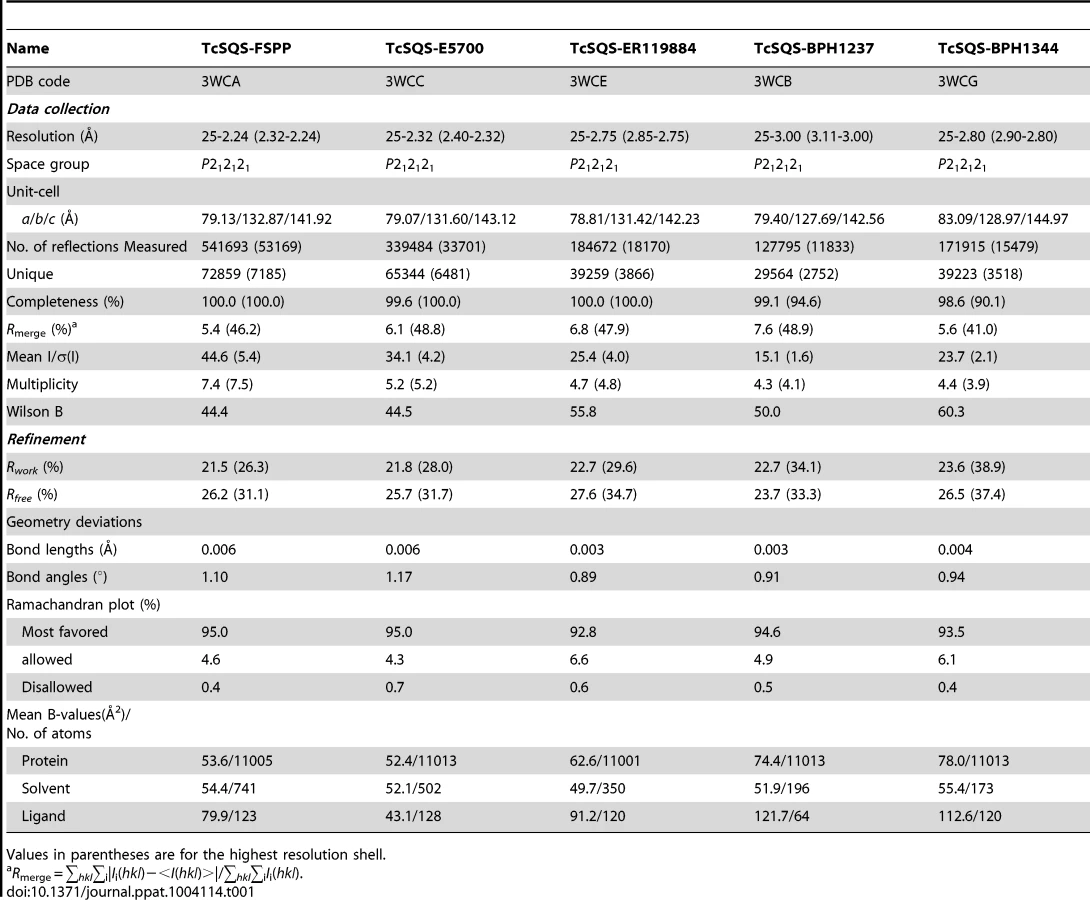
We expressed, purified and crystallized T. cruzi squalene synthase and solved its structure using the method of molecular replacement. TcSQS crystals could only be obtained in the presence of the substrate-like inhibitor, FSPP. Full experimental details are given in Materials and Methods. The construct was the recombinant enzyme described previously [16] in which 24 amino acids from the membrane-spanning N-terminus and 36 from the C-terminus were deleted in order to enable high-level production of active, soluble protein. We also crystallized HsSQS [30] and solved its structure, again in the presence of FSPP. Full crystallographic data acquisition and refinement statistics for both proteins are given in Tables 1 and 2. For the 13 structures investigated the resolution was in the range 2.06–3.00 Å (2.48 Å, on average), Tables 1–3.


All TcSQS crystals investigated belong to the space group P212121 and contain four molecules in the asymmetric unit. The crystals of HsSQS belong to the space group P21 and contain six HsSQS molecules in the asymmetric unit. The two enzymes share 44% amino acid identity and have the same fold (Figure 2A, B). The root-mean-square deviation (RMSD) is 0.87 Å for 298 matched Cα atoms within 2 Å of each other. A superposition of the TcSQS and HsSQS structures is shown in Figure 2C. Two FSPP substrate-analog inhibitor molecules bind to one TcSQS monomer. One FSPP is located in the allylic diphosphate (prenyl acceptor) site S1 previously observed in CrtM, Figure 2D, and the FPP that is expected to bind to this site is involved in the first diphosphate ionization reaction. Electron densities are in Figure S2A and (expanded) in Figure S3A. The other is located in the homo-allylic (prenyl donor) site, S2. The hydrocarbon moiety in the FSPP in S2 interacts with Ala168, Val171, Gly172, Leu175, Thr176 and Tyr179 from helix αG; Met199, Gly200 and Leu203 from helix αH; Phe282, Ser283 and Pro286 from helix αJ, as well as Phe45, Tyr64 and Tyr270. The diphosphate group of FSPP in S2 is located at the N-terminal end of helix αB and interacts with Ser42, Arg43, Ser44, and Arg68, while the diphosphate group from the first FSPP (in S1) is adjacent to the DXXED motif in helix αH. The DXXXD motif is highly conserved in many terpene synthases and cyclases [31] and is involved in binding to, in general, 3 Mg2+ that in turn bind to an allylic diphosphate, the Mg2+ facilitating carbocation formation due to diphosphate loss [32]. In HsSQS, the hydrocarbon moieties of FSPP bind in a similar way to the S1 and S2 sites, Figure 2E, but the orientations of the diphosphate head groups are somewhat different to those found with TcSQS, as can be seen in the ligand superpositions shown in Figure 2E, although the IC50 values for SQS inhibition are quite similar: 31 nM (TcSQS) and 64 nM (HsSQS). Binding appears to be driven by primarily hydrophobic interactions, consistent with limited electrostatic interactions with cationic amino-acid residues and the relatively solvent-accessible positions of the diphosphate group. Isothermal titration calorimetry results for the binding of FSPP to TcSQS are shown in Figure S4 and indicate a Kd of 2.6 µM. All major protein-ligand interactions are shown in Figure S5A–D.

Structures of SQS with quinuclidine inhibitors
We next obtained the structures of two very potent SQS inhibitors: the quinuclidines E5700 and ER119884 (Figure 1B) developed by Eisai Company (Tokyo, Japan) as anti-hypercholesterolemia drug leads that also have ∼1 nM activity against TcSQS and potent antiproliferative activity against T. cruzi [15], [16]. We solved the structures of the quinuclidines bound to both TcSQS and HsSQS using the method of molecular replacement. Full data acquisition and structure refinement details are in Tables 1 and 2 and electron densities are in Figures S2, S6 and S7. In both proteins the inhibitors span both the S1 and S2 isoprenoid binding sites, helping explain the very potent enzyme inhibition seen experimentally. With E5700, the quinuclidine ring occupies the S1 carbocation site (called here S1A) while the opposite end of the inhibitor is located at the bottom end of the S2 binding site, Figure 3A. The quinuclidine is expected to bind as a cationic species (quinuclidine pKa ∼10), and likely mimics one of the carbocations involved in squalene biosynthesis. The phenyl ring in the benzyl group occupies the bottom of the S1 binding site (S1B), as shown in Figure 3A, and the terminal methoxyl group in E5700 (in S2) is closely aligned with the “tail” of the FSPP ligand (in S2). With the ER119884 inhibitor bound to TcSQS (Figure 3B), the quinuclidine again binds to S1A and, as with E5700, the benzyl group occupies S1B, at the terminus of the S1 FPP site. The methyl terminus of the methoxy-propyl group also again aligns with the “tail” of the FSPP in S2 (Figure 3B). Detailed protein-ligand interactions are in Figure S5E–H.

With the HsSQS/quinuclidine structures we were able to obtain crystals by co-crystallization. However, this was not possible with TcSQS and it was necessary to soak TcSQS⋅FSPP crystals with the quinuclidine inhibitors, raising the possibility that there might be incomplete FSPP replacement (even in the presence of 10 mM quinuclidine). We thus show in Figures S3, S5 and S7 expanded and stereo views of the ligands in the TcSQS structures (FSPP, E5700 and ER119884) and for comparison, the HsSQS/quinuclidine densities (Figure S6), all contoured at 3σ. As can be seen in the TcSQS structures (superimposed on the TcSQS⋅FSPP structure), the ligand densities are well defined for all of E5700 (Figure S7A) and for almost all of ER119884 (Figure 3C and S7B).
To begin to see how selective inhibitors might be designed, we obtained the structures of both E5700 and ER119884 bound to HsSQS. Data acquisition and structure refinement details are given in Table 2. We show in Figure 3C a superposition of E5700 bound to TcSQS (cyan) with that of E5700 bound to human SQS (orange). It is clear that the drug binds to the two enzymes in a generally similar way, albeit with a slight tilt of the long axis. The quinuclidinol cage is also rotated by ∼70° between the two structures, changing the OH position by 2.4 Å and the cation center by 1.4 Å. More intriguing is the observation that at the bottom of the S2 ligand-binding pocket, TcSQS has Tyr179/Ser283 while HsSQS has Phe187/Cys289. In the future it may be possible to design an inhibitor that will not bind to Phe187/Cys289 in HsSQS, but will H-bond to Tyr179/Ser283 in TcSQS, since Tyr/Ser are good H-bond acceptors while Cys (and of course Phe) are not [33], [34]. The second quinuclidine, ER119884, binds in basically the same way as does E5700, with the quinuclidinol and benzyl groups in S1 while the methoxypropyl side chain locates to S2, as shown in Figure 3D. And as with the E5700 structures, the ER119884 structures indicate close proximity to the F/Y and C/S residues that may be targets for more selective inhibitors.
Lipophilic bisphosphonates are potent SQS inhibitors
We next sought to find new SQS inhibitors since, although having extremely potent activity against TcSQS and T. cruzi, the quinuclidines have not yet provided parasitological cures in vivo, due most likely to a short mean residence time [15], so new leads are of interest. In earlier work, Amin et al. [18] reported that human SQS was inhibited by nitrogen-containing bisphosphonate drugs such as ibandronate and incadronate, with low nM Ki values. Such bisphosphonates are also potent FPPS inhibitors but are extremely polar (clogP∼−3) and do not readily enter cells with Amin et al. reporting only a ∼60 µM IC50 in cholesterol lowering in J774 cells [19]. In addition, as noted previously, such bisphosphonates bind avidly to bone mineral – a desirable feature for a bone resorption drug but not, in general, an anti-infective. The binding of bisphosphonates to bone involves the interaction of the phosphonate groups, the 1-OH group, as well as of an unhindered cationic side-chain with the bone surface [21]. Replacement of the 1-OH group by a 1-H reduces bone binding, as does introduction of a hydrophobic side-chain which also greatly increases activity in a variety of cell assays including γδ T cell activation (where FPPS targeting is involved) [35], tumor cell killing [22] and inhibition of malaria parasite cell growth [24].
We thus next tested a series of lipophilic bisphosphonates [24], n-alkyl side-chain analogs of zoledronate (Figure 1B), with or without 1-OH groups (part of the “bone-hook” that enables binding to bone mineral) against TcSQS, HsSQS and TcFPPS (HsFPPS inhibition being reported earlier [24]). We also tested a subset of these bisphosphonates for their ability to inhibit the proliferation of the clinically relevant, intracellular, amastigote form of T. cruzi. Results are shown in Table 4 and Figure 4A. As can be seen in Figure 4A, we find with SQS inhibition that there is an increase in potency with increasing chain length up to n = 10–11, but then potency decreases on further chain elongation, for both TcSQS and HsSQS. There is no obvious effect of the 1-OH/1-H group on enzyme inhibition, but for the most active (C11) species, the HsSQS IC50 values are ∼10× higher than with TcSQS, indicating selectivity for the parasite enzyme. With TcFPPS, the increase in activity with chain length is less pronounced (as found also with HsFPPS), and maximum activity is found with a C6 substituent while the longer chain species are notably less active, Figure 4B. Activity in T. cruzi cell growth inhibition increases monotonically with increasing chain length up to the longest (C15) chain species investigated, Figure 4C, as seen also with malaria parasite cell growth inhibition [24], an effect that we attribute [36] in both organisms to the improved logP values (clogP∼3.3–3.8 for the longer chain inhibitors), resulting in enhanced cell permeability. FPP biosynthesis is not likely to be a major target with the most effective cell growth inhibitors (the C10–C15 bisphosphonates) in T. cruzi since they have relatively weak activity against FPPS and there was no “rescue” afforded by farnesol, and only a ∼20% rescue by geranylgeraniol (at 40 µM) with BPH-1218, one of the most potent SQS inhibitors. However, in the presence of BPH-1218 (C10, 4 µM), the content of exogeneously-acquired cholesterol in T. cruzi epimastigotes increased from 51% to 71% of total sterols, while that of endogenous ergosterol and the three major ergosterol related sterols (24-ethyl-5,7,22-cholestatrien-3β-ol, ergosta-5,7-diene-3β-ol and 24-ethyl-5,7 - cholestadien-3β-ol) decreased from 36% to 15% of total sterols, Figure S8A. The lipophilic bisphosphonates are thus potent SQS inhibitors that target sterol biosynthesis and are active in cells. The longer chain compounds are also poorer inhibitors of TcFPPS (Figure 4B), HsFPPS, a Plasmodium FPPS/GGPPS [24] as well as HsGGPPS (IC50 = 2 µM for the C12/1-H species [23], although any ability to inhibit TcFPPS in addition to TcSQS is of potential interest since this could result in enhanced efficacy in growth inhibition because FPPS inhibition would yield less FPP for the SQS-catalyzed reaction. Importantly, the most potent T. cruzi cell growth inhibitors had low cytotoxicity against Vero cells, Table 4.


There could of course be other targets for the lipophilic bisphosphonates, the most likely being solanesyl diphosphate synthase (SPPS) which produces the C45 diphosphate used in UQ9 (ubiquinone-9) biosynthesis [37]. We tested a subset of inhibitors against SPPS finding IC50s as low as 60 nM for the C10 species. However, unlike the situation with SQS, which is involved in production of the essential ergosterol not produced by humans, it is possible that SPPS is not an “essential gene” for T. cruzi in amastigotes, since quinones might be obtained from the host cell. There is, however, a ∼40% decrease in UQ9 biosynthesis with BPH-1218 in epimastigotes, consistent with multi-site targeting.
We thus believe that reduction in ergosterol (and related endogenous sterols) is a major contributor to the inhibition of cell growth. How sterol composition affects cell growth at the molecular level is not known. However, the azole class [38], [39] of drugs that target ergosterol biosynthesis in fungi have been shown to lead to progressive alkalinization of yeast vacuoles and a loss of V-ATPase activity. While not yet examined in trypanosomatids, this may also be the ultimate mode of action of this class of inhibitors, and since the quinuclidines target ergosterol biosynthesis [40], disruption of V-ATPase function, as found with the azoles in fungi, may also be important.
Structures of lipophilic bisphosphonates bound to SQS
We next investigated the crystallographic structures of four representative lipophilic bisphosphonates (BPH-1218; BPH-1237; BPH-1325 and BPH-1344; Figure 1B) bound to TcSQS and/or HsSQS. These compounds contain C10 or C15 side-chains, with or without 1-OH groups on the bisphosphonate head-group (BPH-1218, C10, H; BPH-1237, C10, OH; BPH-1325, C15, OH; BPH-1344, C15, H). Data acquisition and structure refinement details are given in Tables 1 (for TcSQS) and 3 (for HsSQS). For TcSQS, the structure of the shorter-chain (C10) inhibitor (BPH-1237) is shown, superimposed on FSPP from the TcSQS/FSPP structure, in Figure 5A, and that of the longer chain (C15) inhibitor (BPH-1344) is shown superimposed on FSPP in Figure 5B. In both structures the S2 farnesyl-binding site is occupied by the n-alkyl side-chains, and the methyl terminus of each inhibitor reaches the same position, as do the terminal methyl groups in FSPP (as seen also with the quinuclidine inhibitors). With TcSQS, the electron density with the shorter chain inhibitor is well defined (Figure S2) and it is clear that the imidazolium and bisphosphonate groups occupy S2, Figure 5A. With the longer chain inhibitor (BPH-1344), the density is broken in several places, but the n-alkyl side chain spans both S1 and S2 sites, with strong bisphosphonate fragment density. We attribute this to the presence of multiple conformations in the middle part of the side chain.

For HsSQS, co-crystallizations with BPH-1218, BPH-1237, BPH-1325 and BPH-1344 were all successful and strong electron densities were obtained in all cases (Figure S2). With the shorter chain inhibitors, there are several ways in which the bisphosphonate head-groups bind (Figure 5C, E). With the des-hydroxy species BPH-1218, the inhibitor binds to the S2 site in a similar way to that observed in the TcSQS/BPH-1237 complex, with the polar head-group interacting with Arg52, Ser53, Ser51, Tyr73, Asn215 and Arg218. With the 1-OH bearing BPH-1237, the side-chain binds to the S2 site, but the head-group binds to the S1 site where it interacts with Arg77, Asp80 and Asp84 of the first DXXED motif. The imidazolium group of this S1-head/S2-tail BPH-1237 molecule thus occupies the same cationic binding site as seen with the quinuclidines. In the case of the two longer chain (C15) species bound to HsSQS (Figure 5D, F), the last 8 carbons in both chains locate to the S2 site, but the chain then bends and occupies the S1 site, basically in the “bend” region seen in CrtM/PSPP structures [27]. Then, the bisphosphonate polar head-group returns to the S2 region. This arrangement of the long alkyl chain presumably minimizes repulsive interactions with the protein since the binding energies are less than those with the C10 bisphosphonates, though as noted above, their improved hydrophobicity results in better cell growth inhibition. Detailed protein-ligand interactions for all 6 bisphosphonate structures are shown in Figure S5I–N.
Structure prediction algorithms such as Phyre 2 [41] predict that both T. brucei SQS as well as Leishmania amazonensis SQS (LmSQS) will have similar structures to T. cruzi SQS, making it likely that the new inhibitors described here will inhibit these enzymes as well. Moreover, in T. brucei, SQS has been validated as a drug target by use of RNA interference [42]. It has also been reported that LmSQS is potently inhibited by several quinuclidines including ER119884 and E5700, in the nanomolar/sub-nanomolar range, and these compounds are active against the parasites [43]. The selectivity of these drugs was demonstrated with the JC-1 mitochondrial fluorescent label and by trypan blue exclusion tests with macrophages, which showed that the IC50s against the host cells were 4 to 5 orders of magnitude greater that those against intracellular parasites [15], [40], [43].
Structure of thiocyanate inhibitor WC-9 bound to human SQS
In previous work [27] we found that there was close similarity between the structure of the ligand BPH-652 when bound to CrtM and HsSQS, in which the ligand binds primarily to S2 – a 2.0 Å RMSD for the BPH-652 ligand atoms when using the two aligned protein structures and only 0.6 Å for the two aligned ligands. We also reported the structure of the thiocyanate inhibitor WC-9 (Figure 1B) bound to CrtM and WC-9 is also known to be an inhibitor of TcSQS [17]. We were unable to obtain diffraction quality crystals of WC-9 bound to TcSQS, but we did obtain the crystal structure of WC-9 bound to HsSQS, as shown in Figure 6A. The ligand again appears to bind to S2 [44], however, when we compare the CrtM and HsSQS/WC-9 structures, Figure 6B, it is clear that there is a shift in the position of the S2 site (denoted S2* in Figure 6B).

Comparisons between squalene and dehydrosqualene synthase structures
The results described above are also of interest when compared with earlier results on the bacterial dehydrosqualene synthase (CrtM, from Staphylococcus aureus) since they highlight two similar inhibitor structural features. The first observation is that the quinuclidine ring in E5700 as well as in ER119884 occupies a similar (S1A) binding site to that seen in the SQS/CrtM quinuclidine inhibitor BPH-651 (Figure 1B) bound to CrtM (Figure 7A). This site is also occupied by the adamantane ring in the CrtM inhibitor [44] SQ-109 (Figures 1B and 7B) and this site represents a common lipophilic, cationic binding site. The second observation of interest is that there is a hydrophobic pocket (S1B) that is occupied by the benzyl group in E5700 bound to SQS, as well as to the benzyl group in BPH-702 (Figures 1B and 7C) or the benzyl group in BPH-652 (Figures 1B and 7D). These observations suggest that it may be possible to improve the activity of other inhibitors (such as the bisphosphonates) by incorporating benzyl groups that can bind to S1B.

Synergy between E5700 and the lanosterol 14α-demethylase inhibitor posaconazole
The results described above provide information on the molecular basis for the potency of the aryl-quinuclidines as SQS inhibitors and potential future strategies for the design of parasite-specific inhibitors. The potency of these compounds might also be enhanced by using them in combination with other inhibitors, acting at different steps in the ergosterol biosynthesis pathway [45]. We tested this idea using a combination of E5700 and posaconazole (a very potent lanosterol 14α-demethylase (P450) inhibitor in clinical trials for Chagas disease) on intracellular amastigotes cultured inside murine peritoneal macrophages using the methodology described previously to investigate the combined effects of amiodarone and posaconazole [46]. As can be seen in Figure 4D, we obtain a concave isobologram with a fractional inhibitory concentration index (FICI) of 0.36, which indicates synergism since the accepted value for synergistic effects is FICI<0.5 [47].
In summary, in this work we report the structure of squalene synthase from Trypanosoma cruzi, a potentially important target for the development of novel therapeutic agents for the treatment of Chagas disease. The SQS protein structure had two major hydrophobic pockets: The S1 allylic binding site in which FSPP(FPP) binds to a cluster of highly conserved Asp residues via Mg2+, and a homoallylic S2 binding site to which the second FPP binds via basic residues interacting with the FSPP's diphosphate group. We solved the structures of two potent SQS inhibitors, the quinuclidines E5700 and ER119884, bound to both TcSQS as well as HsSQS. The results show that in all four cases, the more distal parts of the hydrophobic side-chains occupy primarily the S2-like FPP-binding site, while the benzyl groups bind to S1B and occupy the same position as the terminal methyl groups in the S1 FPP. The quinuclidinol groups occupy the S1A site where cationic species have been found to bind in the bacterial SQS homolog, CrtM. There were, however, several differences in inhibitor as well as amino-acid side-chain structures between TcSQS and HsSQS, opening up the possibility of selective inhibitor design. We also screened a library of lipophilic bisphosphonates against TcSQS, HsSQS, TcFPPS and T. cruzi amastigote cell growth. Most potent inhibition was found against TcSQS with a C11 side-chain, but the most potent activity in cells was with a C15 compound, due we propose to its enhanced hydrophobicity and hence, better cell penetration. There was no or little rescue from cell growth inhibition by farnesol or geranylgeraniol, but ergosterol biosynthesis, essential for parasite survival, was inhibited, consistent with an SQS target. We then determined the structures of four bisphosphonates bound to HsSQS and two of these bound to TcSQS. The results showed that in all cases ∼8 carbons at the chain terminus bound to S2 and were closely superposed on the more distal parts of the FSPP binding sites while the imidazolium rings bound close to the cationic binding sites identified earlier in CrtM. With the longer chain compounds, the upper part of the n-alkyl side-chain (close to the bisphosphonate head group) occupied the S1A pocket that is occupied by the “kinked” part of the FSPP structure, seen also in CrtM (+FSPP or PSPP) structures [27]. The polar head-groups bound primarily to the S2 head-group region. Both the bisphosphonates as well as the quinuclidine inhibitors thus bind to both the S1 and S2 substrate-binding sites, providing potent inhibition, leading to strong antiparasite activity. We also found strong synergy between the quinuclidine E5700 and and the 14α-demethylase inhibitor posaconazole, suggesting new therapeutic possibilities. Overall, the results presented here are of interest in the context of the future development of squalene synthase inhibitors as anti-infective drug leads against diseases caused by trypanosomatid parasites – the neglected tropical diseases.
Materials and Methods
Protein expression and purification
The constructs of TcSQS and HsSQS were obtained as described previously [16], [30]. TcSQS expression was induced with 0.8 mM isopropyl β-thiogalactopyranoside (IPTG) at 25°C for 18 hours, and HsSQS with 1.0 mM IPTG at 37°C for 6 hours. Cell pastes were harvested by centrifugation at 4,000× g for 15 min and resuspended in a lysis buffer containing 25 mM Tris-HCl, pH 7.5, 20 mM imidazole, and 150 mM NaCl. Cell lysates were prepared with a French pressure cell press (AIM-AMINCO Spectronic Instruments), and then centrifuged at 17,000× g for 30 min to remove cell debris. The cell-free extract was loaded onto lysis buffer-equilibrated Ni-NTA columns, followed by washing with 20 mM imidazole-containing buffer. The His-tagged enzymes were then eluted with an imidazole gradient from 20–250 mM, dialyzed twice against 5 L of 25 mM Tris-HCl, pH 7.5, and loaded onto a 20 ml DEAE Sepharose Fast Flow column (GE Healthcare Life Sciences). The buffer and gradient were 25 mM Tris, pH 7.5, and 0–500 mM NaCl. Purity of the recombinant proteins (>95%) was checked by SDS-PAGE analysis.
Inhibition of SQS, FPPS and SPPS activity
Initial SQS enzyme inhibition assays were carried out, in duplicate, in 96-well plates, with a total of 200 µL of reaction mixture in each well. Reactions were monitored by using a continuous spectrophotometric assay for phosphate releasing enzymes [26]. Reaction buffer contained 50 mM Tris-HCl, 1 mM MgCl2, 450 µM FPP, pH 7.4. The compounds investigated were pre-incubated with 2 µg of SQS for 30 min at 20°C. The IC50 values were obtained by fitting the inhibition data to a normal dose–response curve, using GraphPad PRISM, version 4.0, software for Windows (GraphPad Software Inc., San Diego, CA, www.graphpad.com). We then verified the results by using a radioactivity assay using 3H-labelled FPP as substrate. The reaction mixture (50 µL) had 2.5 nM T. cruzi or H. sapiens SQS, 0.5 µM [1-3H]-FPP, and 250 µM NADPH, in a detergent-containing buffer (50 mM HEPES-KOH, pH 7.5, 100 mM NaCl, 4 mM MgCl2, 0.5% Tween 80, 2 mM CHAPS, and 4 mM DTT). The enzymes were incubated with inhibitor for 10 minutes before addition of the substrates. The reaction was allowed to continue at 37°C for 15 minutes and then terminated with 50 µL of 0.1 M NaOH/methanol. The product was extracted with 500 µL of hexanes and 300 µL of the organic layer was measured by scintillation counting.
For FPPS inhibition we used the methods described previously [22]. The condensation of geranyl diphosphate (100 µM) and isopentenyl diphosphate (100 µM) was monitored at room temperature by using a continuous spectrophotometric assay for phosphate releasing enzymes [48]. The reaction buffer contained 50 mM Tris-HCl, pH 7.4, 1 mM MgCl2, and 0.01% Triton X100. The compounds investigated were pre-incubated with enzyme for 30 min at room temperature. The IC50 values were obtained from fitting dose-response curve using Prism 4.0 (GraphPad Software, Inc., La Jolla, CA, www.graphpad.com).
The activity of T. cruzi SPPS was determined by a radiometric assay [37]. Briefly, 100 µL of assay buffer (100 mM Tris–HCl buffer, pH 7.4, 1 mM MgCl2, 1% (v/v) Triton X-100, 7.07 µM [4-14C]-IPP (10 µCi/µmol)), and 50 µM GGPP was pre-warmed to 37°C. The assay was initiated by the addition of 10–20 ng of recombinant protein. The assay was allowed to proceed for 30 min at 37°C and was quenched by chilling quickly in an ice bath. The reaction products were extracted with 1 mL of 1-butanol saturated with water. The organic layer was washed with water saturated with NaCl, and transferred to a scintillation vial with 4 mL of Ecolume scintillation solution for counting. One unit of enzyme activity was defined as the activity required to incorporate 1 nmol of [4-14C]-IPP into [4-14C]-FPP in 1 min.
T. cruzi cell growth inhibition
Gamma-irradiated (2,000 Rads) Vero cells (3.4×104 cells/well) were seeded in 96 well plates (black, clear bottom plates from Greiner Bio-One) in 100 µL RPMI media (Sigma) with 10% FBS. Plates were incubated overnight at 35°C and 7% CO2. After overnight incubation, Vero cells were challenged with 3.4×105 trypomastigotes/well (CL strain overexpressing a tdTomato red fluorescent protein) in 50 µL volume and incubated for 5 h at 35°C and 7% CO2. After infection, cells were washed once with Hanks solution (150 µL/well) to eliminate any extracellular parasites, and compounds were added in serial dilutions in RPMI media in 150 µL volumes. Each dilution was tested in quadruplicate. Each plate also contained controls with host cells and no parasites (for a background check), and controls with parasites and no drugs (positive control). Drugs were tested at 5 different concentrations (highest 25 µM). For each set of experiments, benznidazole was used as a positive control. After drug addition, plates were incubated at 35°C and 7% CO2. At day 3 post-infection, plates were assayed for fluorescence and IC50 values were determined by non-linear regression analysis using SigmaPlot.
Sterol biosynthesis inhibition
T. cruzi epimastigotes were grown for 72 hours at 28°C in liver infusion tryptose medium [49] supplemented with 10% heat inactivated fetal bovine serum in the absence or presence of 2 µM BPH-1218. Cells from cultures (96 h incubation) were washed with Buffer A with glucose (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM D-glucose, and 50 mM HEPES at pH 7.0) then pelleted and chloroform/methanol (2∶1 v/v) added (∼30 mL of solvent per gram of wet cells) followed by vigorous stirring and overnight incubation at 4°C. The extract was then filtered using Whatman #2 paper to eliminate insoluble material, and the filtrate (a single phase) dried under nitrogen. The extract was then re-extracted using chloroform/methanol 9/1 v/v, dried under nitrogen, dissolved in 1 mL of pure chloroform and re-dried under nitrogen gas. The extracted lipids were saponified with 5 mL of ethanolic-potassium hydroxide (5 g KOH, 7 mL water, 14 mL ethanol) for 1 hour at 80°C. After cooling, 2.5 mL of water and 1.5 mL hexane were added and the phases allowed to separate. The aqueous phase was then re-extracted twice with hexane. The hexane extracts were dried under nitrogen, dissolved in chloroform and converted to the trimethylsilyl derivatives by using bis-trimethylsilyl amide. Both standards and samples (1 µL) were injected in split mode (10∶1) on a GC/MS system, which consisted of an Agilent 6890 gas chromatograph, an Agilent 5973 mass selective detector and an HP 7683B (Agilent Inc, Palo Alto, CA, USA) auto-sampler. Samples were analyzed on a 30 m DB5 column with 0.32 mm I.D. and 0.25 µm film thickness (Agilent Inc, Palo Alto, CA, USA) with an injection port temperature of 300°C, the interface was set to 300°C, the ion source adjusted to 230°C, MS Quadrupole 150°C. The helium carrier gas was set at a constant flow rate of 2.7 mL min−1. The temperature program was: 2 min at 250°C, followed by an oven temperature increase of 25°C min−1 to 320°C, for 10 min. The mass spectrometer was operated in positive electron impact mode (EI) at a 69.9 eV ionization energy in the m/z 50–800 scan range. The spectra of all chromatogram peaks were evaluated by using the HP Chemstation (Agilent, Palo Alto, CA, USA) program. Identifications and quantifications were performed using mass spectra obtained from authentic standards and with NIST08 and W8N08 libraries (John Wiley & Sons, Inc., USA).
Quinone extraction and analysis
For ubiquinone extraction, epimastigotes (1 g wet weight) were washed twice with buffer A with glucose (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 5.5 mM D-glucose, and 50 mM HEPES at pH 7.0). To break the cells, the pellets were frozen in a dry ice-100% ethanol bath (for 7 min) and thawed at 37°C (for 3 min) for a total of 5 times. One volume of water and three volumes of 1-propanol were added to the pellet. The samples were vortexed for 30 sec at room temperature and allowed to stand for one minute. Two volumes of n-hexane were added, the mixture vortexed for 30 sec at room temperature, then centrifuged at 12,000× g for 5 min. The upper phase, containing ubiquinone, was collected into a glass tube, and dried under nitrogen gas, then analyzed using LC-MS.
LC/MS was performed using an Agilent LC/MSD Trap XCT Plus system (Agilent Technologies, Santa Clara, CA) with an 1100 series HPLC system including a degasser, an auto-sampler, a binary pump, and a multiple-wavelength detector. The LC separation used a Phenomenex Gemini 3u C18 110A column (2 mm×100 mm) (Torrence, CA) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The flow-rate was 0.25 mL/min. The linear gradient was as follows: 0–1 min, 100% A; 7–50 min, 0% A; 51–56 min, 100% A. The auto-sampler was set at 5°C and the injection volume was 15 µL. UV signals were recorded at 275 nm. Positive ion mass spectrometry used atmospheric pressure chemical ionization (APCI). The capillary voltage was 4000 V and the nebulizer pressure and drying gas flow were 60 psi, 7 L/min, respectively. Temperatures for drying gas and APCI Vap were 350°C and 400°C, respectively.
Isothermal titration calorimetry (ITC)
ITC experiments were carried out basically as described previously for ligands binding to FPPS [50]. The thermodynamic parameters were: ΔG = −32.1 kJ·mol−1, ΔH = −10.4 kJ·mol−1, ΔS = 74.3 J·mol−1·K−1, and n = 1.86.
Cell growth inhibition by E5700+ posaconazole
The method used to determine possible synergism between the quinuclidine E5700 and the azole posaconazole followed that described by Veiga-Santos et al [46].
Measuring Vero cell proliferation using alamarBlueTM
Vero cells (1.7×104 cells/well) were seeded in 96 well plates (black, clear bottom from Greiner Bio-One Cat#655090) in 100 µL RPMI media (Sigma Cat# R-4130) with 10% FBS. Plates were tested as described by Recher et al [51].
Protein crystallization, data collection and structure determination
Initial crystallization screening was performed using Hampton Research Crystal Screens (Laguna Niguel, CA, USA) with the hanging-drop vapor-diffusion method at room temperature. In general, 2 µL TcSQS protein solution (8 mg/mL TcSQS enzyme, 25 mM Tris-HCl, pH 7.5, 2.5 mM FSPP) was mixed with 2 µL of reservoir solution. Single crystals were obtained in 0.1 M Tris, pH 8.5, 21% PEG 3350. The crystals were mounted in a cryo-loop and flash-frozen in liquid nitrogen with 0.15 M Tris, pH 8.5, 25% PEG3350 and 8% glycerol as a cryo-protectant. For HsSQS, 2 µL protein solution (5 mg/mL HsSQS enzyme, 25 mM Tris-HCl, pH 7.5) was mixed with 2 µL of reservoir solution. Single crystals were obtained in 0.2 M sodium citrate, pH 8.2, 28% PEG 3350. Prior to data collection at 100 K, the crystals were mounted in a cryo-loop and flash-frozen in liquid nitrogen with 0.3 M sodium citrate, pH 8.2, 30% PEG3350 and 2% glycerol as a cryo-protectant.
Crystals of TcSQS could only be obtained in the presence of FSPP, and direct co-crystallization of TcSQS with ER119884 and E5700 was not successful. Instead, we soaked the crystals of the TcSQS-FSPP complex with cryo-protectant solution containing 10 mM ER119884 or E5700 for at least 4 hrs prior to data acquisition. Crystals of (human) HsSQS in complex with the inhibitors FSPP, ER119884, E5700, BPH-1218, BPH-1237 and BPH-1325 were obtained by co-crystallization under similar conditions (0.2 M sodium citrate, pH 8.2, 24–28% PEG 3350, 5 mM inhibitor concentration).
Diffraction data were collected at beam lines BL13B1 and BL13C1 of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan), and processed using the program HKL2000. Prior to structure refinement, 5% randomly selected reflections were set aside for calculating Rfree as a monitor of model quality. The crystal structure of TcSQS was solved by using the molecular replacement (MR) method using the CNS program [52]. The previously determined HsSQS structure (PDB code 3VJA) was used as a search model (since it has 44% identity to TcSQS). The TcSQS crystal belongs to the space group P212121 and contains four protein molecules in an asymmetric unit. The other TcSQS-drug complex structures were solved by using the refined TcSQS (but without the bound FSPP) as a starting model. All crystals of HsSQS belong to the space group P21 and contain six protein molecules in the asymmetric unit. NCS restraints were applied at early stages of refinement and released at later stages. The refinement of every structure started with a temperature factor (B-factor) of 30, but upon rigid-body refinement an overall B was calculated for best scaling, which should be close to the corresponding Wilson B-factor. The individual atomic B-factors were not restrained by NCS and average B values are shown for the protein, ligand and solvent atoms in Tables 1–3. The rmsd between the four monomers of TcSQS ranged from 0.34 to 0.68 Å for 341 Cα atoms. The rmsd between six HsSQS monomers were 0.23–0.80 Å for 324–335 Cα atoms. The rmsd between TcSQS and HsSQS were larger, ranging from 0.82 to 1.28 Å for 264–273 Cα atoms.
For all drug molecules, the descriptions of atoms, structures, chemical bonds and their restraints were compiled into respective “topology” and “parameter” files by using the PRODRG server [53] for structural refinement by using CNS. Manual adjustments of the models and addition of water molecules used the program COOT [54].
Accession numbers
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank, www.pdb.org for T. cruzi SQS complexes with FSPP (3WCA), E5700 (3WCC), ER119884 (3WCE), BPH-1237 (3WCB) and BPH-1344 (3WCG), and human SQS complexes with FSPP (3WC9), E5700 (3WCJ), ER119884 (3WCM), BPH-1218 (3WCF), BPH-1237 (3WCH), BPH-1325 (3WCI), BPH-1344 (3WCL) and WC-9(3WCD).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KobetsT, GrekovI, LipoldovaM (2012) Leishmaniasis: prevention, parasite detection and treatment. Curr Med Chem 19 : 1443–1474 doi:10.2174/092986712799828300
2. CDC-Centers for Disease Control and Prevention (2013) Chagas Disease - Epidemiology & Risk Factors. Available at: http://www.cdc.gov/parasites/chagas/epi.html.
3. CDC-Centers for Disease Control and Prevention (2013) Chagas Disease in the Americas. Available at: http://www.cdc.gov/parasites/chagas/resources/chagasdiseaseintheamericas.pdf.
4. LeeBY, BaconKM, BottazziME, HotezPJ (2013) Global economic burden of Chagas disease: a computational simulation model. Lancet Infect Dis 13 : 342–348 doi:10.1016/S1473-3099(13)70002-1
5. ClaytonJ (2010) Chagas disease: pushing through the pipeline. Nature 465: S12–S15 doi:10.1038/nature09224
6. MoloneyA (2009) Trial renews interest in Chagas' disease. Lancet 374 : 1490.
7. DocampoR, MorenoSNJ, TurrensJF, KatzinAM, Gonzalez-CappaSM, et al. (1981) Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol Biochem Parasitol 3 : 169–180 doi:10.1016/0166-6851(81)90047-5
8. MaldonadoRA, MolinaJ, PayaresG, UrbinaJA (1993) Experimental chemotherapy with combinations of ergosterol biosynthesis inhibitors in murine models of Chagas' disease. Antimicrob Agents Chemother 37 : 1353–1359 doi:10.1128/AAC.37.6.1353
9. BucknerFS, UrbinaJA (2012) Recent developments in sterol 14-demethylase inhibitors for Chagas disease. Int J Parasitol Drugs Drug Resist 2 : 236–242 doi:10.1016/j.ijpddr.2011.12.002
10. MartinMB, ArnoldW, HeathHTIII, UrbinaJA, OldfieldE (1999) Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem Biophys Res Commun 263 : 754–758 doi:10.1006/bbrc.1999.1404
11. OldfieldE (2010) Targeting isoprenoid biosynthesis for drug discovery: bench to bedside. Acc Chem Res 43 : 1216–1226 doi:10.1021/ar100026v
12. GodefroiEF, HeeresJ, Van CutsemJ, JanssenPAJ (1969) Preparation and antimycotic properties of derivatives of 1-phenethylimidazole. J Med Chem 12 : 784–791 doi:10.1021/jm00305a014
13. UrbinaJA, PayaresG, ContrerasLM, LiendoA, SanojaC, et al. (1998) Antiproliferative effects and mechanism of action of SCH 56592 against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob Agents Chemother 42 : 1771–1777.
14. Okada T, Kurusu N, Tanaka K, Miyazaki K, Shinmyo D, et al., inventors; Eisai Co., Ltd., assignee (2003 Jul 29) Quinuclidine compounds and drugs containing the same as the active ingredient. United States Patent 6,599,917.
15. UrbinaJA, ConcepcionJL, CalderaA, PayaresG, SanojaC, et al. (2004) In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob Agents Chemother 48 : 2379–2387 doi:10.1128/AAC.48.7.2379-2387.2004
16. Sealey-CardonaM, CammererS, JonesS, Ruiz-PérezLM, BrunR, et al. (2007) Kinetic characterization of squalene synthase from Trypanosoma cruzi: selective inhibition by quinuclidine derivatives. Antimicrob Agents Chemother 51 : 2123–2129 doi:10.1128/AAC.01454-06
17. UrbinaJA, ConcepcionJL, MontalvettiA, RodriguezJB, DocampoR (2003) Mechanism of action of 4-phenoxyphenoxyethyl thiocyanate (WC-9) against Trypanosoma cruzi, the causative agent of Chagas' disease. Antimicrob Agents Chemother 47 : 2047–2050 doi:10.1128/AAC.47.6.2047-2050.2003
18. AminD, CornellSA, GustafsonSK, NeedleSJ, UllrichJW, et al. (1992) Bisphosphonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis. J Lipid Res 33 : 1657–1663.
19. AminD, CornellSA, PerroneMH, BilderGE (1996) 1-Hydroxy-3-(methylpentylamino)-propylidene-1,1-bisphosphonic acid as a potent inhibitor of squalene synthase. Arzneimittelforschung 46 : 759–762.
20. KavanaghKL, GuoK, DunfordJE, WuX, KnappS, et al. (2006) The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci 103 : 7829–7834 doi:10.1073/pnas.0601643103
21. MukherjeeS, HuangC, GuerraF, WangK, OldfieldE (2009) Thermodynamics of Bisphosphonates Binding to Human Bone: A Two-Site Model. J Am Chem Soc 131 : 8374–8375 doi:10.1021/ja902895p
22. ZhangY, CaoR, YinF, HudockMP, GuoR-T, et al. (2009) Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: an X-ray and NMR investigation. J Am Chem Soc 131 : 5153–5162 doi:10.1021/ja808285e
23. NoJH, Dossin F deM, ZhangY, LiuY-L, ZhuW, et al. (2012) Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity. Proc Natl Acad Sci 109 : 4058–4063 doi:10.1073/pnas.1118215109
24. ZhangY, ZhuW, LiuY-L, WangH, WangK, et al. (2013) Chemo-immunotherapeutic antimalarials targeting isoprenoid biosynthesis. ACS Med Chem Lett 4 : 423–427 doi:10.1021/ml4000436
25. PanditJ, DanleyDE, SchulteGK, MazzalupoS, PaulyTA, et al. (2000) Crystal structure of human squalene synthase, a key enzyme in cholesterol biosynthesis. J Biol Chem 275 : 30610–30617 doi:10.1074/jbc.M004132200
26. LiuC-I, LiuGY, SongY, YinF, HenslerME, et al. (2008) A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319 : 1391–1394 doi:10.1126/science.1153018
27. LinF-Y, LiuC-I, LiuY-L, ZhangY, WangK, et al. (2010) Mechanism of action and inhibition of dehydrosqualene synthase. Proc Natl Acad Sci 107 : 21337–21342 doi:10.1073/pnas.1010907107
28. BlaggBSJ, JarstferMB, RogersDH, PoulterCD (2002) Recombinant Squalene Synthase. A Mechanism for the Rearrangement of Presqualene Diphosphate to Squalene. J Am Chem Soc 124 : 8846–8853 doi:10.1021/ja020411a
29. LiuCI, JengWY, ChangWJ, ShihMF, KoTP, et al. (2014) Structural insights into the catalytic mechanism of human squalene synthase. Acta Crystallogr D Biol Crystallogr 70 : 231–241 doi:10.1107/S1399004713026230
30. SongY, LinF-Y, YinF, HenslerM, Rodrígues PovedaCA, et al. (2009) Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. J Med Chem 52 : 976–988 doi:10.1021/jm801023u
31. OldfieldE, LinF-Y (2012) Terpene Biosynthesis: Modularity Rules. Angew Chem Int Ed Engl 51 : 1124–1137 doi:10.1002/anie.201103110
32. AaronJA, ChristiansonDW (2010) Trinuclear Metal Clusters in Catalysis by Terpenoid Synthases. Pure Appl Chem Chim Pure Appl 82 : 1585–1597 doi:10.1351/PAC-CON-09-09-37
33. ZhouP, TianF, LvF, ShangZ (2009) Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins 76 : 151–163 doi:10.1002/prot.22327
34. Desiraju GR, Steiner T (2001) The weak hydrogen bond: in structural chemistry and biology. Oxford: Oxford University Press.
35. ZhangY, CaoR, YinF, LinF-Y, WangH, et al. (2010) Lipophilic pyridinium bisphosphonates: potent γδ T cell stimulators. Angew Chem Int Ed 49 : 1136–1138 doi:10.1002/anie.200905933
36. MukkamalaD, NoJH, CassLM, ChangT-K, OldfieldE (2008) Bisphosphonate inhibition of a Plasmodium farnesyl diphosphate synthase and a general method for predicting cell-based activity from enzyme data. J Med Chem 51 : 7827–7833 doi:10.1021/jm8009074
37. FerellaM, MontalvettiA, RohloffP, MirandaK, FangJ, et al. (2006) A solanesyl-diphosphate synthase Localizes in glycosomes of Trypanosoma cruzi. J Biol Chem 281 : 39339–39348 doi:10.1074/jbc.M607451200
38. ZhangY, RaoR (2012) The V-ATPase as a target for antifungal drugs. Curr Protein Pept Sci 13 : 134–140.
39. ZhangY-Q, GamarraS, Garcia-EffronG, ParkS, PerlinDS, et al. (2010) Requirement for Ergosterol in V-ATPase Function Underlies Antifungal Activity of Azole Drugs. PLoS Pathog 6: e1000939 doi:10.1371/journal.ppat.1000939
40. UrbinaJA, ConcepcionJL, RangelS, VisbalG, LiraR (2002) Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol Biochem Parasitol 125 : 35–45.
41. KelleyLA, SternbergMJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4 : 363–371 doi:10.1038/nprot.2009.2
42. Pérez-MorenoG, Sealey-CardonaM, Rodrigues-PovedaC, GelbMH, Ruiz-PérezLM, et al. (2012) Endogenous sterol biosynthesis is important for mitochondrial function and cell morphology in procyclic forms of Trypanosoma brucei. Int J Parasitol 42 : 975–989 doi:10.1016/j.ijpara.2012.07.012
43. Fernandes RodriguesJC, ConcepcionJL, RodriguesC, CalderaA, UrbinaJA, et al. (2008) In vitro activities of ER-119884 and E5700, two potent squalene synthase inhibitors, against Leishmania amazonensis: antiproliferative, biochemical, and ultrastructural effects. Antimicrob Agents Chemother 52 : 4098–4114 doi:10.1128/AAC.01616-07
44. LinF-Y, LiuY-L, LiK, CaoR, ZhuW, et al. (2012) Head-to-head prenyl tranferases: anti-infective drug targets. J Med Chem 55 : 4367–4372 doi:10.1021/jm300208p
45. UrbinaJA (2010) New insights in Chagas disease treatment. Drugs Future 35 : 409 doi:10.1358/dof.2010.035.05.1484391
46. Veiga-SantosP, BarriasES, SantosJFC, de Barros MoreiraTL, de CarvalhoTMU, et al. (2012) Effects of amiodarone and posaconazole on the growth and ultrastructure of Trypanosoma cruzi. Int J Antimicrob Agents 40 : 61–71 doi:10.1016/j.ijantimicag.2012.03.009
47. HallanderHO, DornbuschK, GezeliusL, JacobsonK, KarlssonI (1982) Synergism between aminoglycosides and cephalosporins with antipseudomonal activity: interaction index and killing curve method. Antimicrob Agents Chemother 22 : 743–752 doi:10.1128/AAC.22.5.743
48. WebbMR (1992) A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci U S A 89 : 4884–4887.
49. BonéGJ, SteinertM (1956) Isotopes incorporated in the nucleic acids of Trypanosoma mega. Nature 178 : 308–309 doi:10.1038/178308a0
50. YinF, CaoR, GoddardA, ZhangY, OldfieldE (2006) Enthalpy versus entropy-driven binding of bisphosphonates to farnesyl diphosphate synthase. J Am Chem Soc 128 : 3524–3525 doi:10.1021/ja0601639
51. RecherM, BarbozaAP, LiZ-H, GalizziM, Ferrer-CasalM, et al. (2013) Design, synthesis and biological evaluation of sulfur-containing 1,1-bisphosphonic acids as antiparasitic agents. Eur J Med Chem 60 : 431–440 doi:10.1016/j.ejmech.2012.12.015
52. BrungerAT (2007) Version 1.2 of the Crystallography and NMR system. Nat Protoc 2 : 2728–2733 doi:10.1038/nprot.2007.406
53. SchüttelkopfAW, van AaltenDMF (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr 60 : 1355–1363 doi:10.1107/S0907444904011679
54. EmsleyP, CowtanK (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 : 2126–2132 doi:10.1107/S0907444904019158
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases