
The Downregulation of GFI1 by the EZH2-NDY1/KDM2B-JARID2 Axis and by Human Cytomegalovirus (HCMV) Associated Factors Allows the Activation of the HCMV Major IE Promoter and the Transition to Productive Infection
Human cytomegalovirus (HCMV) is a significant pathogen that belongs to the herpesvirus family. Here we show that the histone H3K27 methyltransferase EZH2 and its regulators JARID2 and NDY1/KDM2B are required for the establishment of productive infection. Mechanistically, the EZH2-NDY1/KDM2B-JARID2 axis downregulates GFI1, a repressor of the HCMV major-immediate-early promoter (MIEP) and inhibition of this axis upregulates GFI1 and interferes with the activation of the MIEP and HCMV infection. GFI1 is rapidly downregulated during infection in both wild-type and EZH2, NDY1/KDM2B, JARID2 knockdown cells. However, since the starting levels of GFI1 in the latter are significantly higher, they remain high despite the virus-induced GFI1 downregulation, preventing the infection. Following the downregulation of GFI1 immediately after virus entry, HCMV initiates an EZH2-NDY1/KDM2B-JARID2-JMJD3-dependent program to maintain the low expression of GFI1 throughout the infection cycle. The knockdown of EZH2 also modulates the accumulation of histone H3K27me3 and H3K4me3 in the immediate-early region of HCMV, and by doing so, it may contribute directly to the MIEP repression induced by the knockdown of EZH2. These data show that HCMV uses multiple mechanisms to allow the activation of the HCMV MIEP and to prevent cellular mechanisms from blocking the HCMV replication program.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004136
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004136
Summary
Human cytomegalovirus (HCMV) is a significant pathogen that belongs to the herpesvirus family. Here we show that the histone H3K27 methyltransferase EZH2 and its regulators JARID2 and NDY1/KDM2B are required for the establishment of productive infection. Mechanistically, the EZH2-NDY1/KDM2B-JARID2 axis downregulates GFI1, a repressor of the HCMV major-immediate-early promoter (MIEP) and inhibition of this axis upregulates GFI1 and interferes with the activation of the MIEP and HCMV infection. GFI1 is rapidly downregulated during infection in both wild-type and EZH2, NDY1/KDM2B, JARID2 knockdown cells. However, since the starting levels of GFI1 in the latter are significantly higher, they remain high despite the virus-induced GFI1 downregulation, preventing the infection. Following the downregulation of GFI1 immediately after virus entry, HCMV initiates an EZH2-NDY1/KDM2B-JARID2-JMJD3-dependent program to maintain the low expression of GFI1 throughout the infection cycle. The knockdown of EZH2 also modulates the accumulation of histone H3K27me3 and H3K4me3 in the immediate-early region of HCMV, and by doing so, it may contribute directly to the MIEP repression induced by the knockdown of EZH2. These data show that HCMV uses multiple mechanisms to allow the activation of the HCMV MIEP and to prevent cellular mechanisms from blocking the HCMV replication program.
Introduction
Human cytomegalovirus (HCMV) is a double stranded DNA virus that belongs to the beta-herpesvirus subfamily of the herpesvirus family. Other members of this subfamily are the human herpes viruses 6 and 7 (HHV-6 and HHV-7). HCMV seroprevalence varies widely among populations residing in different geographical regions and among different socioeconomic and age groups [1]. The virus infects many cell types, including fibroblasts, hematopoietic, endothelial, epithelial, smooth muscle and neuronal cells [2]. Most otherwise healthy individuals that are infected with HCMV, experience few if any symptoms. However, some may present symptoms similar to mononucleosis, including fatigue, fever and muscle aches [1]. After the initial infection, the virus enters life-long latency in hematopoietic and endothelial cells, during which the viral genome is maintained as a low-copy number extrachromosomal plasmid. During latency, the productive viral transcription program is almost entirely repressed, with only a subset of latency-associated transcripts being expressed [3]. The Immediate-Early (IE) genes whose expression is a prerequisite for the onset and progression of productive infection remain silenced, and as a result, there is no production of infectious virions. Under specific conditions, the viral genomes can undergo sporadic reactivation, re-initiating a full replicative cycle, which results in virus production and dissemination. Latently-infected individuals are typically asymptomatic. Reactivation of the virus is frequently observed in HIV-infected individuals and in patients undergoing treatment with immunosuppressive or chemotherapeutic drugs [1], [3], [4], although it may also occur in immunocompetent hosts [3]. Virus reactivation may be responsible for debilitating or life-threatening illnesses [1], [3], [4].
The genome of HCMV consists of unique short (US) and unique long (UL) segments both of which are flanked by inverted repeats [1]. Viral gene expression, during HCMV infection, occurs in a temporally regulated manner and it is characterized by three sequential and interdependent waves of transcription. The first wave includes the robust transcription of the immediate-early (IE) genes IE1-72 KDa and IE2-86 KDa, which antagonize and inactivate the host defenses while in addition they induce the expression of the early viral genes. The early genes, expressed in the course of the second wave of transcription, contribute to viral DNA replication, a prerequisite for the activation of the late genes. The latter encode viral structural proteins and are required for virion assembly and virion release from the infected cells. To initiate the transcription of the immediate-early genes, the virus employs cellular transcriptional activators and inhibits cellular transcriptional repressors targeting the major immediate-early promoter (MIEP) [5]. One of the transcriptional repressors targeting this promoter is Growth factor independence 1 (GFI1), a zinc finger protein with a SNAG repressor domain [6], [7]. GFI1 was originally identified as a transcription factor that contributes to the transition of IL-2-dependent T cell lymphoma lines to IL-2 Independence [8]. Today, we know that GFI1 is an important regulator of hematopoietic cell differentiation, contributing to multiple steps in hematopoiesis and lymphopoiesis, (reviewed in [9], [10]). In addition, we know that GFI1 regulates the functional response of macrophages and dendritic cells to Toll like receptor (TLR) signals [11]. At the molecular level, it has been shown that GFI1 is part of a large nuclear complex that includes CoREST, lysine-specific demethylase-1 (LSD1), and HDACs 1 and 2. CoREST and LSD1 associate with GFI1 by binding the GFI1 SNAG repression domain [12].
Immediate-early gene transcription during HCMV infection, or virus reactivation in latently infected cells, depends on the state of differentiation of the target cells [3], [13]-[19]. Undifferentiated cells tend to resist productive infection, suggesting that epigenetic mechanisms, including chromatin modifications and DNA methylation may alter the permissiveness to the virus. Earlier studies addressing this hypothesis, confirmed that immediate-early gene transcription can be altered by the acetylation status of histone H3 associated with the major immediate-early promoter [14], [16], [20]–[25]. In the present study, we focus our attention on the role of histone methylation in HCMV infection and we show that changes in viral infectivity caused by modulation of the chromatin modification machinery of the cell are due to changes in the transcription of the immediate-early genes. Unlike earlier studies however, the present study focuses on the regulation of cellular transcription factors that control the expression the HCMV immediate-early region.
Methylation of histone tails in the promoter region, or the body of a gene, plays a major role in the regulation of gene expression. Histones undergo lysine mono-, di-, or tri-methylation at multiple sites and the functional consequences of histone methylation are site-dependent. Thus, tri-methylation of promoter-associated histone H3 at K4 is a feature of active chromatin, while di-methylation and tri-methylation of histone H3 at K9, or tri-methylation at K27 are features of inactive chromatin. Moreover, mono-, di - and tri-methylation at other sites, such as K36 in the body of a gene, may affect transcriptional elongation and/or RNA splicing (reviewed in [26]). Methylation of core histones at different sites is catalyzed by a host of site-specific methyltransferases. For example, tri-methylation of histone H3 at K27 is catalyzed by EZH2, a component of the polycomb repressor complex 2 (PRC2) [27], [28], whose activity is regulated by several co-factors, including the jumonji domain-containing proteins JARID2 [29] and NDY1/KDM2B [30].
Histone methylation is reversible, with demethylation being catalyzed by a host of site-specific histone demethylases. The first histone demethylase to be identified (LSD1) removes H3K4me1 and H3K4me2 methyl groups, through an oxidative reaction that uses FAD as a co-factor and produces an unstable imine intermediate [31]. The large family of jumonji domain-containing histone demethylases removes lysine methyl groups from a variety of mono-, di - or tri-methylated sites, through an oxidative reaction that uses Iron (FeII) and α-ketoglutarate as co-factors and produces an unstable hydroxymethyl intermediate. Demethylation of histone H3K27me3 is catalyzed by the jumonji domain demethylases, UTX/KDM6A, its homolog UTY, and JMJD3/KDM6B (Reviewed in [32]).
The results of the present study showed that immediate-early gene transcription and HCMV infection of human foreskin fibroblasts (HFFs) depend on histone H3K27 trimethylation, which is under the control of EZH2, JARID2, NDY1/KDM2B and the histone demethylase JMJD3. The EZH2/NDY1/ /JARID2/JMJD3 axis silences GFI1, a repressor of the MIEP of HCMV. Inhibition of this axis therefore, upregulates GFI1 and interferes with the activation of the MIEP and HCMV infection. Immediately after virus entry in virus-infected cells, UV-sensitive virus-associated factors facilitate MIEP activation by promoting the rapid downregulation of GFI1 in both wild-type and NDY1/KDM2B, EZH2 or JARID2 knockdown cells. However, since the levels of GFI1 in the latter cells prior to the infection are significantly higher than in wild-type cells, the HCMV-induced GFI1 degradation fails to downregulate GFI1 to levels permissive for MIEP activation and viral infection. To maintain the silencing of GFI1, the virus also initiates an NDY1/EZH2/JARID2/JMJD3-dependent program, which represses GFI1 throughout the infection cycle. The knockdown of EZH2 may contribute to the repression of the MIEP, also by modulating the accumulation of histone H3K27me3 and H3K4me3 in the immediate early region of HCMV in the first three hours from the start of the viral infection. We conclude that HCMV infection depends on EZH2NDY1/ /JARID2/JMJD3-dependent and independent mechanisms which are activated by the virus and control the expression of GFI1, a transcriptional repressor of the immediate-early region of HCMV. EZH2-dependent mechanisms also control histone modifications in the immediate-early region of HCMV that may contribute to the activation of the MIEP very early in infection.
Results
NDY1/KDM2B and EZH2 are selectively required for the infection of human foreskin fibroblasts by HCMV. The role of histone H3K27 trimethylation
To determine whether histone methylation plays a role in the efficiency of viral infection and replication, human foreskin fibroblasts (HFFs) were transduced with pLKO.1-based lentiviral constructs of shRNAs of NDY1/KDM2B, EZH2, PHF2 and RBP2/JARID1A/KDM5A, or with the empty vector, prior to infection with HCMV. NDY1/KDM2B, PHF2 and JARID1A/RBP2/KDM5A are Jumonji domain-containing histone demethylases that target histone H3K36me2/me1 (NDY1/KDM2B), K9me1 (PHF2) and K4me3/me2/me1 (RBP2) [32], [33]. EZH2 is a SET domain histone methyltransferase that promotes histone H3K27 tri-methylation [27], [28]. It is important to note that H3K27 trimethylation is also promoted by the demethylase NDY1/KDM2B, which upregulates the expression of EZH2 and contributes to its functional activation [30], [34]. Furthermore, NDY1/KDM2B and EZH2 function in concert on a subset of promoters, which cannot be repressed by either of the two acting alone [30]. The results of the experiment revealed that, whereas the knockdown of PHF2 and RBP2 have no effect on the ability of the virus to infect HFFs, the knockdown of NDY1/KDM2B and EZH2 almost completely block the infection (Fig. 1A). Transduction of HFFs with pBabe-puro-based retroviral constructs of the same histone modifying enzymes, or with the empty vector, had no effect on the efficiency of HCMV infection (Fig. 1B).

To determine whether the resistance of shNDY1/KDM2B and shEZH2-transduced cells to HCMV infection indicates a block or a mere delay of the infection, we monitored the virus titer in the supernatants of infected cells every other day for up to 9 days post-infection. The results confirmed that the knockdown of NDY1/KDM2B or EZH2, but not PHF2 or RBP2, block the infection (Fig. 1C).
The preceding experiments revealed that NDY1/KDM2B and EZH2 are both required for HCMV infection and replication, while other histone modifying enzymes are not. Given that NDY1/KDM2B and EZH2 operate in concert to upregulate EZH2 and histone H3K27 trimethylation [30], [34], we hypothesized that it is histone H3K27 trimethylation that is required for efficient infection by HCMV. To address this hypothesis, human foreskin fibroblasts were transduced with a pBabe-puro-based retroviral construct of the H3K27me3 demethylase JMJD3, or with a pLKO.1-based lentiviral shRNA construct of the same enzyme. Cells transduced with these constructs or with the empty vectors, were infected with HCMV and the efficiency of infection was determined qualitatively, as well as quantitatively, using a plaque assay for virus titration. The results showed that whereas shJMJD3 did not interfere with viral infection and replication, JMJD3, which demethylates histone H3K27me3, did (Fig. 1D).
EZH2 is known to form a complex with the prototype Jumonji domain protein JARID2 and to bind chromatin in concert with JARID2 [29]. Through this interaction JARID2 regulates the EZH2 methyltransferase activity. We therefore proceeded to address the role of JARID2 in HCMV infection and replication in HFFs. The experiments in Figure 1D showed that whereas tranduction with a pLKO.1-based lentiviral construct of shJARID2 interferes with HCMV infection and replication, transduction with a pBabe-based construct of JARID2 does not.
If histone H3K27 tri-methylation is required for HCMV infection, as suggested by the preceding experiments, replacement of the endogenous EZH2 with its SET domain mutant, which lacks histone methytransferase activity, should fail to rescue the permissiveness of shEZH2-transduced HFFs to HCMV. To address this hypothesis, we knocked down the endogenous EZH2 with an shRNA that targets sequences in the 3′ UTR of the endogenous EZH2 mRNA, and we replaced it with wild-type exogenous EZH2 or with a catalytically-inactive ΔSET mutant of EZH2. Titration of HCMV in these cells confirmed the hypothesis (Fig. 1E). We conclude that the histone methyltransferase activity of EZH2 is required for infection of human foreskin fibroblasts by HCMV.
NDY1/KDM2B, EZH2 and histone H3K27 tri-methylation are required for the transcriptional activation of the major immediate-early promoter of HCMV and the initiation of HCMV infection
To explore the mechanism by which histone H3K27 tri-methylation controls viral infection and replication, we first examined whether the knockdown of NDY1/KDM2B, EZH2 or JARID2, inhibits viral entry. To this end, HFFs transduced with pLKO.1 or pLKO.1-based shNDY1/KDM2B, shEZH2 or shJARID2 constructs were infected with the recombinant virus UL32-EGFP-HCMV-TB40, which expresses the capsid-associated tegument protein pUL32 (pp150) as a fusion with EGFP (MOI 10 PFU per cell), producing fluorescent virions as well as tegument puncta in the cytoplasm and the nucleus of infected cells [35]. One hour after infection, the cells were fixed and the viral entry was visualized by EGFP fluorescence, which monitors the UL32-EGFP-containing viral particles. Quantitative analysis of the UL32-EGFP showed that the efficiency of viral entry in cells transduced with the empty vector and in cells transduced with the shRNA constructs was similar (Fig. S1A and S1B). In a repeat of the experiment with HCMV AD169, the viral strain used in all other experiments in this study, we examined the efficiency of viral entry by monitoring intracellular pp65 by immunofluorescence. The results confirmed that viral entry is not affected by the knockdown of EZH2, NDY1/KDM2B or JARID2 (Fig. S1C and S1D). Parallel experiments employing quantitative real time PCR to measure HCMV genomic equivalents in nuclear and cytoplasmic lysates of HFFs transduced with the same shRNA constructs and isolated 6 hours after infection, also revealed no differences (Fig. S1E and S1F). The preceding data combined, suggest that H3K27 tri-methylation does not affect viral entry and thus, the resistance to infection caused by the inhibition of H3K27 trimethylation is due to a barrier at a different step of the infection cycle.
To determine whether histone modifying enzymes regulate the activation of the immediate-early gene promoter, cells transduced with shRNAs or expression constructs of these enzymes, or with the corresponding empty vectors, were infected with HCMV and 5 hours later, they were stained for IE1 and counterstained with DAPI. Flow cytometry and fluorescence microscopy of the stained cells illustrated that NDY1/KDM2B and EZH2 are required for HCMV immediate-early gene expression (Fig. 2A and Fig.S2A). The knockdown of PHF2 and RBP2, which are not required for HCMV infection, also had no effect on IE1 promoter activity (Fig. 2A). The results of these experiments suggested that H3K27 trimethylation is required for HCMV immediate-early gene expression. Parallel experiments provided additional support to this conclusion by showing that both the knockdown of JARID2 and the overexpression of the histone H3K27me3 demethylase JMJD3 also inhibit the activation of the HCMV MIEP (Fig. 2B and Fig.S2B).To determine whether blocking histone H3K27 trimethylation inhibits or simply delays the expression of IE1, we knocked down EZH2, or NDY1/KDM2B, in HFFs and we examined the expression of IE1 at multiple time points from the start of the infection. The results demonstrated that these manipulations interfere with the expression of IE1, at all the time points (Fig. 2C). In agreement with these data, treatment of human foreskin fibroblasts with the EZH2 inhibitor 3-Deazaneplanocin A (DZNep) [36]-[38] inhibited both the expression of immediate-early genes and the infection by HCMV (Fig. 2D–F).

In parallel experiments, we monitored the abundance of EGFP-positive cells and the intensity of EGFP fluorescence by fluorescence microscopy, or flow cytometry in HeLa cells transduced with shEZH2 or shNDY1/KDM2B lentiviral constructs, and transfected with a reporter construct expressing EGFP from the MIEP of HCMV. The results showed that the knockdown of either NDY1/KDM2B or EZH2 inhibits the activity of the MIE promoter even in cells not infected with HCMV (Fig.S3). We conclude that H3K27 trimethylation is both necessary and sufficient for the activation of this promoter.
The knockdown of NDY1/KDM2B, EZH2 and JARID2 and the overexpression of JMJD3 selectively upregulate GFI1
Since H3K27me3 normally represses transcription [39], we hypothesized that it may activate the major immediate-early promoter by repressing a transcriptional repressor. To address this hypothesis, we examined whether the knockdown of NDY1/KDM2B, EZH2, or JARID2 and the overexpression of JMJD3 in HFFs, alter the expression of known transcriptional regulators of the MIEP (Fig.3A). Real time RT-PCR revealed that GFI1 is the only MIEP repressor significantly upregulated in these cells (Fig. 3A). Probing western blots of lysates of the same cells with an anti-GFI1 antibody showed that GFI1 is upregulated also at the protein level (Fig. 3A).

GFI1 represses the MIE promoter of HCMV by binding to two sites within the promoter [6] (Fig.3B, Upper panel). The binding of GFI1 to these sites was tested with experiments using foreskin fibroblasts transduced with pLKO.1 or with the pLKO.1-based shNDY1/KDM2B, shEZH2, or shJARID2 constructs, which de-repress GFI1 in these cells (Fig. 3A). These cells were infected with HCMV AD169. ChIP experiments, using cell lysates harvested 1 hour after infection, confirmed the binding of GFI1 to both GFI1 binding sites in the MIE promoter, but not in the MIE coding region and showed that treatments promoting the upregulation of GFI1 by inhibiting H3K27 trimethylation increase the binding (Fig 3B Lower panel). To determine the functional role of GFI1 binding, we mutated both sites by site-directed mutagenesis (AATC mutated to AACT and AAGT, respectively) of an MIEP-EGFP reporter construct. The wild-type and mutant constructs were transfected into HEK 293T cells that had been stably transduced with pLKO1, shEZH2, shJARID2, shNDY1/KDM2B, GFI1, or shGFI1. Analyzing the cells by fluorescence microscopy, revealed that whereas the knockdown of EZH2, JARID2, or NDY1/KDM2B, and the overexpression of GFI1 inhibit the wild-type promoter, they have no effect on the mutant promoter (Fig 3C). These data combined, confirmed that histone H3K27 tri-methylation represses GFI1. Inhibiting H3K27 trimethylation de-represses GFI1, which binds and represses the MIE promoter of HCMV.
The preceding data raised the question of the mechanism by which the tri-methylation of histone H3 at K27 regulates the GFI1 promoter. This question was addressed with ChIP assays designed to measure the relative abundance of H3K27me3 at five promoter sites (from position −1044 bp to position −209 bp), in cells transduced either with the empty lentiviral vector, or with shRNA lentivirus constructs targeting EZH2, NDY1/KDM2B or JARID2. P16INK4a was used as the positive control. The most 5′ of the five promoter sites (site #1, located between −1044 bp and −956 bp), maps within a repressive domain [40]. These experiments revealed that the knockdown of any of these chromatin regulators induced a significant decrease in the abundance of H3K27me3 at this site (Fig. 3D). We conclude that EZH2, NDY1/KDM2B and JARID2 promote histone H3K27 trimethylation within a negative regulatory domain of the GFI1 promoter and may be responsible for the transcriptional repression function previously mapped within this domain [40].
Based on the data in the preceding paragraphs, we conclude that GFI1 is a direct repressor of the HCMV MIEP. However, it is possible that GFI1 may regulate the MIEP by additional indirect mechanisms. One such mechanism is via p21CIP1/WAF1 which is a known target of the GFI1 transcriptional repressor [41]. The repression of GFI1 by NDY1/EZH2/JARID2 may lead to the upregulation of p21CIP/WAF1, which in turn, may inhibit the progression from the G1 phase to the S phase of the cell cycle. Since the accumulation of cells in G1 favors the expression of the IE genes of HCMV [42], [43], GFI1 may regulate the MIEP not only directly, but also indirectly via p21CIP/WAF1. To address this question, HFFs were transduced with pLKO1-based lentiviral constructs of shEZH2 or shNDY1/KDM2B, or with the empty pLKO1 lentiviral vector. Western blots of lysates of these cells harvested before, and at various time points after infection and probed with an anti-p21CIP/WAF1 antibody, revealed that the expression of p21CIP/WAF1 is not affected by the knockdown of either EZH2 or NDY1 (Fig.S4). These data suggest that p21CIP/WAF1 is not involved in the regulation of the MIEP of HCMV by NDY1/EZH2/JARID2.
Earlier studies had shown that the resistance to HCMV caused by the repression of the MIEP can be overcome by infection at a high moi [44]–[47]. Based on this observation, we predicted that HCMV infection of shEZH2, shNDY1, shJARID2, or JMJD3-transduced HFFs at an moi of 5 could overcome their resistance to infection. The results confirmed the prediction (Fig. S5), providing additional support to the hypothesis that the HCMV phenotype induced by the knockdown or overexpression of these molecules is caused by the repression of the MIEP.
HCMV infection promotes the immediate and rapid downregulation of GFI1. It also promotes the gradual deregulation of EZH2, NDY1/KDM2B, JARID2 and JMJD3
The preceding data showed that by repressing the MIE promoter, GFI1 can block HCMV infection. Based on these data, we hypothesized that HCMV may down-regulate GFI1, to increase the permissiveness of the cells to the incoming virus. Experiments addressing this hypothesis showed that GFI1 is indeed down-regulated rapidly in HCMV-infected cells, both at the RNA and protein levels (Fig. 4A and 4B). The rapid downregulation of GFI1 in HCMV-infected cells suggested that the incoming virus may induce the degradation of both the GFI1 mRNA and protein. This hypothesis was addressed with the experiments in Figure 4B. Monitoring the levels of the GFI1 mRNA in Actinomycin D-treated HFFs by real time RT-PCR, confirmed that virus infection accelerates the degradation of the GFI1 mRNA (Fig. 4C). Similarly, monitoring the levels of the GFI1 protein in MG132-treated cells by western blotting showed that MG132 stabilizes the expression of GFI1 in virus-infected cells (Fig. 4D). These data suggested that the incoming virus renders the cells permissive to infection by rapidly degrading GFI1 both at the RNA and the protein levels, and that protein degradation is mediated through the ubiquitin-proteasome pathway. However, since inhibition of the proteasome is also known to stabilize hDaxx [17], [44], it is possible that MG132 may block the degradation of GFI1 by inhibiting the degradation of hDaxx and viral infection.

The fact that both the GFI1 RNA and the GFI1 protein were degraded rapidly after virus infection, suggests that both processes are initiated by factors entering the cells with the incoming virus. To determine the nature of these factors, we infected the cells with UV-irradiated virus and we examined the expression of GFI1 at 0, 0.5, 1 and 2.5 hours from the start of the exposure to the virus. The results showed that the UV-irradiated virus induced the degradation of hDaxx as expected [44] (FigS6A), but failed to downregulate GFI1 at both the RNA and protein levels (Fig.S6B and S6C). The downregulation of the GFI1 mRNA may be mediated by virion-associated UV-sensitive non-coding RNAs that target GFI1. The downregulation of the GFI1 protein may be mediated by another UV-sensitive virion-associated molecule, whose nature remains to be determined. However, we have not formally excluded that a de novo expressed protein may be responsible for the phenotype.
The regulation of GFI1 by EZH2, NDY1/KDM2B or JARID2, prompted us to investigate whether the HCMV-induced GFI1 downregulation is EZH2/NDY1/JARID2-dependent. To address this question, HFFs were transduced with the lentiviral vector pLKO.1, or with pLKO.1-based constructs of shNDY1, shEZH2, or shJARID2, and 48 hours later, they were infected with HCMV. Western blotting of uninfected and HCMV-infected cell lysates, harvested two hours after the infection, revealed that the downregulation of GFI1 was not prevented by the knockdown of any of these chromatin regulators. However, since the starting levels of GFI1 prior to the infection in shEZH2 shNDY1, or shJARID2-transduced cells were significantly higher than in control cells, GFI1 continued to be expressed, even after the infection (Fig.4E). Given the inhibitory effects of the NDY1/EZH2/JARID2 knockdown on the activity of the MIEP, we conclude that the levels of GFI1 detected in these cells are sufficient to repress the promoter.
Although the rapid downregulation of GFI1 in the initial stages of viral infection may be independent of the NDY1/EZH2/JARID2 axis however, the initial downregulation of GFI1 may be maintained via the activation of this axis throughout the infection cycle. Real time RT-PCR indeed showed that the mRNA levels of NDY1/KDM2B, EZH2 and JARID2 increase gradually, while the RNA levels of JMJD3 decrease in the course of the viral infection (Fig.4F). UV-irradiated virus, which cannot establish a productive infection, had no effect on the expression of these epigenetic regulators (Fig.S7).
The expression of the HCMV immediate-early genes and HCMV infection depend on the EZH2-mediated repression of GFI1, but not GFI1B
To determine whether HCMV infection depends on the down-regulation of GFI1, human foreskin fibroblasts were transduced with pBabe-puro-based retroviral constructs of GFI1 or GFI1B, a GFI1-related gene, also encoding a SNAG domain-containing transcriptional repressor, or with the empty vector. The transduced cells were infected with HCMV. Infection was monitored by light microscopy 5 days later (Fig.S8) and the progeny virus was harvested 12 days later and titrated by a plaque assay (Fig.S8). Alternatively, transduced cells were infected with HCMV and they were stained for IE1 expression 5 hours post-infection. Stained cells were analyzed by flow cytometry (Fig.S8). The results revealed that GFI1 inhibits the activity of the MIEP and HCMV infection as expected, while GFI1B does not. Since GFI1B also represses p21Cip/WAF1 [48], these results provide additional support to the conclusion that p21CIP/WAF1 is not involved in the regulation of the MIEP of HCMV by NDY1/EZH2/JARID2/GFI1 (see above).
In parallel experiments, we used a TRIPZ-based doxycycline-inducible shRNA construct of EZH2 and a pLKO1-based constitutive shRNA construct of GFI1 to knock down these genes in HFFs, separately or in combination. The knockdown of EZH2 and GFI1 were confirmed by western blotting of lysates harvested from these cells prior to HCMV infection, before and after treatment with doxycycline (Fig.5A1). Infection of these cells with HCMV showed that whereas the knockdown of EZH2 inhibits IE1 expression (Fig. 5A2 lanes 1 and 3) and permissiveness to infection (Fig5A3, first and third bar), the knockdown of GFI1 does not affect either (Fig.5A2, lanes 1 and 2 and5A3, bars 1 and 2). However, the knockdown of GFI1, partially restored IE1 expression and permissiveness to viral infection in cells in which EZH2 was also knocked down (Fig.5A2, lanes 3 and 4 and Fig5A3, bars 3 and 4). We conclude that EZH2 contributes to HCMV infection by inhibiting the expression of GFI1. The fact that the restoration of IE1 expression and permissiveness to viral infection were only partial, suggests that EZH2 may have additional GFI1-independet effects on IE1 expression.

EZH2 regulates histone H3K27 and H3K4 trimethylation in the IE region of HCMV in the immediate-early stages of HCMV infection
Next we examined the effects of the EZH2 knockdown on histone H3K27 and H3K4 trimethylation in the enhancer, the cis repression sequence (crs) and intron 1 in the immediate-early region of HCMV (Fig.6). The peak of H3K27 trimethylation in the enhancer and in intron 1 in HFFs transduced with a lentiviral shControl construct was observed at 1.5 hours from the start of the exposure to the virus and declined to very low levels at the 3 hour time point. H3K27 trimethylation in the crs increased more rapidly (0.5 hours) and remained high throughout the observation period. Knocking down EZH2 delayed H3K27 trimethylation in the crs, and perhaps in intron 1, with low levels of H3K27me3 at the 0.5 hour time point, and in the enhancer, with low levels of H3K27me3 at the 1.5 hour time point. More important, in the shEZH2 cells H3K27 trimethylation remained high at the three hour time point in all three sites. These changes in the abundance of H3K27me3 were associated with parallel changes in the abundance of H3K4me3. In HFFs transduced with the shControl construct, the abundance of H3K4me3 increased in all three sites throughout the three hour observation period. However, in the shEZH2-transduced cells, its abundance in the enhancer region increased more slowly than in control cells. Moreover, in the crs and the intron 1 regions its abundance declined at the three hour time point, with the decline in intron 1, being dramatic. These data are consistent with the chromatin modification data in the IE region of the murine CMV in the immediate-early stage of the infection [49]. In addition, they are consistent with earlier observations suggesting an initial cell-mediated MIEP repression that precedes viral gene expression in HCMV-infected cells [16]. More important, these data suggest that H3K27 and H3K4 trimethylation in the regulatory elements of the IE region of HCMV are co-ordinatelly regulated. However, the rules of their co-ordinate regulation are not yet known and they will require additional work to be determined. One additional question that remains is how the regulatory elements of the IE region of HCMV undergo delayed H3K27 trimethylation, when EZH2 is knocked down. We hypothesize that this may be happening because of residual EZH2 activity, remaining after the EZH2 knockdown. Alternatively, it may be mediated by EZH1. This question will be addressed in future studies.

Discussion
Data presented in this report, showed that NDY1/KDM2B, EZH2 and JARID2 synergize to repress GFI1, a SNAG domain-containing transcriptional repressor [6]–[8]. In addition, they confirmed that GFI1 represses the MIEP of HCMV by binding to two sites, 159–163 and 105–109 base pairs upstream of the transcription start site, and that the knockdown of NDY1/KDM2B, EZH2 or JARID2 results in the upregulation of GFI1 and in the GFI1-dependent dramatic repression of the MIEP of HCMV. During HCMV infection, the GFI1 protein and mRNA are downregulated rapidly, most likely via degradation, both in control cells and in cells in which NDY1/KDM2B, EZH2 or JARID2 was knocked down. However, the pre-infection levels of GFI1 in the latter cells are significantly higher than in the control cells, and the degradation is not sufficient to extinguish GFI1 expression, which is required for the establishment of HCMV infection. As a result, cells in which NDY1/KDM2B, EZH2 or JARID2 were knocked down, are resistant to HCMV infection. Following the initial degradation of GFI1, the virus reprograms the epigenetic machinery of the cell, by up-regulating NDY1/KDM2B, EZH2 and JARID2 and by down-regulating the histone H3K27me3 demethylase JMJD3. This reprogramming is expected to maintain the expression of GFI1 at low levels throughout the infection cycle (Fig. 7).

The combination of NDY1/KDM2B, EZH2 and JARID2 promotes histone H3K27 trimethylation, a chromatin mark associated with transcriptional repression [50], [51]. EZH2, the enzyme responsible for histone H3K27 trimethylation, binds JARID2, a non-canonical jumonji domain protein that regulates the EZH2 methyltransferase activity [29]. NDY1/KDM2B, which can be induced by growth factors such as FGF2 [30], binds some EZH2 target genes and demethylates histone H3K36(me2) and H3K36(me1) [34]. The latter promotes EZH2 binding to the same genes and transcriptional repression [32]. In this report, we presented evidence that GFI1, a transcriptional repressor of the major immediate-early promoter of HCMV [6], [52], is one of the genes targeted by the H3K27(me3) methyltransferase EZH2 and its regulators JARID2 and NDY1/KDM2B, as well as the histone H3K27(me3) demethylase JMJD3.
HCMV initiates viral infection by targeting GFI1 via multiple mechanisms. Immediately after exposure to the virus, the GFI1 mRNA and protein are rapidly downregulated, most likely via degradation. The rapid drop in the levels of these molecules immediately after exposure to the virus suggested that they may be degraded by virion-associated factor(s). Their downregulation was UV-sensitive, suggesting that it may be due to degradation by virion-associated nucleic acids [53], [54]. The GFI1 mRNA may be a direct target of virion-associated non-coding RNAs [55], [56]. The GFI1 protein may be degraded via the proteasome, which is known to play an important role in the transcription of the HCMV immediate early genes [57], [58]. However, the mechanism by which virion-associated nucleic acids may regulate the proteasomal degradation of the GFI1 protein remains to be determined. Over the years, the main focus of studies addressing the activation of the proteasome by HCMV is on the tegument protein pp71 [59]. pp71 interacts with hDaxx in PML bodies to inhibit hDaxx-mediated silencing by promoting its degradation [60]-[63]. However, GFI1 cannot be a target of pp71, because the latter is not UV-sensitive.
The rapid downregulation of the GFI1 mRNA and protein, which occurs immediately after exposure to the virus, is followed by epigenetic reprogramming, which is expected to downregulate GFI1 throughout the infection cycle. Thus, in the early stages of infection, HCMV employs mechanisms that have not yet been determined to up-regulate NDY1/KDM2B, EZH2 and JARID2 and to down-regulate JMJD3. These chromatin modifiers target an inhibitory domain within the GFI1 promoter and enhance the trimethylation of histone H3 at K27 (Fig.3D). It is not yet known how these chromatin regulators are targeted to the GFI1 promoter. Potentially, they may function as GFI1 co-repressors and they may be targeted to the GFI1 promoter by GFI1 itself. This is suggested by earlier findings showing that GFI1 represses its own promoter [64] and by the observation that within the inhibitory domain of the human GFI1 promoter [40], there is one GFI1 binding site, containing the characteristic AATC core.
The effects of the knockdown of EZH2, NDY1/KDM2B and JARID2, along with the effects of the overexpression of JMJD3, on the activity of the MIEP and viral infection were counterintuitive. One would expect that these genetic manipulations would lead to a decrease in H3K27 trimethylation, both globally and regionally in the MIEP, and that this would result in an increase in MIEP activity. The fact that we see the opposite would suggest that either these genetic manipulations fail to alter the balance of repressive and activating epigenetic marks in the MIEP, or that tipping the balance toward the activating marks is not sufficient to override the effects of the GFI1 repressor. Of course, it is also possible that changes in the pattern of MIEP-associated chromatin modifications, induced by these genetic manipulations, promote the binding of GFI1, facilitating the MIEP repression. To address these questions, we surveyed the effects of the EZH2 knockdown on the abundance of H3K27me3 (a repressive mark) and H3K4me3 (an activating mark) in the enhancer, crs and intron 1 in the IE region of HCMV in the first three hours from the start of the infection. The results showed a delay in H3K27 trimethylation. The slow kinetics of this process resulted in an increase in the abundance of H3K27me3 in the IE enhancer and intron 1 at the three hour time point, when the abundance H3K27me3 normally decreases. The rapid increase in the abundance of H3K27me3, which we observed in HCMV-infected control cells in the very early stages of the infection, is consistent with the results of earlier studies showing that MIEP activation during infection by murine CMV and HCMV is preceded by an increase in the abundance of repressive histone marks. Our data also showed that the increase in the abundance of in H3K27me3 at the three hour time point in shEZH2-transduced cells is paralleled by a significant decrease in the abundance of H3K4me3,.suggesting that repressive and activating marks are co-ordinately regulated. The rules of this coordinate regulation and the potential involvement of these epigenetic modifications in the recruitment of GFI1 remain to be determined.
Overall, the data presented in this report identify a novel pathway of epigenetic regulation of cellular gene expression that regulates the expression of HCMV immediate-early genes and viral infection. Inhibition of the pathway may have preventive or therapeutic applications in viral infection, while selective activation of the pathway may have therapeutic applications in cancer.
Materials and Methods
Cells, viruses and small molecules
Human Foreskin Fibroblasts (HFFs) were used for HCMV infection (kind gift from Dimitrios Iliopoulos, Harvard Medical School) and HEK 293T cells were transfected to package lentivirus and retrovirus constructs. HEK 293T cells or HELA cells were used for transfection of HCMV MIEP reporter constructs in experiments addressing the regulation of the HCMV major immediate-early promoter by chromatin modifying enzymes. All cell lines were maintained in Dulbecco's modified Eagle's minimal essential medium (DMEM) supplemented with 10% fetal bovine serum, penicillin/streptomycin, L-glutamine and non-essential amino acids.
The wild-type laboratory strain of HCMV we used was the AD169 strain. The recombinant UL32-EGFP-HCMV-TB40 virus, which expresses the capsid-associated tegument protein pUL32 (pp150), fused to EGFP [35] was used for some experiments addressing viral entry.
To determine the role of the EZH2 enzymatic activity on immediate-early gene transcription and HCMV infection, virus-infected cells were treated with the EZH2 inhibitor 3-deazaneplanocin A (DZNep) (Cayman Chemical Company, MI), at the final concentration of 10 µM. DZNep was dissolved in dimethyl sulfoxide (DMSO).
HCMV infection and titration and viral entry
To infect HFFs with HCMV, cell monolayers were incubated with the virus at a multiplicity of infection (MOI) of 0.5 PFU/cell, or at variable multiplicities in virus titration experiments.Unless otherwise specified, the cells were exposed to the virus for 2 hours at 37°C. Subsequently, the virus was removed and replaced with fresh medium. Plaque assays for virus titration were performed on HFFs according to standard protocols [65]. To monitor the growth of HCMV in HFFs transduced with shEZH2, shNDY1/KDM2B, shPHF2 and shRBP2 lentiviral constructs, cells were infected with HCMV AD169 at an MOI of 0.5 PFU/cell. Viral supernatants harvested from these cultures every two days for 9 days were titrated, using plaque assays.
To measure the efficiency of viral entry, HFFs transduced with pLKO.1-based lentiviral constructs of shEZH2, shNDY1/KDM2B, shJARID2, or with the empty vector, were infected with the UL32-EGFP-HCMV-TB40 recombinant virus at an MOI of 10 PFU/cell, as previously described [66]. One hour after infection, the cells were fixed and viral entry was visualized by monitoring intracellular EGFP fluorescence via fluorescence microscopy. Fluorescence intensities of UL32-EGFP were calculated with the Zeiss LSM image examiner software. To correct for background fluorescence, we deduced from the fluorescence of infected cells the fluorescence of adjacent non-infected cells. Alternatively, viral entry was monitored by real-time PCR of viral DNA in cell lysates harvested at six hours from the start of the exposure to the wild-type HCMV AD169 virus [45].
Cloning and site-directed mutagenesis
The retrovirus and lentivirus constructs we used are listed in Table 1. Human JARID2 was cloned into the pLenti-CMV-puro-DEST vector (Addgene, cat no 17452) using the LR Clonase II Plus enzyme mix (Invitrogen, cat no 12538120) according to the manufacturer's instructions. Human HA-PHF2 was cloned into the EcoRI site of pBABEpuro, and human FLAG-JMJD3-HA was cloned between the BamHI and XhoI sites of the same vector. The rest of the lentiviral and retroviral constructs were either purchased or kindly provided by others (Table 1). shEZH2 was induced in TRIPZ-shEZH2-transduced cells with Doxycycline (1 µg/ml)

Jarid2 cDNA was PCR-amplified using the pCMV-SPORT6-JARID2 (Open Biosystems, cat. no. MHS1010-99622028) as template, cloned in the pENTR/TOPO vector (Invitrogen, cat no K2400-20). The sequence was verified and was recombined to the pLentipuro vector (Addgene, cat no. 17452) using the Gateway LR Clonase II Plus kit (Invitrogen, cat no. 12538-120).
To determine the effects of NDY1/KDM2B, EZH2 JARID2 and GFI1 on the activity of the HCMV MIEP in the absence of viral infection, a MIEP-EGFP reporter construct (pEGFP-C1) (Clontech) was transfected into HEK 293T cells or their derivatives in which NDY1/KDM2B, EZH2 or JARID2 were knocked down or GFI1 was overexpressed and the expression of EGFP was monitored by fluorescence microscopy or flow cytometry. The same cells were also transfected with a derivative of pEGFP-C1 in which the two GFI1 binding sites in the MIEP [7] were inactivated by point mutation. The mutant construct was generated by site-directed mutagenesis, as previously described [67], using primers: CMVmut1 : 5'-GGGTGGAGACTTGGAAAGTCCCGTGAGTCAAACCG-3′ and CMVmut2 : 5'-ATTTTGGAAAGTCCCGTTAGTTTTGGTGCCAAAACAAAC-3′. All products of PCR mutagenesis were sequenced after cloning, to ensure that no additional mutations were generated.
Lentiviral and retroviral packaging and transduction
HEK 293T cells were transiently co-transfected with retroviral constructs and the amphotropic packaging construct (Ampho-pac). Alternatively, HEK 293T cells were transiently co-transfected with lentiviral constructs and pCMV/VSV-G (where VSV-G is vesicular stomatitis virus protein G) and pCMV-dR8.2 dvpr. Transfection was carried out using Fugene 6 (Roche Applied Science).
To transduce HFFs with the packaged viruses, early passage cells were incubated with viral supernatants in the presence of 5 µg/ml polybrene (Sigma-Aldrich, Deisenhofen, Germany) for 24 hours. Forty-eight hours later, cells were selected with puromycin (2 µg/ml) or hygromycin B (200 µg/ml). Cells infected with multiple retrovirus or lentivirus constructs, were selected for these constructs sequentially.
Cell lysis and western blotting
Cells were washed twice in ice-cold PBS and they were lysed in Triton X-100 lysis buffer [50 mM Tris (pH 7.5), 200 mM NaCl, 1% Triton X-100, 0.1% SDS, 10 mM Na3VO4, 50 mMNaF, 1 mM β-glycerophosphate, 1 mM sodium pyrophosphate, 1 mM EDTA, 1 mM EGTA, and 1 mM PMSF supplemented with a mixture of protease inhibitors]. The lysates were sonicated in a Misonix 3000 sonicator for 5 seconds at power level 1.5, and they were centrifuged for 20 min at 13,000×g. Western blots of the supernatants (soluble whole-cell lysates) were probed with the EZH2 rabbit monoclonal antibody (no. 4905, Cell Signaling), the GFI1 mouse monoclonal antibody (2.5D17, Sigma), the KDM2B goat polyclonal antibody (sc-69477, Santa Cruz), the p21WAF1/CIP1 human monoclonal antibody (no. 05-345, Cell Signaling), the RBP2 (JARID1B) rabbit polyclonal antibody (no. ABE239, Millipore), the PHF2 rabbit polyclonal antibody (3497, Cell Singaling), the JMJD3 rabbit polyclonal antibody (no. 3457, Cell Singaling), the JARID2 rabbit polyclonal antibody (ab48137, Abcam), the HA-Tag mouse monoclonal antibody (no. 2367, Cell Signaling), the myc-Tag rabbit monoclonal antibody (no. 2278, Cell Signaling), the pp71 (2H10-9) antibody or the IE1 mouse monoclonal antibody (BS500) [68]. Anti-mouse as well as anti-rabbit horseradish peroxidase-conjugated secondary antibodies, obtained from Sigma, were diluted in 5% milk in TBS-T and incubated with the blots for 1 h at room temperature. The bound secondary antibodies were detected with ECL-plus detection reagent (Amersham Biosciences) or the ECL SuperSignal (Pierce). Digital images of the proteins were acquired using the LAS-4000 luminescent image analyzer (Fujifilm Life Science).
Immunofluorescence
To monitor the activation of the MIE promoter, 0.8×105 HFFs were plated on coverslips and they were infected with HCMV [69]. Five hours later, they were fixed and immunostained with an anti-IE1 monoclonal antibody (BS500) as previously described [65]. Anti-mouse as well as anti-rabbit Alexa 488-conjugated secondary antibodies were purchased from Molecular Probes (Invitrogen). The number of IE1-positive cells/coverslip was determined by epifluorescence microscopy. Each experiment was performed in triplicate.
Flow cytometry
HFFs cultured in 12-well plates were infected with HCMV AD169. HELA or HEK 293T cells, also seeded in 12-well plates, were transfected with pEGFP-C2 (Clontech) using Fugene 6 (Roche Applied Science). Infected and transfected cells were harvested, using a cell dissociation buffer (Molecular Probes). Harvested cells were fixed in paraformaldehyde (3% vol/vol in PBS). The HCMV infected cells were first permeabilized with 0.1% saponin in PBS, also supplemented with 2% calf serum, and then washed and resuspended in 100 µl of the same buffer, containing a 1/100 dilution of a mouse anti-IE1 antibody (BS500) [68] Following incubation with the antibody at room temperature for 1 h, the cells were washed twice and then incubated with a fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse secondary antibody (Sigma) (dilution 1∶1000) for 1 h. Transfected HELA cells were stored in PBS supplemented with 2% calf serum, after they were fixed. Virus and mock-infected, as well as transfected and non-transfected samples, were analyzed on a CyAn LX High Performance Flow Cytometer.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using a Chromatin Immunoprecipitation assay kit (Millipore, cat no. 17-295). Chromatin cross-linking was achieved via a 10 minute treatment of nuclear extracts with 1% formaldehyde at 37°C. Cross-linked lysates were sonicated to shear the DNA to an average length of 300 to 1000 base pairs. Following sonication, the lysates were pre-cleared via incubation with a 50% slurry of salmon sperm DNA/Protein A Agarose for 30 minutes. The pre-cleared supernatants were incubated with the primary antibodies anti-H3K27me3 (no. 9756; Cell Signaling), anti-H3K4me3 (Abcam ab8580) and total anti-H3 (Abcam ab1791) (1∶50 dilution) overnight and with salmon sperm DNA/Protein A Agarose beads at 4°C for 1 h. Following multiple washes, the DNA-protein complexes were eluted and the DNA was recovered by reversing the cross-linking with NaCl and proteinase K. The DNA was then extracted using the Qiaquick PCR Purification Kit (Qiagen, cat. no 28106) and it was analyzed by SYBR-Green real-time qPCR, along with the input DNA. The primer sets used to amplify the GFI1 and the p16Ink4a loci as well as the HCMV MIEP are listed in the Table 2.

Real-time RT-PCR
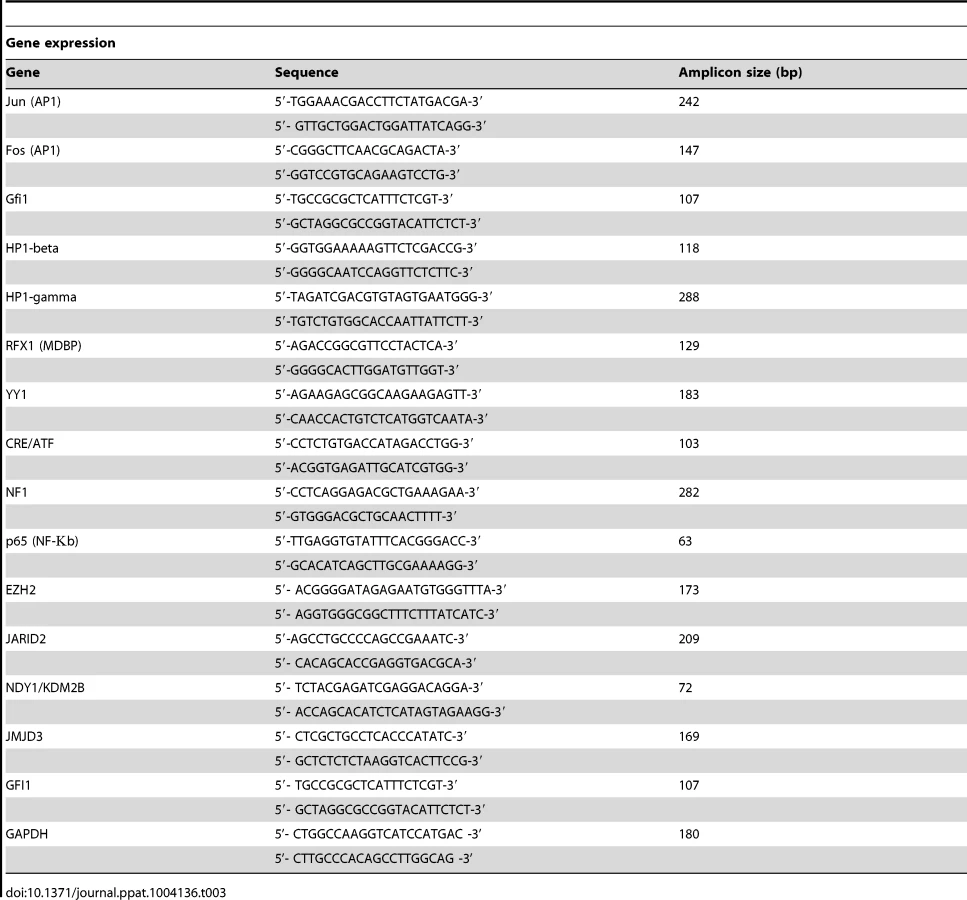
Total cell RNA was isolated, using Trizol (Invitrogen). cDNA was synthesized from 1.0 µg of total RNA, using oligo-dT priming and the Retroscript reverse transcription kit (Ambion, cat no. AM1710). The genes analyzed and the primers used are listed in Table 3. Real-time PCR was performed in triplicate using the Universal SYBR Green PCR master mix kit (Exiqon) and a 7500 Real-Time System (Applied Biosystems). mRNA levels were normalized to GAPDH, which was used as an internal control. All data are from 3 independent experiments, each performed in triplicate.
Cell fractionation and HCMV qPCR
Nuclear and cytoplasmic fractions were isolated from HFFs cells using the Nuclear/Cytosolic Fractionation kit (Cat No AKR-171, Cell Biolabs, Inc.) according to the manufacturer's instructions. Purified DNA from each fraction was amplified by real-time PCR and HCMV genomes were quantified using the CMV Real-TM Quant kit (Cat No V7-100/2FRT, Sacacce).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BoeckhM, GeballeAP (2011) Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest 121 : 1673–1680.
2. SinzgerC, GrefteA, PlachterB, GouwAS, TheTH, et al. (1995) Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol 76 (Pt 4): 741–750.
3. SinclairJ (2008) Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J Clin Virol 41 : 180–185.
4. StreblowDN, NelsonJA (2003) Models of HCMV latency and reactivation. Trends Microbiol 11 : 293–295.
5. CastilloJP, KowalikTF (2002) Human cytomegalovirus immediate early proteins and cell growth control. Gene 290 : 19–34.
6. Zweidler-MckayPA, GrimesHL, FlubacherMM, TsichlisPN (1996) Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol 16 : 4024–4034.
7. GrimesHL, ChanTO, Zweidler-McKayPA, TongB, TsichlisPN (1996) The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol 16 : 6263–6272.
8. GilksCB, BearSE, GrimesHL, TsichlisPN (1993) Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol 13 : 1759–1768.
9. van der MeerLT, JansenJH, van der ReijdenBA (2010) Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia 24 : 1834–1843.
10. PhelanJD, ShroyerNF, CookT, GebeleinB, GrimesHL (2010) Gfi1-cells and circuits: unraveling transcriptional networks of development and disease. Curr Opin Hematol 17 : 300–307.
11. Sharif-AskariE, VassenL, KosanC, KhandanpourC, GaudreauMC, et al. (2010) Zinc finger protein Gfi1 controls the endotoxin-mediated Toll-like receptor inflammatory response by antagonizing NF-kappaB p65. Mol Cell Biol 30 : 3929–3942.
12. SalequeS, KimJ, RookeHM, OrkinSH (2007) Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell 27 : 562–572.
13. SinclairJH, BaillieJ, BryantLA, Taylor-WiedemanJA, SissonsJG (1992) Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J Gen Virol 73 (Pt 2): 433–435.
14. MurphyJC, FischleW, VerdinE, SinclairJH (2002) Control of cytomegalovirus lytic gene expression by histone acetylation. Embo J 21 : 1112–1120.
15. SinclairJ (2010) Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta 1799 : 286–295.
16. GrovesIJ, ReevesMB, SinclairJH (2009) Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic 'pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 90 : 2364–2374.
17. SaffertRT, KalejtaRF (2007) Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81 : 9109–9120.
18. YeeLF, LinPL, StinskiMF (2007) Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 363 : 174–188.
19. ReevesMB, LehnerPJ, SissonsJG, SinclairJH (2005) An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86 : 2949–2954.
20. LusserA (2002) Acetylated, methylated, remodeled: chromatin states for gene regulation. Curr Opin Plant Biol 5 : 437–443.
21. IoudinkovaE, ArcangelettiMC, RynditchA, De ContoF, MottaF, et al. (2006) Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384 : 120–128.
22. ReevesMB, SinclairJH (2010) Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91 : 599–604.
23. ReevesMB, MacAryPA, LehnerPJ, SissonsJG, SinclairJH (2005) Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102 : 4140–4145.
24. NitzscheA, PaulusC, NevelsM (2008) Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol 82 : 11167–11180.
25. Cuevas-BennettC, ShenkT (2008) Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol 82 : 9525–9536.
26. SuganumaT, WorkmanJL (2011) Signals and combinatorial functions of histone modifications. Annu Rev Biochem 80 : 473–499.
27. MullerJ, HartCM, FrancisNJ, VargasML, SenguptaA, et al. (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111 : 197–208.
28. KuzmichevA, NishiokaK, Erdjument-BromageH, TempstP, ReinbergD (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16 : 2893–2905.
29. HerzHM, ShilatifardA (2010) The JARID2-PRC2 duality. Genes Dev 24 : 857–861.
30. KottakisF, PolytarchouC, FoltopoulouP, SanidasI, KampranisSC, et al. (2011) FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell 43 : 285–298.
31. ShiY, LanF, MatsonC, MulliganP, WhetstineJR, et al. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119 : 941–953.
32. KampranisSC, TsichlisPN (2009) Histone demethylases and cancer. Adv Cancer Res 102 : 103–169.
33. WenH, LiJ, SongT, LuM, KanPY, et al. (2010) Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J Biol Chem 285 : 9322–9326.
34. TzatsosA, PfauR, KampranisSC, TsichlisPN (2009) Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci U S A 106 : 2641–2646.
35. SampaioKL, CavignacY, StierhofYD, SinzgerC (2005) Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol 79 : 2754–2767.
36. SnoeckR, AndreiG, NeytsJ, ScholsD, CoolsM, et al. (1993) Inhibitory activity of S-adenosylhomocysteine hydrolase inhibitors against human cytomegalovirus replication. Antiviral Res 21 : 197–216.
37. TanJ, YangX, ZhuangL, JiangX, ChenW, et al. (2007) Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 21 : 1050–1063.
38. De ClercqE (1990) Selective virus inhibitors. Microbiologica 13 : 165–178.
39. SchonrockN, BouveretR, LeroyO, BorghiL, KohlerC, et al. (2006) Polycomb-group proteins repress the floral activator AGL19 in the FLC-independent vernalization pathway. Genes Dev 20 : 1667–1678.
40. LiuS, CowellJK (2000) Cloning and characterization of the TATA-less promoter from the human GFI1 proto-oncogene. Ann Hum Genet 64 : 83–86.
41. DuanZ, ZarebskiA, Montoya-DurangoD, GrimesHL, HorwitzM (2005) Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol 25 : 10338–10351.
42. SalvantBS, FortunatoEA, SpectorDH (1998) Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72 : 3729–3741.
43. FortunatoEA, SanchezV, YenJY, SpectorDH (2002) Infection of cells with human cytomegalovirus during S phase results in a blockade to immediate-early gene expression that can be overcome by inhibition of the proteasome. J Virol 76 : 5369–5379.
44. SaffertRT, KalejtaRF (2006) Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80 : 3863–3871.
45. TavalaiN, PapiorP, RechterS, LeisM, StammingerT (2006) Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol 80 : 8006–8018.
46. CantrellSR, BresnahanWA (2006) Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J Virol 80 : 6188–6191.
47. WoodhallDL, GrovesIJ, ReevesMB, WilkinsonG, SinclairJH (2006) Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem 281 : 37652–37660.
48. TongB, GrimesHL, YangTY, BearSE, QinZ, et al. (1998) The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol Cell Biol 18 : 2462–2473.
49. AbrahamCG, KuleszaCA (2012) Polycomb repressive complex 2 targets murine cytomegalovirus chromatin for modification and associates with viral replication centers. PLoS One 7: e29410.
50. CaoR, WangH, HeJ, Erdjument-BromageH, TempstP, et al. (2008) Role of hPHF1 in H3K27 methylation and Hox gene silencing. Mol Cell Biol 28 : 1862–1872.
51. NekrasovM, KlymenkoT, FratermanS, PappB, OktabaK, et al. (2007) Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J 26 : 4078–4088.
52. White EA, Spector DH (2007) Early viral gene expression and function. In: Arvin A C-FG, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editor. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press.
53. BresnahanWA, ShenkT (2000) A subset of viral transcripts packaged within human cytomegalovirus particles. Science 288 : 2373–2376.
54. GreijerAE, DekkersCA, MiddeldorpJM (2000) Human cytomegalovirus virions differentially incorporate viral and host cell RNA during the assembly process. J Virol 74 : 9078–9082.
55. SarcinellaE, BrownM, TellierR, PetricM, MazzulliT (2004) Detection of RNA in purified cytomegalovirus virions. Virus Res 104 : 129–137.
56. TerhuneSS, SchroerJ, ShenkT (2004) RNAs are packaged into human cytomegalovirus virions in proportion to their intracellular concentration. J Virol 78 : 10390–10398.
57. ProschS, PriemerC, HoflichC, LiebenthafC, BabelN, et al. (2003) Proteasome inhibitors: a novel tool to suppress human cytomegalovirus replication and virus-induced immune modulation. Antivir Ther 8 : 555–567.
58. TranK, MahrJA, SpectorDH (2010) Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J Virol 84 : 3079–3093.
59. BaldickCJJr, MarchiniA, PattersonCE, ShenkT (1997) Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J Virol 71 : 4400–4408.
60. HofmannH, SindreH, StammingerT (2002) Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J Virol 76 : 5769–5783.
61. CantrellSR, BresnahanWA (2005) Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol 79 : 7792–7802.
62. HwangJ, KalejtaRF (2007) Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367 : 334–338.
63. HwangJ, KalejtaRF (2009) Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J Virol 83 : 6591–6598.
64. DoanLL, PorterSD, DuanZ, FlubacherMM, MontoyaD, et al. (2004) Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res 32 : 2508–2519.
65. DimitropoulouP, CaswellR, McSharryBP, GreavesRF, SpandidosDA, et al. (2010) Differential relocation and stability of PML-body components during productive human cytomegalovirus infection: detailed characterization by live-cell imaging. Eur J Cell Biol 89 : 757–768.
66. VanarsdallAL, WisnerTW, LeiH, KazlauskasA, JohnsonDC (2012) PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog 8: e1002905.
67. IgneaC, TrikkaFA, KourtzelisI, ArgiriouA, KanellisAK, et al. (2012) Positive genetic interactors of HMG2 identify a new set of genetic perturbations for improving sesquiterpene production in Saccharomyces cerevisiae. Microb Cell Fact 11 : 162.
68. PlachterB, BrittW, VornhagenR, StammingerT, JahnG (1993) Analysis of proteins encoded by IE regions 1 and 2 of human cytomegalovirus using monoclonal antibodies generated against recombinant antigens. Virology 193 : 642–652.
69. EverettRD, SourvinosG, OrrA (2003) Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J Virol 77 : 3680–3689.
70. PasiniD, HansenKH, ChristensenJ, AggerK, CloosPA, et al. (2008) Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev 22 : 1345–1355.
71. Montoya-DurangoDE, VeluCS, KazanjianA, RojasME, JayCM, et al. (2008) Ajuba functions as a histone deacetylase-dependent co-repressor for autoregulation of the growth factor-independent-1 transcription factor. J Biol Chem 283 : 32056–32065.
72. AggerK, CloosPA, RudkjaerL, WilliamsK, AndersenG, et al. (2009) The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene - and stress-induced senescence. Genes Dev 23 : 1171–1176.
73. CarettiG, Di PadovaM, MicalesB, LyonsGE, SartorelliV (2004) The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18 : 2627–2638.
74. KloseRJ, YanQ, TothovaZ, YamaneK, Erdjument-BromageH, et al. (2007) The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128 : 889–900.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases