
Disruption of an Membrane Protein Causes a Magnesium-dependent Cell Division Defect and Failure to Persist in Mice
The success of Mycobacterium tuberculosis (Mtb) as a human pathogen is due to ability to persist in chronic infection, despite a robust adaptive immune response by the host. The mechanisms by which Mtb achieves this are, however, poorly understood. Here we show that a novel integral membrane protein, Rv0955/PerM, is essential for Mtb persistence during chronic mouse infection. The perM mutant required increased magnesium compared to wild type Mtb for replication and survival in culture and elongated in media with reduced magnesium concentration. Transcriptomic, electron microscopy and live cell imaging approaches provided evidence that PerM is involved in cell division. The survival defects of the perM mutant in reduced magnesium and during chronic mouse infection are consistent with the hypothesis that magnesium deprivation constitutes an IFN-γ dependent host defense strategy. This work also has potential clinical implications, as disruption of PerM renders Mtb susceptible to β-lactam antibiotics, which are commonly used to treat non-mycobacterial infections.
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004645
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004645
Summary
The success of Mycobacterium tuberculosis (Mtb) as a human pathogen is due to ability to persist in chronic infection, despite a robust adaptive immune response by the host. The mechanisms by which Mtb achieves this are, however, poorly understood. Here we show that a novel integral membrane protein, Rv0955/PerM, is essential for Mtb persistence during chronic mouse infection. The perM mutant required increased magnesium compared to wild type Mtb for replication and survival in culture and elongated in media with reduced magnesium concentration. Transcriptomic, electron microscopy and live cell imaging approaches provided evidence that PerM is involved in cell division. The survival defects of the perM mutant in reduced magnesium and during chronic mouse infection are consistent with the hypothesis that magnesium deprivation constitutes an IFN-γ dependent host defense strategy. This work also has potential clinical implications, as disruption of PerM renders Mtb susceptible to β-lactam antibiotics, which are commonly used to treat non-mycobacterial infections.
Introduction
With an estimated one-third of the world’s population latently infected with Mycobacterium tuberculosis (Mtb), the question remains: how is this pathogen able to persist in vivo? In the mouse model, Mtb infection is characterized by an acute phase of logarithmic bacterial growth lasting approximately three weeks, followed by a plateau in bacterial burden, persisting as a chronic infection. The transition from acute to chronic infection—from logarithmic bacterial growth to stable bacterial counts—results from the onset of the adaptive immune response and activation of host macrophages by CD4+ T cell-derived IFN-γ [1,2]. IFN-γ enhances the antimicrobial capacity of macrophages by numerous mechanisms including promotion of phagosome maturation and acidification via induction of the GTPase Irgm1 and production of reactive nitrogen and oxygen species mediated by nitric oxide synthase and phagocyte oxidase [3–6]. However, IFN-γ induces hundreds of genes in macrophages [7] and the array of environmental modifications occurring within these macrophages and leading to control of Mtb growth is not entirely understood.
Mtb persistence mutants (per mutants) are a unique class of strains that are competent for replication during acute infection, but attenuated during chronic infection [8]. Several previously identified per mutants provide information about the processes required for survival in the activated macrophage following the onset of adaptive immunity. For example, a per phenotype was observed for an Mtb mutant lacking isocitrate lyase-1, an enzyme involved in the glyoxylate shunt and methylcitrate cycle, as well as a mutant lacking the cholesterol transporter Mce4, indicating that cholesterol and fatty acids are carbon sources required by Mtb to survive during chronic infection [9,10].
Macrophage activation promotes phagosomal maturation and intraphagosomal acidification [6,11,12]. In a screen for Mtb transposon mutants hypersusceptible to acid stress, we previously identified 21 genes whose interruption lead to reduced viability in low pH [13]. The majority of these genes are annotated to have functions related to cell wall processes. These included two independent transposon mutants of the previously uncharacterized Mtb gene rv0955, a 1,368 base pair open reading frame, which is annotated to encode an integral membrane protein with a predicted topology of ten transmembrane helices (S1 Fig.) [14–16]. Rv0955 is highly conserved among mycobacteria and actinobacteria, but has no known homologues in other species, and no conserved sequence motifs to predict its function. It is included among the 219 mycobacterial “core” genes noteworthy for their conservation among mycobacterial species, including Mtb and M. leprae [17]. These core genes lack homologues in other bacteria, suggesting that their function may be unique to mycobacteria, and making them potential targets for mycobacteria-specific drugs.
Here, we investigated the function of the previously uncharacterized Mtb Rv0955 protein. Disruption of rv0955 resulted in a striking persistence defect in chronic mouse infection with a 300-fold decline in bacterial burden in the lungs. We therefore named this gene perM, encoding a persistence-associated integral membrane protein. As Vandal et al. noted, the acid susceptibility of the perM mutant—similar to many of the mutants identified in the screen—was detergent-dependent, observed only when the bacteria were exposed to a combination of low pH and Tween-80 detergent [13]. We thus sought to investigate mechanisms beyond protection from acid, which might account for the strong attenuation of the mutant in vivo.
We found that the perM mutant required increased magnesium (Mg2+) compared to wild type (wt) Mtb for replication and survival in culture. Mg2+ is among the most abundant divalent cations in both prokaryotic and eukaryotic cells, and is essential for bacterial growth. In bacteria, Mg2+ serves a wide range of roles: it functions as a cofactor with ATP in numerous enzymatic reactions, enables the formation of tRNA and ribosomal tertiary structure, and regulates stability of the cell wall and membrane [18–20]. Mg2+ also impacts virulence in Salmonella enterica by regulating the PhoP/PhoQ two-component system [21].
In Mtb, two Mg2+-dependent mutants have been identified: Mtb∆phoP and Mtb∆mgtC [22,23]. PhoP shows high similarity to the PhoP response regulator of Salmonella enterica and is required in Mtb for the synthesis of several complex cell wall lipids as well as replication in macrophages and mice [22,24,25]. MgtC is required for virulence of both Mtb and Salmonella enterica and inhibits the bacterial F1F0 ATP synthase to maintain physiological ATP levels and intrabacterial pH [23,26].
Mg2+ restriction remains a plausible but unconfirmed antimycobacterial mechanism employed by the host. In media with low Mg2+ concentrations, the perM mutant elongated and upregulated expression of cell division and cell wall biosynthesis genes. Furthermore, Mtb PerM accumulated at the putative division septa in the closely related M. smegmatis. Disruption of perM resulted in pronounced hypersusceptibility to beta-lactam antibiotics, including cephalexin and piperacillin, which are specific inhibitors of the cell division-associated peptidoglycan synthesis protein FtsI. This work characterizes a novel mycobacterial protein necessary for persistence in vivo and implicated in cell division, and is consistent with the hypothesis that Mtb has reduced access to Mg2+ during chronic infection.
Results
PerM is required for Mtb persistence in vivo
PerM was previously identified in a screen for Mtb genes required for acid resistance [13]. To examine the role of PerM in vivo, we monitored replication and survival of a perM transposon mutant, perM::tn, in wild type mice. PerM::tn established infection and replicated during the acute phase, with only a 5-fold reduction in peak bacterial burden, measured by colony forming units (CFU), compared to wt (P = 0.032) at 21 days (Fig. 1A). However, perM::tn exhibited a severe persistence defect in chronic infection, with a 300-fold reduction in CFU in the lungs at fourteen weeks post-infection.

In agreement with these growth patterns, histological analysis revealed markedly fewer and smaller lesions in perM::tn-infected lung tissue compared to wt Mtb-infected mice, a difference observed at the 148 day post-infection time point, but not at the end of acute infection (Fig. 1B). Genetic complementation of the perM mutant with a wild-type copy of the gene expressed chromosomally under control of the hsp60 promoter restored persistence and increased granulomatous inflammation, indicating that attenuation in vivo was due to disruption of perM.
The adaptive immune response to Mtb is characterized by IFN-γ mediated activation of host macrophages. To examine whether death of perM::tn in vivo was dependent on host IFN-γ, we infected IFN-γ knockout mice, which are unable to control replication of wt Mtb [1,2]. PerM::tn replicated in IFN-γ knockout mice, but at a slower rate than wt Mtb (Fig. 1C). IFN-γ knockout mice infected with wt Mtb had to be sacrificed at day 50, because they were moribund, in contrast to IFN-γ knockout mice infected with perM::tn, which remained healthy through the end of the experiment (day 106). These results indicate that killing of perM::tn in wt mice requires host IFN-γ, while the mutant also exhibits an IFN-γ-independent replication defect.
PerM::tn stimulates a hyperinflammatory cytokine response in infected macrophages ex vivo
Since later-occurring persistence defects like that of perM::tn often depend on the adaptive immune response of the host, we hypothesized that PerM might cause a more robust immune response than wt Mtb. To examine this possibility, we infected bone marrow derived mouse macrophages with equal numbers of wt, perM::tn and complemented mutant and measured cytokine concentrations in macrophage culture supernatants 24 hours later. Supernatants of macrophages infected with perM::tn contained elevated levels of proinflammatory cytokines, including TNF-α, IL-6, IL-12 p70, the anti-inflammatory cytokine IL-10, and the chemokine KC (Fig. 2A).

To assess the immune response to perM::tn during mouse infection we measured cytokine transcripts in mouse lungs by quantitative real-time polymerase chain reaction (qRTPCR), focusing on pro-inflammatory cytokines required to attenuate Mtb growth in vivo [1,27]. We did not observe significant differences in IFN-γ or TNF-α mRNA levels at 2 weeks post-infection, when bacterial titers of perM::tn were 3-fold lower than those of wt (S2A–S2B Fig.). In an independent experiment, IL-12p40 protein levels in lung homogenates from mice infected with wt or perM::tn were similar at 1 or 2 weeks post-infection and increased in wt compared to mutant infected lungs or 3 weeks post-infection (S2C–S2D Fig.). Differences in CFU confounded interpretation of these data, as bacterial counts of perM::tn were 3-, 5 - and 6-fold reduced compared to wt at weeks 1, 2 and 3, respectively; however, these results suggested that attenuation of the mutant in vivo was not exclusively due to a more robust immune response that preceded in perM::tn-infected mice.
We sought to better understand the properties of perM::tn leading to the increased innate immune response by macrophages infected ex vivo. In a mixed-strain Mtb infection of macrophages, TNF-α production was similar to that induced by infection with perM::tn alone at the same total multiplicity of infection (MOI) (Fig. 2B). The dominance of the mutant suggested that the difference in response to these strains was due to an immunostimulatory effect of the mutant, as opposed to a suppressive effect of intact PerM protein produced by wt Mtb. The stimulatory effect of perM::tn was reproduced by exposure of macrophages to formalin-killed Mtb (Fig. 2C) and to cell-free Mtb-conditioned culture media (Fig. 2D), indicating that the stimulatory component(s) were shed or secreted by live perM::tn, but did not require viable bacteria for production or release during macrophage infection. In the absence of NOD and TLR2 signaling, perM::tn still elicited higher levels of TNF-α than wt (Fig. 2E). NOD and TLR2 are required for the macrophage response to bacterial peptidoglycan and triacylated lipoproteins, respectively, suggesting that the hyperinflammatory phenotype of perM::tn is not tied specifically to one of these cell wall components. TNF-α production was, however, significantly lower in cultures from knockout macrophages compared to wt macrophages, indicating that these receptors are important for TNF-α production following infection with both strains. Together, these data suggest that a combination of cellular components, both released into the medium during growth and expressed on the surface of killed perM::tn, function to stimulate increased inflammatory signaling in macrophages.
PerM is necessary for growth and survival in low magnesium
In liquid media, perM::tn replicated at a near-normal rate (Fig. 3A), but formed a loose aggregate during growth (Fig. 3B). Unlike previously described mycobacterial biofilms [28], these aggregates formed on the bottom of standing cultures, rather than at the liquid-air interface, and could be readily dispersed by shaking or pipetting. These aggregates suggested a perturbation of the perM::tn cell envelope. Since extracellular magnesium (Mg2+) has been shown to overcome phenotypes of mutants with cell envelope defects [22], we asked whether reduction of Mg2+ would affect growth or survival of perM::tn. Strains were cultured in nominally Mg2+-free Sauton’s minimal media, and supplemented with Mg2+ at a range of concentrations up to 2000 μM, the normal concentration in Sauton’s media (Fig. 3C). Wt Mtb died in nominally Mg2+-free media, but survived and replicated at Mg2+ concentrations of 25 μM and higher. In contrast, perM::tn exhibited death, observed by decreasing CFU counts, and lysis, observed by decreasing absorbance, at Mg2+ concentrations 100 μM and below. At 250 and 500 μM Mg2+, perM::tn replicated, but at a slower rate than wt and the complemented mutant.
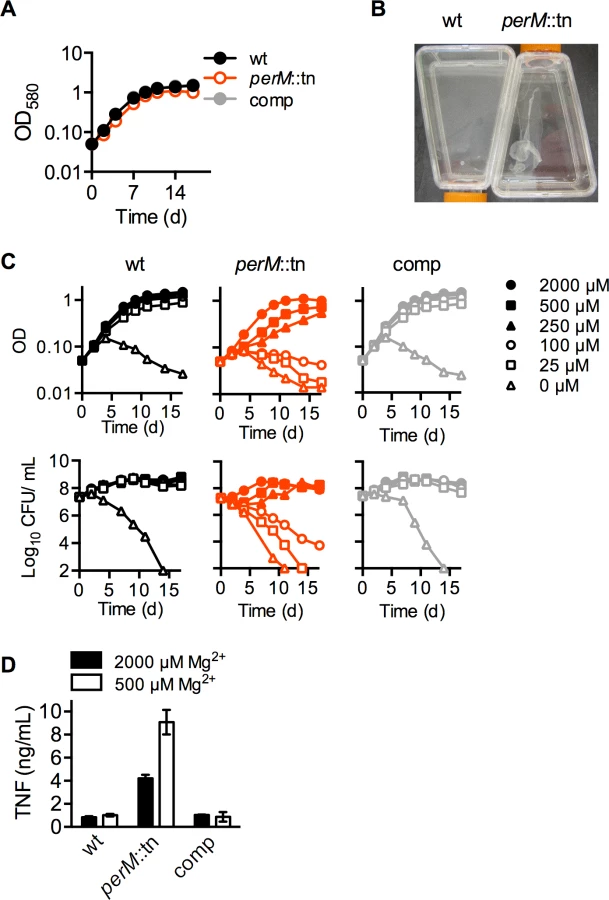
The requirement for additional Mg2+ was specific, as other cations, including Mn2+, Ca2+, Zn2+, and Fe3+, could not restore growth of perM::tn in reduced (100 or 250 μM) Mg2+ media (S3 Fig.). For further experiments, Mtb was grown in modified Sauton’s media containing 250 or 500 μM Mg2+ (“reduced” Mg2+), concentrations at which perM::tn displayed a growth defect without apparent death or lysis, or 2000 μM (“high”) Mg2+.
Within the IFN-γ-activated macrophage, Mtb is subject to numerous stresses, including low pH, reactive nitrogen intermediates, reactive oxygen species, and nutrient limitation [29,30], and it has been postulated that Mg2+ restriction may be an additional stress encountered by intraphagosomal pathogens including Mtb [23,31,32]. The inability of perM::tn to replicate and survive in low Mg2+ raised the possibility that the persistence defect in vivo might follow depletion of intraphagosomal Mg2+ in activated macrophages. We infected resting and IFN-γ-activated macrophages with wt, perM::tn and the complemented mutant following growth in high (2 mM) and reduced (250 μM) Mg2+. The mutant displayed a growth defect in resting macrophages, which was larger when it was pre-cultured in reduced magnesium. Survival of perM::tn in IFN-γ-activated macrophages was impaired in comparison to wt and complemented mutant, but only following pre-culture in reduced magnesium (S4 Fig.). We examined whether perM::tn was more susceptible than wt Mtb to a range of stresses in vitro, including exposure to hydrogen peroxide, lysozyme, detergent, acidified sodium nitrite, free fatty acid, zinc, and copper, as well as carbon starvation, iron depletion, and a multi-stress assay combining a fatty acid carbon source, reduced pH, hypoxia, and sodium nitrite (S5 Fig.). The mutant survived at wt levels under all conditions, indicating that perM::tn does not have a general viability defect, but rather, appears to be specifically vulnerable to reduced Mg2+.
The increased Mg2+ requirement of perM::tn suggested a possible role for PerM in Mg2+ transport. However, analysis of total Mg2+ content by inductively coupled mass spectrometry (ICP-QQQ) showed no significant differences in strains grown in either high (2000 μM) or reduced (250 and 25 μM) Mg2+ (S6 Fig.), suggesting that PerM is not required for Mg2+ acquisition. Given our data, along with work in Salmonella suggesting a redundancy of Mg2+ transporters that ensures significant Mg2+ uptake [33], the persistent defect of perM::tn is unlikely the result of impaired Mg2+ transport.
In the context of the host response, infection of mouse macrophages with Mtb pre-grown in reduced (500 μM) Mg2+ media resulted in a 2-fold increase in TNF-α production by macrophages infected with perM::tn, but not wt or complemented strains (Fig. 3D). This suggests that either the immunostimulatory component(s) of perM::tn are more highly produced, secreted, or shed in reduced Mg2+; or that Mtb growth in reduced Mg2+ leads to increased exposure of these components to macrophage pattern recognition receptors and induction of a proinflammatory response.
Increased expression of cell division and cell wall biosynthesis genes in perM::tn in reduced Mg2+
To gain insight into the function of PerM, we compared the transcriptomes of wt and perM::tn Mtb grown in high (2000 μM) and reduced (250 μM) Mg2+ in three independent experiments, using a p-value of 0.05 and 2-fold cutoff to identify differentially regulated genes. In reduced Mg2+, 41 genes were differentially expressed between strains, all of which except one were upregulated in the mutant (Table 1). Sixteen of these genes are annotated with predicted or possible roles in cell division and/or cell wall biosynthesis. Upregulation of a subset of these genes was confirmed by qRTPCR analysis (Fig. 4A).


Genes listed were differentially expressed at least 2-fold in perM::tn compared to wt grown for 5 days in media supplemented with 250 μM Mg2+. Fold change values are averages of three independent experiments, P<0.05. Annotations adapted from TB Database (tbdb.org), TubercuList (tuberculist.epfl.ch) and PATRIC (patricbrc.org). FC, fold change in perM::tn compared to wt. Genes also regulated greater than 2-fold between strains in 2000 μM Mg2+ are marked with *.
Cell division genes more highly expressed in the mutant compared to wt under reduced Mg2+ included ftsK and xerC, involved in chromosome segregation; ftsI, necessary for peptidoglycan crosslinking during division; and ftsW, whose product likely translocates peptidoglycan precursors across the cell membrane [34] and interacts with both FtsI as well as cell division initiator FtsZ in mycobacteria [35]. Also upregulated in the mutant were genes encoding four putative penicillin binding proteins (FtsI, DacB1, Rv2864c, and Rv1433), enzymes which carry out the transpeptidation necessary for crosslinking of cell wall peptidoglycan strands; Rv3717, a possible peptidoglycan amidase with a role in cell wall remodeling; and Rv0519c, a possible mycolyltransferase involved in mycolic acid processing [36]. Secreted fibronectin-binding protein C (FbpC), a possible trehalose mycolyltransferase thought to have both antigenic and cell wall biosynthesis roles, also showed increased expression in the mutant. Notably, expression of ftsZ, encoding the cytosolic, tubulin-like initiator of cell division, was not increased in the mutant at either Mg2+ concentration, nor was expression of genes in the cell wall biosynthetic gene cluster (rv3779-rv3809c) contributing to mycolic acid, arabinogalactan, and LAM synthesis [37], pointing towards a specific response rather than a global induction of all cell division and cell wall biosynthesis genes in the mutant.
Seven genes were upregulated in the mutant compared to wt in both high and reduced Mg2+, with more pronounced differences in expression between strains in reduced Mg2+ (Tables 1, S1), suggesting that Mg2+ reduction exacerbates differential transcriptional responses that are also present in high Mg2+.
Comparison of gene expression in wt Mtb in reduced versus high Mg2+ revealed only two genes meeting the 2-fold cutoff: pe20 was upregulated in reduced Mg2+ and fadD5 was downregulated (S2 Table). This transcriptional response was far less pronounced than that previously identified consisting of 24 genes differentially regulated in wt Mtb grown in media with or without Mg2+ [22], suggesting that the transcriptional response to Mg2+ starvation in wt Mtb was not triggered at 250 μM Mg2+, used in our experiment. The gene expression pattern of perM::tn in 250 μM Mg2+ (S2 Table) did not resemble Mg2+-starved wt Mtb [22], contrary to what might be expected if Mg2+ uptake were impaired in the mutant.
PerM accumulates at the septum and is necessary for division in reduced Mg2+
The increased expression of cell division and cell wall biosynthesis genes in the mutant suggested a possible defect in these processes. To examine the impact of perM disruption on cell morphology, Mtb was grown in a range of Mg2+ concentrations, fixed, and imaged by scanning electron microscopy (SEM). PerM::tn exhibited Mg2+-dependent defects in morphology and division. Median cell length increased as the concentration of Mg2+ decreased, and some mutant bacilli exhibited bulging at the poles in reduced Mg2+ (Fig. 4B,C).
To examine localization of PerM, GFP-tagged Mtb PerM protein was expressed in wt Mycobacterium smegmatis, a non-pathogenic species closely related to Mtb and itself containing an PerM homolog with 73% identity. Mtb requires containment within a biosafety level 3 facility, which prevented us from performing live cell imaging experiments in Mtb. Live cell imaging of recombinant M. smegmatis revealed that PerMMtb localized to the membrane and it accumulated at the mid-cell division site (Fig. 5), similar to mycobacterial cell division proteins, such as FtsI and FtsZ, as well as peptidoglycan synthesis enzymes, such as penicillin binding protein 1 [38–40].

PerM::tn is hypersensitive to beta lactam antibiotics
We next compared sensitivity of perM::tn and wt Mtb to a range of compounds targeting cell wall biosynthesis, as well as drugs with other established targets. The majority of compounds assayed exhibited a similar minimum inhibitory concentration (MIC) in wt and perM::tn (Table 2 and S7 Fig.), with a shift of 2-fold or less considered insignificant. However, perM::tn was acutely sensitive to growth inhibition by β-lactam antibiotics, which target penicillin binding proteins that carry out the transpeptidation reaction resulting in crosslinking of cell wall peptidoglycan, a final step in peptidoglycan synthesis. The shift in MIC was most pronounced for cephalexin and piperacillin, β-lactams that specifically inhibit FtsI, the transpeptidase required for peptidoglycan crosslinking during bacterial cell division [41–43]. β-lactamase activities in wt and perM::tn were not significantly different (S3 Table) excluding the possibility that impaired β-lactamase activity caused the mutant’s increased susceptibility to β-lactams. Notably, the MICs of vancomycin and D-cycloserine, which inhibit earlier steps in peptidoglycan synthesis than do β-lactams, were similar for wt and perM::tn. Furthermore, there was little to no shift in MIC of isoniazid and ethambutol, which inhibit production of other cell wall components (mycolic acids and arabinogalactan, respectively), indicating that the perM::tn is not broadly hypersusceptible to interference with cell wall biosynthesis.

Minimum inhibitory concentration (MIC) of various drugs against wt and perM::tn Mtb. MIC90 values in μg/mL, determined by minimum concentration at which OD580 was less than 10% that of untreated control. FC, fold change reduction of perM::tn MIC compared to wt MIC.
Discussion
This work implicates a novel mycobacterial membrane protein in cell division and demonstrates its requirement for Mtb persistence in vivo. The persistence defect of the PerM mutant is one of the most dramatic per phenotypes observed to date, and to our knowledge the first noted in a mutant of an Mtb membrane protein. Global gene expression profiling revealed increased expression of cell division and cell wall biosynthesis genes in the mutant, and these increases exacerbated during growth in reduced Mg2+. Several additional observations support the hypothesis that PerM plays a role in cell division. First, the mutant elongated in reduced Mg2+, with additional morphological changes at very low Mg2+. Second, the mutant exhibited hypersusceptibility to β-lactam antibiotics, which inhibit the enzymes necessary for crosslinking of cell wall peptidoglycan. In particular, the mutant was hypersusceptible to piperacillin and cephalexin, β-lactams that specifically target the cell division-associated peptidoglycan transpeptidase, FtsI [42–44]. Third, PerM localized to the mid-cell region in M. smegmatis, similarly to previously studied mycobacterial proteins involved in cell division and peptidoglycan biosynthesis [38–40]. The mutant hyperstimulated mouse macrophages ex vivo, a phenotype exacerbated after culture in reduced Mg2+, which may be related to shedding of cell wall components during a compromised cell division process.
The inability of perM::tn to replicate and survive at low Mg2+ suggested that PerM may play a role in Mg2+ acquisition, could be necessary for the adaptive response of Mtb to low Mg2+, or that Mg2+ might serve a compensatory function to mask physiological defects caused by the absence of PerM. We examined the first possibility by ICP-QQQ analysis, which revealed perM::tn and wt Mtb to contain the same total Mg2+, even when grown in reduced Mg2+ media. Furthermore, gene expression data from the mutant showed a regulation pattern distinct from that of Mg2+-starved wt Mtb [22]. The second possibility, that PerM is a component of the bacterial response to low Mg2+, is similarly not supported by the gene expression profile of wt Mtb grown in low Mg2+ [22]. However, it is possible that PerM, constitutively expressed, is required for a successful adaptive response to Mg2+ starvation, perhaps through interaction with Mg2+ response proteins. Future protein interaction studies may shed light on this question.
Our work supports the third possibility, that Mg2+ serves a compensatory function in the mutant through stabilization of a weakened cell envelope; in particular, our data suggest that the mutant cell envelope may be especially vulnerable during cell division. While the role of Mg2+ in cell wall stability is widely acknowledged, the mechanism by which this occurs is not entirely clear. In Salmonella, outer membrane permeability decreased in high Mg2+, and a phoP Salmonella mutant with lipopolysaccharide alterations displayed increased permeability and susceptibility to numerous antibiotics in low Mg2+, but behaved like wt Salmonella when Mg2+ was high [45]. This suggests a role for Mg2+ in stabilizing the outer membrane, perhaps through interaction with negatively-charged lipopolysaccharide. In the Gram-positive B. subtilis, which lacks an outer membrane, high Mg2+ partially suppressed the growth defect of a mutant lacking teichoic acid suggesting that Mg2+ might be able to compensate for loss of teichoic acid in the cell wall [46]. On the other hand, high concentrations of Mg2+ may serve to stabilize an otherwise vulnerable peptidoglycan sacculus. B. subtilis mutants lacking MreB, RodB and PonA—proteins thought to be involved in peptidoglycan synthesis—display morphological and growth defects that were rescued by high Mg2+ [47–49]. Of note, peptidoglycan synthesis decreased and peptidoglycan precursors accumulated in Mg2+-deprived B. subtilis [50], and in Salmonella, lipid A acylation increased in response to Mg2+ deprivation [20], suggesting that the influence of Mg2+ on peptidoglycan integrity may occur by several mechanisms, both structural and regulatory. It has also been proposed that Mg2+ might affect the degree of peptidoglycan crosslinking that occurs; stabilize or regulate important cell-wall synthases or hydrolases; or serve to stiffen the cell envelope [45,51].
The upregulation of cell division genes in perM::tn, combined with the hypersensitivity of the mutant to specific inhibitors of FtsI, suggests a role for PerM in peptidoglycan synthesis or remodeling during cell division. The perM mutant was not hypersusceptible to all peptidoglycan synthesis inhibitors: the MICs of vancomycin and cycloserine were similar for mutant and wt Mtb. Cycloserine, an analog of D-alanine, blocks synthesis of cytoplasmic peptidoglycan precursors [52], while vancomycin prevents both the early transglycosylation step necessary for incorporation of peptidoglycan monomer into the sacculus, as well as the final crosslinking of peptidoglycan by transpeptidases [53]. The specific vulnerability of perM::tn to β-lactams, which target the transpeptidation step, suggests that PerM may play a role in late peptidoglycan biosynthesis during cell division. Interestingly, a conditional mutant of ripA, which encodes an essential mycobacterial peptidoglycan hydrolase, was similarly hypersusceptible to a β-lactam, carbenicillin, but not to cycloserine following ripA depletion [54].
While its specific mechanism of action remains to be determined, it is plausible that PerM, as an integral membrane protein with 10 transmembrane helices, could serve a structural role, recruiting or anchoring key cell division proteins, such as peptidoglycan transpeptidases or hydrolases, to the division site. It may serve to bridge cytoplasmic proteins, such as FtsZ or early peptidoglycan synthesis machinery, with cell division proteins in the periplasm, or it could be involved in transport of cell envelope components.
The perM mutant withstood numerous stresses in vitro, including reactive oxygen and nitrogen species, cell wall-perturbing detergent, and carbon starvation, showing a specific vulnerability to a low-Mg2+ environment. Surprisingly, the perM mutant was not more susceptible than wt to exposure to arachidonic acid at pH 5.5, despite its sensitivity to Tween-80 at pH 4.5, which suggested that free oleic acid might be toxic to the mutant at low pH. It is plausible that oleic acid released from Tween-80 and arachidonic acid cause toxicity by different mechanisms. In addition, the lower pH of the Tween-80 containing medium may have contributed to the enhanced killing of the mutant.
Survival of perM:tn in IFN-γ activated macrophages was impaired, when the bacteria were pre-grown in reduced Mg2+. IFN-γ activated, Mtb infected macrophages have a limited lifespan ex vivo, which prevented extending the time course of the ex vivo infection to better mimic the mouse infection. It is possible that pre-growth in reduced Mg2+ has the same impact as replication in the acute phase of mouse infection, but interpretation of the ex vivo macrophage infection data is difficult and does not allow direct conclusions about the intraphagosomal availability of Mg2+.
Previous work revealed that macrophage activation by IFN-γ results in changes in the intraphagosomal concentrations of several metals, but Mg2+ was not measured [55]. The Salmonella-containing phagosome was estimated to contain 10 to 50 μM Mg2+, based on strong induction of the Mg2+-regulated mgtB gene in Salmonella in both low Mg2+ media and upon uptake by mammalian cells [56,57]; however, measurement of intraphagosomal Mg2+ using nanosensor particles showed the concentration to be approximately 1 mM in the first two hours of infection [58]. The intraphagosomal concentration of Mg2+ in vivo, after days or weeks of Mtb infection, remains a topic of speculation. Our work lends support to the hypothesis of an Mg2+-depleted environment in the Mtb-containing activated macrophage in vivo. Unfortunately, measuring intraphagosomal Mg2+ concentrations is extremely challenging. Purification of Mtb infected phagosomes is difficult and it is unknown if the purification process alters phagosomal ion concentrations. Fluorescent Mg2+ reporters exhibit much higher affinity for Ca2+ and also bind Zn2+, while PEPPLE (probe encapsulated by biologically localized embedding) technology suffers from low magnesium affinity [59]. Future development of novel and better sensors for magnesium is required to overcome these obstacles.
The PerM mutant stimulated a hyperinflammatory cytokine response in infected macrophages ex vivo. While we did not detect elevated cytokine levels in lungs of mice infected with the perM mutant compared to wt-infected mice, we cannot rule out the possible contribution of a hyperinflammatory response to the persistence defect. The cytokine measurements might have been confounded by differences in bacterial loads, and even a small, perhaps difficult to quantify, difference in the host immune response could synergize with Mg2+ restriction to result in killing of perM::tn; or the response may be localized, with lesion-centric inflammation contributing to killing perM::tn, but little impact on total cytokine levels in the lungs.
Growth of Mtb in reduced Mg2+ prior to macrophage infection resulted in an augmented response to the mutant, but no difference in response to wt Mtb, suggesting that cell wall instability of the mutant may contribute to the hyperinflammatory phenotype. In light of other evidence linking PerM to cell division, it is plausible that the mutant sheds multiple components of the cell wall during a stalled or otherwise compromised division process, resulting in increased stimulation of macrophage response pathways.
The bacterial cell wall is the target of many drugs in current use. The remarkable sensitivity of perM::tn to cephalexin and piperacillin, antibiotics routinely and safely used in clinical practice, suggests an exciting possibility of PerM as a co-target. An inhibitor of PerM could potentially be used to sensitize Mtb to β-lactam antibiotics, extending their use to mycobacterial infections.
Materials and Methods
Ethics statement
Mouse studies were performed following National Institutes of Health guidelines for housing and care of laboratory animals and performed in accordance with institutional regulations after protocol review and approval by the Institutional Animal Care and Use Committee of Weill Cornell Medical College (protocol # 2008–0006, pH homeostasis in Mycobacterium tuberculosis).
Bacterial strains and media
PerM::tn, the Mtb H37Rv transposon mutant of gene perM (rv0955) containing a ΦMycoMarT7 transposon insertion at nucleotide 701, was isolated in a screen for acid-sensitive mutants described previously [13]. Mtb strains were grown in a humidified incubator at 37°C with 5% CO2 in Sauton’s media with 0.05% Tween 80 or 0.05% tyloxapol; Middlebrook 7H9 medium (Difco) containing 0.2% glycerol, 0.5% bovine serum albumin, 0.2% dextrose, 0.085% NaCl, and 0.05% Tween 80; or Middlebrook 7H11 agar (Difco) containing 10% OADC supplement (Becton Dickinson) and 0.5% glycerol. Nominally magnesium-free Sauton’s media was prepared with 0.8 mM citric acid, 9 mM sodium citrate, 3 mM potassium phosphate, 30 mM L-asparagine, and 6% glycerol, chelated overnight with 20 g/L Chelex 100 resin (Bio-Rad), filtered to remove Chelex, supplemented with 0.2 mM ferric ammonium citrate and 5 μM zinc sulfate, and adjusted to pH 7.4. Before use, 0.05% Tween 80 and 2 mM MgCl2 were added unless otherwise indicated. We call this medium “nominally magnesium-free” as trace residual magnesium is likely present. Hygromycin B (50 μg/ml), kanamycin (15 μg/ml) and streptomycin (20 μg/ml) were included when required for selection.
Complementation of perM::tn
Rv0955 was PCR amplified from H37Rv genomic DNA and cloned behind the hsp60 promoter into a plasmid that integrates into the chromosomal phage integration attL5 site. For localization studies, GFP was fused to the C-terminus of Rv0955 and expressed from the hsp60 promoter on an integrative plasmid.
Mouse infections
Female C57BL/6, or IFN-γ-/- mice (Jackson Laboratory) were infected using an inhalation exposure system (Glas-Col) with early-log-phase Mtb to deliver approximately 100 bacilli per mouse. Bacterial numbers were enumerated by plating serial dilutions of lung or spleen homogenates on 7H11 agar plates for CFU. Upper left lung lobes were fixed in 10% buffered formalin, embedded in paraffin and stained with hematoxylin and eosin.
Macrophage infection experiments
Bone marrow derived macrophages were harvested and differentiated as previously described [13] and seeded at 4x106 cells/mL, with or without 50 ng/mL murine IFN-γ (R&D Systems). Approximately sixteen hours later, macrophages were infected at a multiplicity of infection (MOI) of 0.1 with a single cell suspensions of log-phase Mtb grown for 6 days in 250 or 2000 μM MgCl2. Monolayers were washed with PBS 4 hours post-infection to remove extracellular bacteria. After 4 hours, 3 days, or 6 days, macrophages were lysed with 0.5% Triton X-100 and bacteria were enumerated by plating serial dilutions on 7H11 agar plates. Half of the media in each well was replaced with fresh media after 3 days.
Measurement of cytokine production by infected macrophages ex vivo and in vivo
Bone marrow derived macrophages from C56BL/6 or TLR2-/- mice (Jackson Laboratories) were harvested and differentiated as previously described [13]. Immortalized macrophage cell lines from wild type, and Nod1/2-/- mice [60] were a gift from M. A. Kelliher at the University of Massachusetts. Macrophages were seeded at 4x105 cells/ml (wt macrophages) or 6x105 cells/ml (knockout macrophages). After 16 hours, they were infected at the indicated MOI with a single cell suspension of log-phase Mtb. For experiments using dead bacteria, Mtb was fixed in 10% formalin for 16 hours, washed twice in PBS, and added to Mtb at an MOI of 20. For exposure of macrophages to Mtb-conditioned culture media, Mtb was grown for 8 days in detergent-free Sauton’s media containing 2 mM MgCl2, then culture supernatant was passed through a 0.2 μm filter, concentrated approximately 10-fold in Amicon Ultra-15 Centrifugal Filter Units (Millipore), and added to macrophages at a volume equivalent to 10 μg protein. Supernatants were collected after 24 hours, passed through a 0.2 μm filter, and stored at -80°C. Cytokine levels were quantified using BD OptEIA ELISA kits for mouse TNF or IL-12p40 (BD Biosciences), or a multiplex ELISA Mouse ProInflammatory 7-Plex Tissue Culture Kit (Meso Scale Discovery). Tissue processing, RNA isolation, and real-time PCR were performed as previously described [61].
In vitro stress susceptibility assays
Mtb was grown to log phase in Sauton’s media containing 2000 μM MgCl2 prior to each experiment. Single cell suspensions were prepared in assay medium by centrifugation at 800 rpm for 12 minutes, then diluted to OD 0.02–0.05 and incubated in the following conditions: 3 days in pH 4.5 in media containing 0.05% Tween 80 or tyloxapol; 5 weeks in PBS with 0.05% tyloxapol; 24 hours in 7H9 media with 0.05% Tween 80 and 2.5 mg/ml lysozyme; 5 hours in 7H9 media with 0.05% Tween 80 and 0.1% SDS; 3 days in 7H9 media at pH 5.5 with 0.05% tyloxapol and 5 mM NaNO2; 3 days in 7H9 media at pH 5.5 with 0.05% tyloxapol and 50 μM arachidonic acid; 3 hours in 7H9 media containing 10 mM H2O2. For the multi-stress survival assay, Mtb was incubated in 1% oxygen for 14 days in modified Sauton’s media at pH 5.5 containing 0.05% tyloxapol, 0.05% butyrate, 0.5 mM sodium nitrite, 2000 μM MgCl2, and without glycerol. The exposure times to different stress conditions were selected so that viability of wt Mtb was reduced by approximately 5 - to 10-fold. For conditions in which wt Mtb survived without significant or very slow death (carbon starvation, multi-stress model) extended incubation times were chosen. To determine viability, serial dilutions of cultures were plated on 7H11 plates.
Cation dose response experiments
Mtb was grown to log phase, washed twice in assay medium, and diluted to OD580 0.02 in plates containing two-fold serial dilutions of MgCl2, MgSO4, ZnCl2, MnCl2, CaCl2, CuCl2, or ferric ammonium citrate. For experiments testing various cations as substitutes for Mg2+, a basal level of either 100 or 250 μM MgCl2 was added as indicated. For experiments involving ZnCl2 or ferric ammonium citrate, modified Sauton’s medium was prepared without the respective cation.
ICP-QQQ analysis
Mtb was washed twice in nominally Mg2+-free Sauton’s media, diluted to OD 0.1 in Sauton’s media containing 250 or 2000 μM added MgCl2. After 5 days, cultures were washed twice in PBS with 0.05% Tween 80. To determine the impact of very low Mg2+, cultures were grown in Sauton’s media containing 2000 μM added MgCl2 until mid-log phase, then washed twice in nominally Mg2+-free Sauton’s media and incubated in Sauton’s media containing 25 μM added MgCl2. Pellets were collected at 0 hour, 3 hours and 10 hours post inoculation. After normalizing for biomass, pellets were heated at 80°C for 1 hour to kill Mtb, then resuspended in 200 μL 70% nitric acid, trace element grade (Fisher) and heated at 80°C for 2h before ICP-QQQ processing and analysis. Samples were analyzed on an Agilent 8800 ICP-QQQ running in MS/MS mode. Instrument daily performance qualification and method specific tuning was achieved by the expert AutoTune function of the MassHunter software (B.01.02). Typical sample introduction parameters for direct injection were used; RF Power 1550W, sample depth 8 mm, carrier gas 0.95 L/min, and dilution gas was set at 0.15 L/min. These parameters resulted in an oxide ratio of 0.8% (CeO/Ce). Prior to analysis, samples were diluted to a final volume of 2 ml and analyzed against multi-element external calibration standards (Agilent, Wilmington, DE). NIST 1643e was used as a standard reference material for calibration verification and monitor any possible drift during the analytical run.
RNA isolation and gene expression analysis
Mtb was grown standing for 5 days in Sauton’s media containing 250 or 2000 μM MgCl2 and 0.05% Tween 80. Flasks were shaken for 5 hours prior to harvest. Cultures were mixed with an equal volume of GTC buffer containing guanidinium thiocyanate (4 M), sodium lauryl sulfate (0.5%), trisodium citrate (25 mM), and 2-mercaptoethanol (0.1 M) and pelleted by centrifugation. Bacterial RNA was isolated as previously described [62]. For microarray experiments, RNA was labeled using a Low Input Quick Amp Labeling Kit (Agilent). Microarrays were custom-designed (Genotypic Technology, Bangalore, India). Analysis was performed using Agilent GeneSpring software. The complete Microarray data sets have been submitted to the Gene Expression Omnibus (GEO) database.
For gene expression analysis by quantitative real-time PCR, cDNA was generated using MuLV Reverse Transcriptase (Invitrogen) and quantified using Roche Light Cycler 480 Real-Time PCR System with primers and TaqMan probes designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0). Primer and probe sequences are available upon request.
Electron microscopy and length measurement
Mtb was grown for 5 days in Sauton’s media containing 25, 250, 500, or 2000 μM MgCl2 and 0.05% Tween 80 before fixation, processing, and imagining by scanning electron microscopy as previously described [13]. Cell lengths were measured using Adobe Photoshop software.
Antibiotic sensitivity assays
Mtb was grown to early log phase and diluted to an optical density of 0.02 in Sauton’s medium containing 2 mM MgCl2 and 0.05% Tween 80. Bacteria were then exposed to twofold dilutions of piperacillin, cephalexin, ampicillin, meropenem, rifampicin, polymyxin B, DCCD, vancomycin, D-cycloserine, streptomycin, chloramphenicol, isoniazid, and ethambutol (Sigma-Aldrich). For assays of meropenem, DCCD, isoniazid, chloramphenicol, and rifampicin, all wells contained 0.5% DMSO. For the assay of cephalexin, all wells contained 4 mM NH4OH. The MIC was recorded as the minimum concentration at which growth, measured by optical density (OD580), was inhibited by at least 90%, as compared to a control containing no antibiotic, after approximately 2 weeks.
Fluorescence microscopy
M. smegmatis expressing PerM-GFP was sealed in B04A microfluidic flow chamber plates (Cell Asic, part of EMD Millipore) and perfused with Middlebrook 7H9 broth at 37°C. Cells were visualized by fluorescence microscopy using an inverted Olympus IX-70 microscope equipped with a GFP filter set, a Photometrics CoolSnap QE cooled CCD camera, and an Insight SSI 7 color solid state illumination system. Snapshots were captured every 15 minutes.
ß-lactamase activity assay
The chromogenic cephalosporin nitrocefin (Fisher) was used to assay ß-lactamase activity as previously described [63] in whole-cell lysates of Mtb saturated cultures grown in Sauton’s media with 2 mM MgCl2.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, et al. (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med 178 : 2249–2254. 7504064
2. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, et al. (1993) Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med 178 : 2243–2247. 8245795
3. Nathan CF, Murray HW, Wiebe ME, Rubin BY (1983) Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med 158 : 670–689. 6411853
4. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, et al. (1997) Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA 94 : 5243–5248. 9144222
5. Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, et al. (2011) A Family of IFN—Inducible 65-kD GTPases Protects Against Bacterial Infection. Science 332 : 717–721. doi: 10.1126/science.1201711 21551061
6. MacMicking JD, Taylor GA, Mckinney JD (2003) Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302 : 654–659. doi: 10.1126/science.1088063 14576437
7. Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, et al. (2001) Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med 194 : 1123–1140. 11602641
8. Glickman MS, Jacobs WR (2001) Microbial pathogenesis of Mycobacterium tuberculosis: dawn of a discipline. Cell 104 : 477–485. 11239406
9. McKinney J, zu Bentrup K, Muñoz-Elías E, Miczak A, Chen B, et al. (2000) Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406 : 735–738. 10963599
10. Pandey AK, Sassetti CM (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA 105 : 4376–4380. doi: 10.1073/pnas.0711159105 18334639
11. Schaible UE, Sturgill-Koszycki S, Schlesinger PH, Russell DG (1998) Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J Immunol 160 : 1290–1296. 9570546
12. Via LE, Fratti RA, McFalone M, Pagan-Ramos E, Deretic D, et al. (1998) Effects of cytokines on mycobacterial phagosome maturation. J Cell Sci 111 (Pt 7): 897–905.
13. Vandal OH, Pierini LM, Schnappinger D, Nathan CF, Ehrt S (2008) A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat Med 14 : 849–854. doi: 10.1038/nm.1795 18641659
14. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 : 537–544. 9634230
15. Krogh A, Larsson B, Heijne von G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305 : 567–580. doi: 10.1006/jmbi.2000.4315 11152613
16. Spyropoulos IC, Liakopoulos TD, Bagos PG, Hamodrakas SJ (2004) TMRPres2D: high quality visual representation of transmembrane protein models. Bioinformatics 20 : 3258–3260. doi: 10.1093/bioinformatics/bth358 15201184
17. Marmiesse M, Brodin P, Buchrieser C, Gutierrez C, Simoes N, et al. (2004) Macro-array and bioinformatic analyses reveal mycobacterial “core” genes, variation in the ESAT-6 gene family and new phylogenetic markers for the Mycobacterium tuberculosis complex. Microbiology 150 : 483–496. 14766927
18. Draper DE, Grilley D, Soto AM (2005) Ions and RNA folding. Annu Rev Biophys Biomol Struct 34 : 221–243. doi: 10.1146/annurev.biophys.34.040204.144511 15869389
19. Smith RL, Maguire ME (1998) Microbial magnesium transport: unusual transporters searching for identity. Mol Microbiol 28 : 217–226. doi: 10.1046/j.1365–2958.1998.00810.x 9622348
20. Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, et al. (1998) Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 95 : 189–198. 9790526
21. García Véscovi E, Soncini FC, Groisman EA (1996) Mg2+ as an extracellular signal: environmental regulation of Salmonella virulence. Cell 84 : 165–174. 8548821
22. Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffé M, et al. (2006) The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol 60 : 312–330. doi: 10.1111/j.1365–2958.2006.05102.x 16573683
23. Buchmeier
N, Blanc-Potard A, Ehrt S, Piddington D, Riley L, et al. (2000) A parallel intraphagosomal survival strategy shared by Mycobacterium tuberculosis and Salmonella enterica. Mol Microbiol 35 : 1375–1382. 10760138
24. Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, et al. (2009) Evolution of a Bacterial Regulon Controlling Virulence and Mg2+ Homeostasis. PLoS Genet 5: e1000428. doi: 10.1371/journal.pgen.1000428.g005 19300486
25. Gonzalo Asensio J, Maia C, Ferrer NL, Barilone N, Laval F, et al. (2006) The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem 281 : 1313–1316. doi: 10.1074/jbc.C500388200 16326699
26. Lee E-J, Pontes MH, Groisman EA (2013) A Bacterial Virulence Protein Promotes Pathogenicity by Inhibiting the Bacterium’s Own F1Fo ATP Synthase. Cell 154 : 146–156. doi: 10.1016/j.cell.2013.06.004 23827679
27. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, et al. (2002) TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 168 : 4620–4627. 11971010
28. Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, et al. (2008) Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol 69 : 164–174. doi: 10.1111/j.1365–2958.2008.06274.x 18466296
29. Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, et al. (2003) Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med 198 : 693–704. doi: 10.1084/jem.20030846 12953091
30. Stallings CL, Glickman MS (2010) Is Mycobacterium tuberculosis stressed out? A critical assessment of the genetic evidence. Microbes Infect 12 : 1091–1101. doi: 10.1016/j.micinf.2010.07.014 20691805
31. Blanc-Potard AB, Groisman EA (1997) The Salmonella selC locus contains a pathogenicity island mediating intramacrophage survival. EMBO J 16 : 5376–5385. doi: 10.1093/emboj/16.17.5376 9311997
32. Lavigne J-P, O’callaghan D, Blanc-Potard A-B (2005) Requirement of MgtC for Brucella suis intramacrophage growth: a potential mechanism shared by Salmonella enterica and Mycobacterium tuberculosis for adaptation to a low-Mg2+ environment. Infect Immun 73 : 3160–3163. doi: 10.1128/IAI.73.5.3160–3163.2005 15845525
33. Froschauer E, Kolisek M, Dieterich F, Schweigel M, Schweyen R (2004) Fluorescence measurements of free [Mg2+] by use of mag‐fura 2 in Salmonella enterica. FEMS Microbiol Lett 237 : 49–55. 15268937
34. Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun AE, et al. (2011) Identification of FtsW as a transporter of lipid - linked cell wall precursors across the membrane. EMBO J 30 : 1425–1432. doi: 10.1038/emboj.2011.61 21386816
35. Datta P, Dasgupta A, Singh AK, Mukherjee P, Kundu M, et al. (2006) Interaction between FtsW and penicillin-binding protein 3 (PBP3) directs PBP3 to mid-cell, controls cell septation and mediates the formation of a trimeric complex involving FtsZ, FtsW and PBP3 in mycobacteria. Mol Microbiol 62 : 1655–1673. doi: 10.1111/j.1365–2958.2006.05491.x 17427288
36. Lew JM, Kapopoulou A, Jones LM, Cole ST (2011) TubercuList—10 years after. Tuberculosis 91 : 1–7. doi: 10.1016/j.tube.2010.09.008 20980199
37. Kaur D, Guerin ME, Scaronkovierovaacute H, Brennan PJ, Jackson M (2009) Chapter 2: Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv Appl Microbiol 69 : 23–78. doi: 10.1016/S0065–2164(09)69002-X 19729090
38. Plocinski P, Ziolkiewicz M, Kiran M, Vadrevu SI, Nguyen HB, et al. (2011) Characterization of CrgA, a New Partner of the Mycobacterium tuberculosis Peptidoglycan Polymerization Complexes. J Bacteriol 193 : 3246–3256. doi: 10.1128/JB.00188–11 21531798
39. Rajagopalan M, Maloney E, Dziadek J, Poplawska M, Lofton H, et al. (2005) Genetic evidence that mycobacterial FtsZ and FtsW proteins interact, and colocalize to the division site in Mycobacterium smegmatis. FEMS Microbiol Lett 250 : 9–17. doi: 10.1016/j.femsle.2005.06.043 16040206
40. Hett EC, Chao MC, Rubin EJ (2010) Interaction and Modulation of Two Antagonistic Cell Wall Enzymes of Mycobacteria. PLoS Pathog 6: e1001020. doi: 10.1371/journal.ppat.1001020.s002 20686708
41. Eberhardt C, Kuerschner L, Weiss DS (2003) Probing the Catalytic Activity of a Cell Division-Specific Transpeptidase In Vivo with ß-Lactams. J Bacteriol 185 : 3726–3734. 2003. doi: 10.1128/JB.185.13.3726–3734 12813065
42. Botta GA, Park JT (1981) Evidence for involvement of penicillin-binding protein 3 in murein synthesis during septation but not during cell elongation. J Bacteriol 145 : 333–340. 6450748
43. Hedge PJ, Spratt BG (1985) Resistance to beta-lactam antibiotics by re-modelling the active site of an E. coli penicillin-binding protein. Nature 318 : 478–480. 3906408
44. Slayden RA, Belisle JT (2009) Morphological features and signature gene response elicited by inactivation of FtsI in Mycobacterium tuberculosis. J of Antimicrob Chemother 63 : 451–457. doi: 10.1093/jac/dkn507 19109339
45. Murata
T, Tseng W, Guina T, Miller S, Nikaido H (2007) PhoPQ-mediated regulation produces a more robust permeability barrier in the outer membrane of Salmonella enterica serovar typhimurium. J Bacteriol 189 : 7213. 17693506
46. D’Elia MA, Millar KE, Beveridge TJ, Brown ED (2006) Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol 188 : 8313–8316. doi: 10.1128/JB.01336–06 17012386
47. Formstone A, Errington J (2005) A magnesium-dependent mreB null mutant: implications for the role of mreB in Bacillus subtilis. Mol Microbiol 55 : 1646–1657. doi: 10.1111/j.1365–2958.2005.04506.x 15752190
48. Murray T, Popham DL, Setlow P (1998) Bacillus subtilis cells lacking penicillin-binding protein 1 require increased levels of divalent cations for growth. J Bacteriol 180 : 4555–4563. 9721295
49. Rogers HJ, Thurman PF, Buxton RS (1976) Magnesium and anion requirements of rodB mutants of Bacillus subtilis. J Bacteriol 125 : 556–564. 812869
50. Garrett AJ (1969) The effect of magnesium ion deprivation on the synthesis of mucopeptide and its precursors in Bacillus subtilis. Biochem J 115 : 419–430. 4982084
51. Chastanet A, Carballido-Lopez R (2012) The actin-like MreB proteins in Bacillus subtilis: a new turn. Front Biosci (Schol Ed) 4 : 1582–1606. 22652894
52. Prosser GA, de Carvalho LPS (2013) Metabolomics Reveal d-Alanine:d-Alanine Ligase As the Target of d-Cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett 4 : 1233–1237. doi: 10.1021/ml400349n 24478820
53. Reynolds PE (1989) Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8 : 943–950. 2532132
54. Hett EC, Chao MC, Deng LL, Rubin EJ (2008) A Mycobacterial Enzyme Essential for Cell Division Synergizes with Resuscitation-Promoting Factor. PLoS Pathog 4: e1000001. doi: 10.1371/journal.ppat.1000001 18463693
55. Wagner
D, Maser
J, Lai B, Cai Z, Barry CE, et al. (2005) Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J Immunol 174 : 1491–1500. 15661908
56. Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JCD (2002) Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol 47 : 103–118. doi: 10.1046/j.1365–2958.2003.03313.x
57. Portillo FGD, Foster JW, Maguire ME, Finlay BB (1992) Characterization of the micro-environment of Salmonella typhimurium-containing vacuoles within MDCK epithelial cells. Mol Microbiol 6 : 3289–3297. doi: 10.1111/j.1365–2958.1992.tb02197.x 1484485
58. Martin-Orozco N, Touret N, Zaharik ML, Park E, Kopelman R, et al. (2006) Visualization of vacuolar acidification-induced transcription of genes of pathogens inside macrophages. Mol Biol Cell 17 : 498–510. doi: 10.1091/mbc.E04–12–1096 16251362
59. Trapani V, Farruggia G, Marraccini C, Iotti S, Cittadini A, et al. (2010) Intracellular magnesium detection: imaging a brighter future. Analyst 135 : 1855. doi: 10.1039/c0an00087f 20544083
60. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, et al. (2009) NOD2, RIP2 and IRF5 Play a Critical Role in the Type I Interferon Response to Mycobacterium tuberculosis. PLoS Pathog 5: e1000500. doi: 10.1371/journal.ppat.1000500.g008 19578435
61. McBride A, Bhatt K, Salgame P (2011) Development of a secondary immune response to Mycobacterium tuberculosis is independent of Toll-like receptor 2. Infect Immun 79 : 1118–1123. doi: 10.1128/IAI.01076–10 21173309
62. Ehrt S, Voskuil MI, Schoolnik GK, Schnappinger D (2002) Genome-wide expression profiling of intracellular bacteria: the interaction of Mycobacterium tuberculosis with macrophages. Immunology of Infection: 169–180.
63. Flores
AR, Parsons LM, Pavelka MS (2005) Genetic analysis of the ß-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to ß-lactam antibiotics. Microbiol 151 : 521–532.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- Dimorphism in Fungal Pathogens of Mammals, Plants, and Insects
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging