
A Specific A/T Polymorphism in Western Tyrosine Phosphorylation B-Motifs Regulates CagA Epithelial Cell Interactions
As the dominant bacterium living in the human stomach, Helicobacter pylori has mixed roles in host health. One significant pathogenic risk factor is the CagA protein, which interferes with multiple host cell signaling pathways through its EPIYA tyrosine phosphorylation motifs (TPMs). Through database searching and silico analysis, we reveal a strong non-random distribution of the EPIYA B motif polymorphisms (including EPIYT and EPIYA) in Western H. pylori isolates, and provide evidence that the EPIYT are significantly less associated with gastric cancer than the EPIYA. By constructing a series of H. pylori cagA isogenic mutants and isogenic complementation plasmids, generating specific antibodies, co-culturing with human AGS cells, performing biochemical and modeling analysis, we demonstrate that CagA B-motif phosphorylation status is essential for its interaction with host PI3-kinase during colonization and that CagA with an EPIYT B-motif had significantly attenuated induction of interleukin-8 and the hummingbird phenotype, had higher affinity with PI3-kinase, and enhanced induction of AKT compared to the EPIYA. These findings provide insight into how Western H. pylori CagA regulates cancer-related activity inside host cells through the A/T polymorphisms at the functionally important B motif.
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004621
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004621
Summary
As the dominant bacterium living in the human stomach, Helicobacter pylori has mixed roles in host health. One significant pathogenic risk factor is the CagA protein, which interferes with multiple host cell signaling pathways through its EPIYA tyrosine phosphorylation motifs (TPMs). Through database searching and silico analysis, we reveal a strong non-random distribution of the EPIYA B motif polymorphisms (including EPIYT and EPIYA) in Western H. pylori isolates, and provide evidence that the EPIYT are significantly less associated with gastric cancer than the EPIYA. By constructing a series of H. pylori cagA isogenic mutants and isogenic complementation plasmids, generating specific antibodies, co-culturing with human AGS cells, performing biochemical and modeling analysis, we demonstrate that CagA B-motif phosphorylation status is essential for its interaction with host PI3-kinase during colonization and that CagA with an EPIYT B-motif had significantly attenuated induction of interleukin-8 and the hummingbird phenotype, had higher affinity with PI3-kinase, and enhanced induction of AKT compared to the EPIYA. These findings provide insight into how Western H. pylori CagA regulates cancer-related activity inside host cells through the A/T polymorphisms at the functionally important B motif.
Introduction
Helicobacter pylori, a spiral-shaped, microaerophilic gram-negative bacterium, persistently colonizes the human gastric mucosa [1,2]. H. pylori is carried by about 50% of the world’s population, and it exhibits extensive genetic diversity and distinct phylogeographic features [3,4]. Colonization increases risk of peptic ulcer disease and gastric carcinoma [5,6], and has been associated with diminished risk for esophageal inflammatory and neoplastic lesions [7,8], and childhood-onset asthma [9,10]. In 1995, the cytotoxin-associated gene A (CagA) protein of H. pylori was first associated with increased risk of gastric cancer [11], and since then, its pathogenic effects have been intensely studied [1,12].
The 120–145 kDa CagA protein is encoded by the cagA gene, located within the ∼40 kb H. pylori cag pathogenicity island (cagPAI) [13,14], along with a type IV secretion system that injects it into host gastric epithelial cells [15]. The carboxy-terminal region of CagA has several Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs which are strongly correlated to gastric disease outcomes [16,17]. The carboxy-terminal region of CagAs exhibit geographical, structural, and functional diversity, which is the result of the evolution of this protein through various modes of recombination mechanism [18].
In host cells, CagA molecules are associated with the inner surface of the plasma membrane and are dimerized via the carboxy-terminal EPIYA motif-containing regions [19,20]. CagA molecules undergo tyrosine phosphorylation (pCagA) at the EPIYA motifs by host Src-family kinases (SFKs) and Abl kinase [21–23]. CagA interacts with multiple host signaling factors through its EPIYA TPMs in a phosphorylation-dependent or -independent manner [24,25], affecting cell proliferation, motility, polarity, apoptosis, inflammation and nuclear responses, which may promote gastric carcinogenesis [26–28]. By mimicking host substrates through its C-terminal sequence, CagA inhibits PAR1/MARK family kinase pathways [29], and by association with the human tumor suppressor apoptosis-stimulating protein of p53-2 (ASPP2) through its N-terminal sequence, CagA inhibits apoptosis of host cells co-colonized with H. pylori [30]. Through phosphorylated EPIYA TPMs, pCagA binds to the Src homology 2 (SH2) domains of host signaling factors [26,28]. In this way pCagA activates the tyrosine phosphatase Shp2, which affects cell proliferation by inducing the ERK MAP kinase cascade [31–33], and also leads to cell elongation (producing the hummingbird phenotype) by inhibition of focal adhesion kinase (FAK) [34–36]. Phosphorylated TPMs also facilitate CagA interactions with C-terminal Src kinase (CSK), which inhibits SFK activity and negatively regulates CagA-Shp2 interaction [37]. The phosphorylated CagA TPMs directly bind other tyrosine phosphatases such as Shp1, phosphatidylinositide 3-kinase (PI3-kinase) and GTPase activating protein Ras GAP1, as well as adaptor proteins Crk-I, Crk-II, Crk-L, Grb2, and Grb7 [12,26]. Transgenic mice expressing wild-type CagA but not tyrosine-phosphorylation-resistant CagA developed gastric and small intestinal epithelial hyperplasia and neoplasia and B cell lymphomas and myeloid leukemias [38], supporting a critical role of CagA tyrosine phosphorylation in H. pylori-induced oncogenesis.
In addition to phosphorylation-dependent effects, CagA also associates with the polarity-regulating kinase partitioning-defective 1 (PAR1) protein through its C-terminal CagA-multimerization motif (CM), which overlaps with the EPIYA C - or D-TPM sequences [33,39]. The interaction between CagA and Par1 disrupts gastric epithelial cell tight junctions and apical-basal polarity [33], and enhances CagA TPM-phosphorylation-dependent interactions by stabilizing complex structures such as CagA-Shp2 [20]. In total, both phosphorylation-dependent and -independent [33–35] interactions affect host signaling pathways.
H. pylori has extensive genetic diversity [40–42]; isolates from different populations exhibit distinct biogeographic features, reflecting ancient human migrations [43]; cagA also possesses population-specific polymorphisms with major East Asian and Western groupings [44,45]. Four distinct CagA EPIYA TPMs (A, B, C or D), have conserved flanking sequences [46]. East Asian CagA include A-, B-, and D-TPMs, while Western CagA has A-, B-, and C-TPMs [47]. The East Asian CagAs are more interactive with host cells than Western CagAs, largely due to the higher affinity of the strongly phosphorylated D-TPM to Shp-2 than the Western C-TPMs [47,48]. Western CagA includes one or multiple C-TPMs, while East Asian CagA only has one D-TPM [12].
Regardless of C/D type, most CagA molecules include single A - and B-TPMs that undergo later and not simultaneous tyrosine phosphorylation [25]. The phosphorylated A - or B-TPMs have distinct host interaction partners from C - or D-TPMs and from each other [26], suggesting unique signaling functions. Here we report and characterize the functional importance of a specific A/T polymorphism present only within the Western CagA EPIYA B-TPMs.
Results
Polymorphisms in the CagA tyrosine phosphorylation EPIYA motif
Based on the CagA sequences published in Genbank, we investigated the variability within the C-terminal EPIYAs. A total of 2,561 complete or partial H. pylori CagA protein sequences were analyzed for polymorphisms within the EPIYAs. Our analysis indicated that the CagA B-TPM exhibits the highest variability (Table 1). EPIYA represents only 72.6% of 2,617 B-TPM sequences, with 23 alternative sequences present, including EPIYT, ESIYT, ESIYA and GSIYD; EPIYT is the most frequent alternative. In contrast, very few alternative sequences are identified in the A - and C-TPMs (Table 1), and none in the 1,196 type D-TPMs; only a single alternative sequence (EPIYV) is observed in the 1,620 C-TPMs (Table 1). All 25 independent CagA sequences with the EPIYV C-TPM show the same rare B-TPM (GSIYD). Only one low frequency (0.2%) alternative sequence (EPIYT) is observed in the A-TPMs (Table 1). In total, these results indicate specific polymorphisms in the CagA tyrosine phosphorylation (EPIYA) sequences, especially involving the B-TPMs. Among the 35 fully-sequenced H. pylori genomes and 81 ongoing partially-sequenced H. pylori genomes possessing the cagPAI (based on the NCBI Bioproject Database), EPIYA represents 52.6%, while EPIYT represents 34.5% of the B-TPMs.

The EPIYA - B-TPM of Western type CagAs has alternative sequences
Next, analyzing the B-TPMs in East Asian CagA possessing D-TPMs or Western type CagA possessing C-TPMs, we found that the distributions of the identified alternative sequences were significantly different (Table 2). In the Western CagA EPIYA B-TPMs, there are two major sequences (EPIYA; 55.5% and EPIYT; 32.9%). Other alternative TPMs comprise about 11.6% of the sequences. In contrast, among East Asian CagA B-TPMs, EPIYA is the major sequence (91.1%), EPIYT is at low (1.7%) frequency, and ESIYA is the alternative TPM with the highest frequency (6.0%) (Table 2). In total, Western CagAs have more alternative B-TPM EPIYA sequences than do East Asian CagAs.

Since Western CagAs may have more than one C-TPM, we asked whether the presence of the alternative EPIYA B-TPMs is related to C-TPM number. There was no link between the alternative B-TPM sequences and the number of C-TPMs, except for ESIYT, which co-appears with 2 C-TPMs on the same Western CagAs at significantly high frequency (Table 3). These findings indicate a common previously unrecognized TPM polymorphism with strongly non-random distribution in the available census of strains.

Pathological association with the Western B-domain TPM alternatives
To assess the relationship of B-TPM sequence and clinical outcome, we analyzed a total of 364 Western CagAs, which were reported to be present in patients with defined gastrointestinal pathology, according to the descriptions from Genbank and the indicated publications (Table 4). Compared with gastritis alone, gastric cancer was significantly associated with the EPIYA B-TPMs, whereas duodenal ulcers were significantly associated with the EPIYT B-TPM (Table 4). That these polymorphisms in B-TPM are associated with different diseases suggest that EPIYT and EPIYA may differentially regulate the CagA pathophysiologic roles in Western H. pylori strains that interact with host cells; while the CM and CRPIA motifs are commonly present in both forms of CagA.

Codon usage in the Western B-domain TPMs
To assess whether the EPIYA/T polymorphisms at the protein level were random, we compared the codons in which the A/T polymorphisms were present (S1 Table in S1 Text and Fig. 1). Only one major (91.1%) set of codons (TAT ACT) encodes the YT of EPIYT B-TPM. In contrast, there are two major sets of codons (TAC GCT and TAT GCT) that encode the YA of the EPIYA B-TPMs with similar frequencies (59.4% and 40.4%, respectively). Compared with YA and YT codons in the other loci in the H. pylori 26695 genome, this distribution is significantly non-random (p<0.001). This non-random distribution suggests that the polymorphisms have been selected rather than being stochastic, potentially providing for different CagA functional roles.

Construction of H. pylori isogenic cagA mutants with TPM polymorphisms
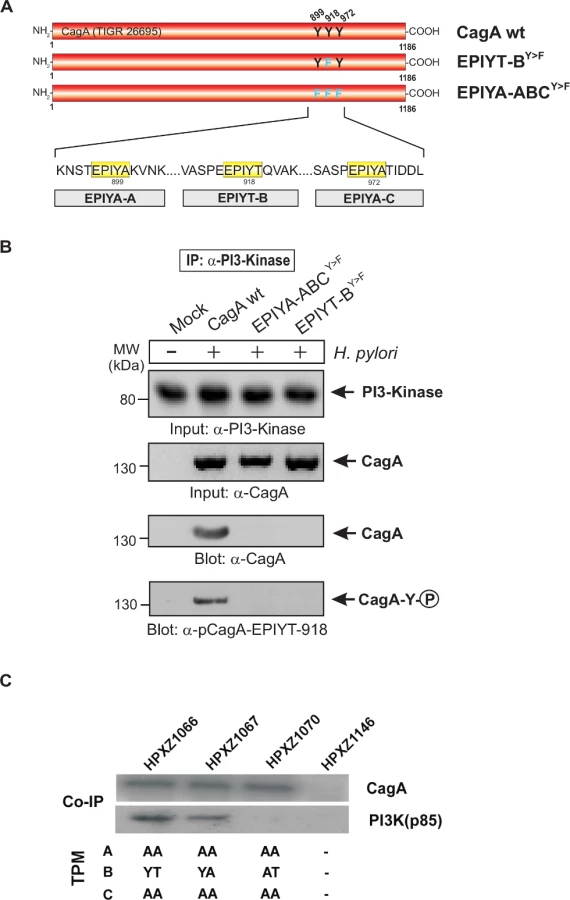
To investigate how the major (EPIYT) alternative TPM affects CagA functions, we created a series of isogenic H. pylori mutants that express a Western CagA with variant TPMs. The Western-type cagA gene from H. pylori strain 147C, originally isolated from a human antrum corpus [44], was used as the CagA parent gene for the constructions, created in a site-directed manner using a recombinant PCR technique (S1A Fig. in S1 Text). This 147C cagA gene possesses one A-, B - and C-TPM. The gene product CagA147C has been previously shown to induce both AGS cell hummingbird phenotype and IL-8 production [44,49]. We replaced the H. pylori 26695 cagA ORF with isogenic cagA genes in its native genetic locus via transformation. The set of 6 isogenic H. pylori 26695 mutants all possess cagA with EPIYA, EPIYT, or EPIAT (as a control: presumed to be inactive) B-TPMs, and with EPIYA or EPIAA forms of the A - or C-TPM (S1A Fig. in S1 Text). These mutants exhibit the same physiological characters, transformation frequency and growth rates in vitro as the parental 26695 strains. Western blotting confirmed that each isogenic mutant expressed a CagA molecule (S1B Fig. in S1 Text), and sequencing of each cagA ORF confirmed that the sequences surrounding the target site(s) were identical.
CagA EPIYT B-TPM is phosphorylated during H. pylori co-culture
To investigate CagA B-TPM phosphorylation status and function during co-culture, we first generated phospho-specific and non-phospho antibodies, α-pCagA-EPIYT-918 (phospho) and α-CagA-EPIYT-918 (non-phospho) against the EPIYT B-TPM motif of CagA, corresponding to the amino acid residues derived from strain 26695 (S2 Fig. in S1 Text). Our analysis indicated that the α-pCagA-EPIYT-918 (phospho) or α-CagA-EPIYT-918 (non-phospho) antibodies recognize the B-TPM including both EPIYT B-TPM and EPIYA B-TPM, but not the A-TPM or C-TPM (control) peptides (S2 Fig. in S1 Text). To investigate whether this CagA EPIYT B-TPM can be phosphorylated during co-culture, AGS cells were co-incubated for 6 h with a set of clinical H. pylori strains, which vary in their CagA carboxy-terminal TPM sites. Phosphorylation of CagA at B-TPM was examined using the now-confirmed phospho-specific α-pCagA-EPIYT-918 antibody. Results indicated that the EPIYT-motif can be phosphorylated during co-culture, but to varying extents (Fig. 2).

The alternative B-TPM EPIYT has enhanced PI3-kinase/AKT pathway induction
To investigate how this alternative B-TPM affects the role of CagA in host signaling pathways, we co-cultured the isogenic H. pylori mutants with AGS cells for 24 h and analyzed cell lysates for CagA-mediated protein binding and signal activation by immunoblotting and co-immunoprecipitation. Co-culture with the EPIYT isogenic strain induced the phosphorylation of serine/threonine kinase AKT (also called protein kinase B, PKB) at threonine residue 308(T-308) 2.4 ± 0.10 fold, compared with 1.7 ± 0.14 fold for the EPIYA strain (Fig. 3), indicating intensified induction of PI3-kinase/AKT activation. Neither co-culture with the isogenic H. pylori strain possessing a B-domain with an abolished tyrosine phosphorylation site, nor co-culture with the cagA knockout strain significantly increased AKT phosphorylation at T-308 (Fig. 3), suggesting that CagA-positive H. pylori can activate the PI3-kinase/AKT pathway with activity dependent on B-domain TPM phosphorylation.

The EPIYT site at B-TPM of CagA is necessary for interaction with PI3-kinase
To investigate the interaction between the CagA B-TPM and PI3-kinase, we first used α-CagA antibodies to perform immunoprecipitation after co-culture of AGS cells with isogenic H. pylori 26695 strains with cagA variations. Western blotting using α-PI3-kinase antibody revealed that only wild-type CagA can bind to PI3-kinase but not EPIYA-ABCY>F or EPIYT-BY>F mutants (S3 Fig. in S1 Text). These data indicate that EPIYT B-TPM, but not EPIYA A - or EPIYA C-TPMs, is necessary for this interaction. The α-pY-99 control blot shows that the wild-type CagA is phosphorylated, while phosphorylated CagA with the EPIYT-BY>F mutation cannot interact with PI3-kinase (S3 Fig. in S1 Text). In a similar experiment, AGS cells were co-cultured with several isogenic CagA-expressing H. pylori strains including the EPIYT-ACY>F and EPIYT-BY>F mutants, followed by α-CagA immunoprecipitation. For both CagA variants (EPIYT-ACY>F and EPIYT-BY>F), the phosphorylation signal was as expected. Western blotting using α-PI3-kinase antibody revealed that only CagA (wt) and EPIYT-ACY>F (with intact B-TPM) can bind to PI3-kinase, but not the EPIYT-BY>F mutant (Fig. 4). These findings indicate that EPIYT B-motif can be phosphorylated, which is necessary for the PI3-kinase-CagA B-TPM interaction. To further confirm our observation, CagA presence and phosphorylation at the EPIYT-site was examined using phospho-specific α-pCagA-EPIYT-918 and α-CagA antibodies when all samples contained similar amounts of PI3-kinase (Fig. 5A and B). Only the lane with H. pylori expressing wild-type CagA revealed a signal for CagA and phosphorylation at EPIYT B-TPM in the immunoprecipitation. These findings provide further evidence that phosphorylated EPIYT B-TPM is necessary for the interaction with PI3-kinase.

The alternative B-TPM EPIYT has enhanced PI3-kinase affinity
Co-immunoprecipitation assays indicated that in AGS cells, CagAs possessing the EPIYA or EPIYT B-TPM interacted with PI3-kinase (p85), while CagA possessing EPIAT did not (Fig. 5C), suggesting that CagA activates PI3-kinase/AKT signaling pathways by interacting with the kinase via a functional B-motif. In AGS cells, CagA molecules possessing the B-domain EPIYT had higher affinity for PI3-kinase than those with EPIYA (Fig. 5C). These results suggest that the CagA interaction with PI3-kinase has activating, rather than inhibiting effects on its major downstream effector, AKT. The CagA molecules without a C-TPM sequence (as in CagA147A) did not induce AKT phosphorylation at T-308, indicating that the C-TPM sequence is necessary for expression of the B-domain function. The C-domain enhancement of B-domain activation of the PI3-kinase/AKT pathway is not dependent on the C-domain tyrosine phosphorylation since the B-domain EPIYT (in HPXZ1066) and B-domain EPIYA (in HPXZ1067) without C-domain activity activated the PI3-kinase/AKT pathway. This suggests the importance of maintaining the CagA dimerization state via the CagA multimerization (CM) sequence [39].
Molecular modeling of CagA B-TPM interaction with the PI3-kinase SH2 domain
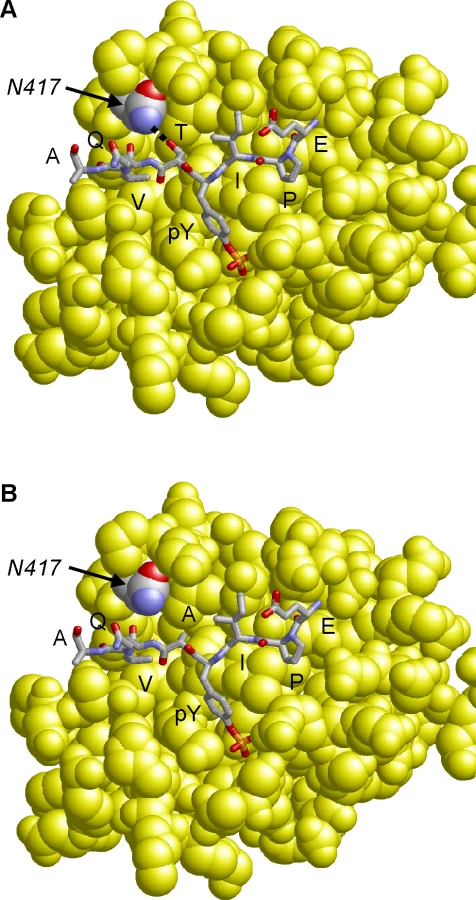
Molecular modeling of the CagA B-TPM EPIYTQVA sequence in complex with the N-terminal PI3-kinase SH2-domain reveals that the threonine residue at the pY+1 position forms a side chain hydrogen bond with an asparagine residue (N-417) of PI3-kinase (Fig. 6A). The respective hydrogen bond cannot be formed for the “EPIYAQVA” motif, because the alanine present at the respective sequence position lacks the hydroxyl group side chain required for an interaction (Fig. 6B). Therefore, a T>A substitution at the pY+1 position is expected to significantly decrease binding affinity of the B-TPM motif to PI3-kinase. This model is also supported by experimental peptide binding studies, that show that threonine at the pY+1 position forms a stronger interaction than alanine with the respective SH2-domain [50]. Notably, PI3-kinase also contains a second SH2 domain, in which the asparagine required for ligand binding is conserved (N-707), suggesting that both PI3-kinase SH2-domains possess similar binding specificity for the pY+1 position.
The alternative B-domain TPM EPIYT attenuates induction of the AGS cell hummingbird phenotype
CagA-positive H. pylori co-cultured with AGS cells induce an elongated cell morphology known as the hummingbird phenotype which is associated with effects on host cell polarity, migration, and adhesion [36,51]. Next we evaluated whether the major alternative B-domain TPM EPIYT affected CagA-induced hummingbird cell formation by co-culturing AGS cells with the isogenic H. pylori 26695 cagA mutants based on a comparable bacterial/host cell population level. We found that the isogenic cagA+ H. pylori strains induced significantly more hummingbird-type AGS cells than did an H. pylori ΔcagA mutant, but abolishing the A - and C - (EPIYA) TPMs significantly decreased hummingbird phenotype, indicating their involvement in hummingbird induction (Fig. 7A and B). In the presence of functional A and C TPMs, CagA with the B-domain EPIYA induced significantly more hummingbird cells than the CagA possessing the B-domain EPIYT or EPIAT (Fig. 7A and B). This observation suggests that the B-domain EPIYT has functional differences as compared to EPIYA. In the presence of non-functional (EPIAA) A - and C-TPMs, strains with CagA possessing EPIYT or EPIYA B-TPM induced significantly more hummingbird cells than the strain possessing the EPIAT B-TPM (p<0.05) (Fig. 7B). B-TPM functions may be affected by the A - and C-TPMs since the significant differential effects of EPIYT and EPIYA B-TPM were lost when we abolished those tyrosine phosphorylation sites.

The alternative B-domain TPM EPIYT attenuates AGS IL-8 induction
Interleukin-8 (IL-8), a neutrophil-activating chemokine [52], may play an important role linking chronic inflammation and carcinogenesis [53]. The IL-8 induction effect is also associated with the number of C domains in Western CagA+ strains [49,54]. Here, we evaluated whether the major alternative B-domain TPM sequence, EPIYT, affects CagA-induced IL-8 production by co-culturing AGS cells with the isogenic H. pylori 26695 cagA mutants based on a comparable bacterial/host cell population level. At 24 h, AGS cells co-cultured with H. pylori ΔcagA mutants had the same IL-8 level as the AGS cells without H. pylori co-culture (control), but co-culture with the isogenic H. pylori cagA variants induced significantly higher IL-8 levels. Under these conditions, the isogenic cagA mutant containing the EPIYA B-TPM induced significantly more IL-8 induction than the mutant with the EPIYT B-TPM (Fig. 7C), a finding indicating differential EPIYA - and EPIYT-B TPM protein functions. The isogenic mutant with an EPIAT B-TPM had significantly decreased IL-8 induction (Fig. 7C). These results confirm that CagA can induce AGS cell IL-8 production via its B-domain TPM, and indicate the differential EPIYT and EPIYA functions.
Discussion
A key host interaction factor of H. pylori, the CagA protein, has multiple polymorphisms which differ in their affinities to host interaction partners and in their regulation of gastric cell signaling cascades. EPIYA TPMs are critically important for CagA regulation of host signaling pathways [28,55], and the four types (A, B, C and D) have different host interaction partners [26] and/or varying affinities to the same partners [31,46], suggesting differential roles in regulation of host signaling pathways.
Matsunari et al. first reported there are three most common types of EPIYA sequences including the EPIYA, EPIYT and ESIYA, and that EPIYT of B-TPM is more predominant in Western CagAs [56]. In this study, we further reveal the A/T polymorphism that specifically occurs within the Western type B-domain, and we provide evidence for the first time that this polymorphism significantly affects CagA functions in host cells. Indeed, the B-domain polymorphisms of the Western strains differed in their correlation with upper GI tract diseases (Table 4), suggesting that a single SNP in a major bacterial interactive factor could decide disease outcome. The isogenic H. pylori cagA mutants expressing from the native genetic locus created for the present investigations may be valuable for further studies.
Through its B-domain, CagA interacts with host partners including the Shp2 phosphatase, Csk and PI3-kinases, as well as adaptor proteins Grb2 and Crk and the Shp1phosphatase, all of which carry SH2-domains [28]. Among these, Shp1 and Shp2 also interact with the A - or C-domains, Csk with the A-domain, Grb2 with C-domain, while PI3-kinase and CrkII only with the B-domain in a tyrosine-phosphorylation-dependent manner [26,28]. Different TPMs have both shared and specific host interaction partners suggesting that different EPIYA motifs could have specific roles in regulating host signaling pathways. Moreover, other motifs aside from the EPIYA TPMs could also be involved in CagA interactions with these host factors. CagA activation of PI3-kinase/AKT appears dependent on the CRPIA sequences [24], but activation of PI3-kinase/AKT also may be CagA-independent [37]. Our finding that H. pylori CagA with functional EPIYA or EPIYT B-domains binds with PI3-kinase confirms and extends the observations by Selbach et al. [28]. Recently, Lind et al. developed a novel strategy to systematically analyse phosphotyrosine antibodies recognizing single phosphorylated CagA EPIYA-motifs utilizing synthesized phospho - and non-phosphopeptides [57]. With this strategy, by generating and analyzing a novel phospho-specific CagA B-motif antibody (anti-pCagA-EPIYT-918) and isogenic CagA mutants with abolished TPMs, we further confirmed the CagA EPIYT-B domain tyrosine-phosphorylation status during H. pylori co-culture with host cells and revealed that tyrosine phosphorylation of the B-domain is necessary for the interaction between CagA and PI3-kinase. We observed that the EPIYT B-domain has higher affinity to PI3-kinase and greater AKT activation than the EPIYA B-domain. Our analysis by structural modeling of the CagA EPIYA and EPIYT B-motifs interacting with the SH2 domain of PI3-kinase further revealed the nature of differential interaction effects caused by the A/T polymorphism.
During gastric colonization, the host tyrosine kinases Src and Abl phosphorylate H. pylori CagA EPIYA motifs [21–23,25], which differ from the classical consensus phosphorylation sites in eukaryotic target factors (E-E-I-Y-E/G-X-F and I/V/L-Y-X-X-P/F of two tyrosine kinases, respectively) [58]. This suggests that the CagA EPIYA motif phosphorylation level in host cells may not be maximal. In that case, we propose that the A/T polymorphism/switch in the EPIYA motif could be important in regulation of the TPM phosphorylation efficiency and stability.
Strain-specific CagA sequence variation involves both conserved and non-conserved regions. CagA147C used in our studies and CagA26695 used by Suzuki et al. share only 87.1% identify and have numerous SNPs flanking each EPIYA/T TPM as well as differing tagging. Considering the complex interactions between CagA with host protein partners as well as the complex signaling network, use of transfected protein-expressing systems and both technical and structural differences may affect signaling. The number of H. pylori CagA molecules within host cells in different assays (e.g. CagA transfection and co-culture with H. pylori with native expressing CagA) could markedly vary with differing kinetics of CagA phosphorylation, leading to different outcomes. Increased AKT activation and decreased IL-8 secretion of AGS cells [59], and PI3-kinase/AKT pathway repression of IL-8 production during Salmonella co-culture with intestinal epithelial cells [60] have been described. Strain - and time-dependent H. pylori CagA-mediated IL-8 induction in AGS cells occurs through the Erk and NF-κB pathways [49,61–64]. CagA-mediated PI3-kinase/AKT activation attenuates IL-8 induction by repressing Erk/NF-κB including through the Shp-2/Erk pathway [49], the Shp2-independent Ras/Raf/Mek/Erk pathway [65], and by Ras-independent Erk activation [66]. Inactivating B-TPM abolished PI3-kinase/AKT activation, but decreased IL-8 secretion. Through B-TPM, CagA also may interact with multiple other proteins in a site-competition and/or time-dependent manner. For example, CagA TPMs interact with Shp2 through phosphorylated EPIYAs [28,34] enhancing Shp2 activity and Erk phosphorylation [31]. The B-TPM could positively regulate IL-8 production through activating the Shp2/Erk/ NF-κB pathway [49]. Abolishing the isogenic single B-TPM inactivated the IL-8-repressing PI3-kinase/AKT, but also inactivated IL-8-stimulating Shp2/Erk. Repression of IL-8 production by the B-TPM-mediated PI3-kinase/AKT effect reflects cross-talk between the PI3-kinase/AKT and Shp2/Erk pathways. Consistent with the overall differential signaling, Western cagA+ H. pylori strains with EPIYT or EPIYA B-TPMs are associated with different patterns of clinical outcomes (Table 4 and Fig. 8), an observation that needs to be confirmed. The clinical outcome of H. pylori colonization results from long-term processes, and therefore, how the B-TPM-mediated PI3-kinase/AKT effect alters host gastric cancer development in long-term H. pylori colonization deserves further investigation. The PI3-kinase/AKT signaling pathway controls many of the hallmarks of cancer, and many tumor tissues have enhanced PI3-kinase/AKT activities [67,68]. However, PI3-kinase/AKT and their effectors are pleiotropic and have complex crosstalk and feedback behaviors in the signaling network, which are not fully known [67,68]. We studied the regulation of CagA on PI3-kinase/AKT pathway in vitro at an early time of bacterial interaction with host cells, while long-term studies in mice or observations in patients at risk for gastric cancer will help resolving the clinical significance of the polymorphism.

H. pylori colonization induces AGS cell scattering and elongation (hummingbird phenotype) through multiple CagA-related mechanisms; CagA binds to Csk through its A - or B-TPMs, inhibiting SFK activity, and binds to Shp2 through its A-, B - or C-TPMs, leading to FAK dephosphorylation [35,69]. Attenuated hummingbird phenotype induction present in the cagA mutant with the EPIYT B-TPM (vs. EPIYA) reflects differential B-domain functional roles, possibly through modulating direct interactions with Csk or Shp2. Inhibition of AKT activation by the PI3-kinase inhibitor LY294002 has no effect on the hummingbird phenotype [36]. However, LY294002 inhibits PI3-kinase catalysis by competing for ATP binding [70], but does not directly affect the p85 binding activity with tyrosine-phosphorylated motifs such as the CagA B-TPM. Such findings suggest that hummingbird induction by the CagA B-TPM relates to the competition between PI3-kinase and Csk or Shp2 for binding at the B-TPM, but is not directly related to PI3-kinase activity.
The A - and B-domains have unique host interacting partners, such as Csk, which do not interact with C - or D-TPMs. These domains could possibly attenuate the C-domain-Shp2-interaction by binding with Csk to inactivate SFK members [71]. In this model, the C - and D-TPMs serve as the primary phosphorylation motifs interacting with host signaling partners, and the A - and B-TPMs serve as secondary sites, phosphorylated after C - or D-TPMs [25], suggesting a potential regulatory role through competition between different TPMs. The EPIYT/EPIYA B-TPM polymorphism that we studied provides a new level of complexity in H. pylori colonization and pathophysiology.
Materials and Methods
Bacterial strains, media and growth conditions
H. pylori strain 26695 was used to construct a series of isogenic cagA mutants, which express CagA variants from the native CagA genetic locus [72]. H. pylori strains 147C and 147A, a pair of naturally occurring isogenic cagA strains with EPIYA ABC and AB motifs, respectively [49], were used as isogenic cagA sequence templates. H. pylori CagA-expressing strains P227, Oki-61, P341, 2002-370 and 26695, also were used for AGS co-culture and evaluation of CagA phosphorylation [73]. The H. pylori strains were grown at 37°C in 5% CO2 on trypticase soy agar (TSA) plates with 5% sheep blood (TSA, BBL Microbiology Systems, Cockeysville MD) or Brucella agar plates (BA, Difco Laboratories, Detroit MI) supplemented with 10% newborn calf serum (NBCS; Serologicals Corporation, Norcross GA) and suitable antibiotics [74]. Antibiotic-resistant isogenic H. pylori strains were selected with kanamycin (Km; 10 μg/mL) or chloramphenicol (Cm; 30 μg/mL), as appropriate. Alternatively, H. pylori strains were grown in thin layers on horse serum GC agar plates supplemented with vancomycin (10 μg/mL), nystatin (1 μg/mL), and trimethoprim (5 μg/mL), and for defined mutants with Cm (6 μg/mL) and/or Km (8 μg/mL) at 37°C for 2 days in an anaerobic jar containing a Campygen gas mixture of 5% O2, 10% CO2, and 85% N2 (Oxoid, Wesel, Germany) [75]. E. coli DH5α was grown in Luria-Bertani (LB) medium at 37°C [76]. Ampicillin (Ap; 100 μg/mL), Cm (30 μg/mL) or Km (50 μg/mL) were used for selecting vectors or the constructs in E. coli during cloning.
Construction of isogenic H. pylori strains
Western type H. pylori strain 147C cagA has an EPIYT B-TPM, as well as one EPIYA -A and -C TPM (cagA147C B:EPIYT A&C:Y) [49]. To evaluate B-TPM A/T polymorphism effects on CagA functions, the threonine site of EPIYT B-TPM was replaced with alanine by recombination-PCR mediated site-directed mutagenesis, leading to the generation of the isogenic cagA, cagA147C B:EPIYA A&C:Y. To evaluate B-TPM A/T polymorphism effects on tyrosine phosphorylation of CagA B-TPM, the two tyrosine phosphorylation sites, A - and C-TPMs of the wild-type cagA (cagA147C B:EPIYT A&C:Y) and the isogenic cagA (cagA147C B:EPIYA A&C:Y), were replaced with alanine, leaving the B-TPM as the only functional tyrosine phosphorylation site. This resulted in two isogenic cagAs: cagA147C B:EPIYT A&C:Y>A and cagA147C B:EPIYA A&C:Y>A. For controls, the tyrosine of B-TPMs of the wild-type and the isogenic cagAs were further replaced with alanine to abolish B-TPM tyrosine phosphorylation function, leading to cagA147C B:EPIAT A&C:Y and cagA147C B:EPIAT A&C:Y>A. Each of the wild-type and isogenic cagA genes was first fused at the 3’ end to the hemagglutinin (HA) tag and then linked to a 617 bp cagA downstream region sequence based on the 26695 genomic sequence [72]. An aphA (KmR) cassette [77] was inserted between the cagA-HA fusion gene and the cagA downstream region sequence as a selection marker for the H. pylori mutant construction. Each construction was cloned into the vector pGEM-T easy (Promega, Madison WI), creating plasmids pXZ476, pXZ465, pXZ468, pXZ471, pXZ472, and pXZ475, which carry cagA147C B:EPIYT A&C:Y, cagA147C B:EPIYA A&C:Y, cagA147C B:EPIAT A&C:Y, cagA147C B:EPIYT A&C:Y>A, cagA147C B:EPIYA A&C:Y>A, and cagA147C B:EPIAT A&C:Y>A, respectively (S2 Table in S1 Text). To replace the cagA26695 sequence of H. pylori strain 26695 with the isogenic cagA147C sequences, a truncated cagA147CN (2445 bp) lacking C-terminal A - B - or C-TPMs was cloned and linked with a cat (CmR) cassette [77] and then with the 617 bp cagA downstream region sequence based on the 26695 sequence using pGEM-T easy, creating pXZ478 (S2 Table in S1 Text). To construct the H. pylori ΔcagA control, the cagA upstream (839 bp) and downstream (1076) region sequences of H. pylori 26695 were linked and inserted with an intervening sacB-cat (CmR) cassette to replace the entire cagA26695 ORF on the same vector, creating plasmid pXZ083 (S2 Table in S1 Text).
To express the series of mutant cagA genes from the cagA native genetic locus in the 26695 genetic background, we first replaced the cagA26695 sequence on the 26695genome with a cagA147Cs via homologous recombination (S1 Fig. in S1 Text). The wild-type H. pylori strain 26695 was transformed by plasmid pXZ478 to CmR to create strain HPXZ1043 with isogenic cagA147CN replacing native cagA26695. DNA sequencing confirmed the replacement of cagA26695 with the cagA147CN sequence in the 26695-derived CmR/KmS strain HPXZ1043, and western blot confirmed the expression of the truncated CagA147CN protein from the native locus and the CagA promoter. Strain HPXZ1043 CmR/KmS was then transformed to KmR/CmS with plasmids, pXZ476, pXZ465, pXZ468, pXZ471, pXZ472, or pXZ475, to create mutants HPXZ1061 (cagA147C B:EPIYT A&C:Y), HPXZ1062 (cagA147C B:EPIYA A&C:Y), HPXZ1065 (cagA147C B:EPIAT A&C:Y), HPXZ1066 (cagA147C B:EPIYT A&C:Y>A), HPXZ1067 (cagA147C B:EPIYA A&C:Y>A) and HPXZ1070 (cagA147C B:EPIAT A&C:Y>A), respectively. The wild-type H. pylori strain 26695 was transformed to CmR with pXZ083 to create the cagA-negative mutant HPXZ1146 (ΔcagA::sacB-cat) (S2 Table in S1 Text). To confirm each construction, sequencing of related regions was performed at Macrogen (Rockville MD), and all sequence analysis was performed using Sequencher (Gene Codes, Ann Arbor MI).
Cloning, complementation and site-directed mutagenesis of CagA using shuttle vector pHel3
To analyse the EPIYA - and EPIYT-motifs in the CagA protein, the complete cagA gene of H. pylori strain 26695 (accession number: AAD07614) containing its promoter was amplified by PCR, cloned into the pCR2.1 vector (Invitrogen) and sequenced [73]. For construction of a complementation vector, this cagA fragment was cloned in the E. coli/H. pylori shuttle vector pHel3 containing the oriT of RP4 and a kanamycin resistance gene cassette (Aph-A3) as a selectable marker, resulting in vector pSB19 [73]. Site-directed mutagenesis of tyrosines Y-899, Y-918 and Y-972 in the CagA sequence was done using the Sculptor mutagenesis kit, as directed (Amersham Pharmacia Biotech) and resulting plasmids were transformed into H. pylori isogenic ΔcagA mutant [78], as described [79].
Synthesis of phospho - and non-phospho CagA TPM peptides
The C-STEPIYAKVNK (EPIYA-A), C-STEPI(pY)AKVNK (phospho-EPIYA-A), C-PEEPIYTQVAK (EPIYT-B), C-PEEPI(pY)TQVAK (phospho-EPIYT-B), C-PEEPIYAQVAK (EPIYA-B), C-PEEPI(pY)AQVAK (phospho-EPIYA-B), C-SPEPIYATIDD (EPIYA-C) and C-SPEPI(pY)ATIDD (phospho-EPIYA-C) amino acid sequences were synthesized by Jerini AG (Berlin, Germany). These 11-mer peptides were chosen because prior studies have shown that α-phosphotyrosine antibodies typically recognize short phosphopeptides, and 11-mer and 9-mer sequences are both necessary and sufficient [57]. Commonly, 11-mer peptides also are used for immunizations to generate phospho-specific antibodies, which then recognize the corresponding phosphopeptides bound to affinity columns and in ELISA (Biogenes, Berlin, Germany). All above EPIYA and EPIYT peptides were purified by HPLC, and full-length synthesis as well as purity of each peptide was confirmed by mass spectrometry by Jerini AG. The peptides were resolved at a concentration of 1 mg/mL in DMSO and stored at −20°C.
Dotblot analysis
Twenty μg of each CagA peptide were mixed in 1 mL of TBST blotting buffer (140 mM NaCl; 25 mM Tris-HCl, pH 7.4; 0.1% Tween-20). These peptide samples were spotted onto Immobilon-P membrane (Merck Millipore, Darmstadt, Germany) using the BioDot SF apparatus (Bio-Rad, Munich, Germany). The resulting Dotblots were dried and subjected to antibody detection as described below for Western blots [57].
AGS culture and co-culture assays
Human gastric epithelial (AGS; ATCC CRL 1739) cells (obtained from American Type Culture Collection) were cultured at 37°C in a humidified atmosphere with 5% CO2 in RPMI 1640 (Invitrogen, Carlsbad CA) with 10% fetal bovine serum (FBS; Invitrogen) with antibiotic-antimycotic mixture (1X; Life Technologies, Grand Island NY) [80]. Before co-culture experiments, AGS cells (2×105cells/well) were transferred to a new 6-well plate and incubated in fresh RPMI 1640 with 10% FBS and antibiotic-antimycotic for 24 h. The attached AGS cells were washed and incubated in the RPMI 1640 media without serum or antibiotic for 16 h. The AGS cells were then co-cultured with PBS-prewashed H. pylori cells, which were collected from 24-h TSA plates at a multiplicity of infection (MOI) of 100 : 1 and from fresh serum - and antibiotic-free RPMI 1640 for 8–24 h.
Cell elongation assays
AGS cultures and the co-culture of AGS and H. pylori were grown on coverglass (Fisher Scientific, Pittsburgh PA) in 6-well plates [81]. After 24-h co-culture, 10 random fields of view for each coverglass were examined at a magnification of ×200 using a Leica DMI 6000B microscope. Alternatively, the AGS cells were co-cultured with H. pylori cells at MOI of 50 for 6 h, when the cells were harvested in ice-cold PBS containing 1 mmol/L Na3VO4 (Sigma-Aldrich). Elongated AGS cells in each experiment were quantitated in 10 different 0.25-mm2 fields using an Olympus IX50 phase contrast microscope [82]. All experiments were performed in triplicate.
IL-8 assay
After 24-h incubation, co-culture media were sampled and centrifuged at 16,000 g, and supernatants collected. AGS cell IL-8 secretion was measured by an enzyme-linked immunosorbent assay using the Human IL-8 ELISA Kit II (BD Biosciences, San Jose CA), in accordance with the manufacturer’s instructions.
Generation of phospho-specific and non-phospho-specific α-EPIYT antibodies
Phospho-specific and non-phospho polyclonal rabbit CagA antibodies were raised against peptides corresponding to the following amino acid residues derived from the B-TPM motif of strain 26695: C-PEEPIYTQVAK (non-phospho-EPIYT-B) and C-PEEPI(pY)TQVAK (phospho-EPIYT-B). For this purpose, both peptides were conjugated to Limulus polyphemus haemocyanin carrier protein and two rabbits each were immunized by Biogenes GmbH (Berlin, Germany), according to standard protocols. The resulting phospho-specific antibodies (α-pCagA-EPIYT-918) were affinity-purified against the corresponding non-phospho peptide bound to a column. The resulting non-phospho antibodies (α-CagA-EPIYT-918) were affinity-purified against the corresponding phospho-peptide. Both antibodies were prepared and purified by Biogenes GmbH (Berlin, Germany). Their specificity was confirmed by dot blotting against the phospho - and non-phospho peptides (S2 Fig. in S1 Text).
Immunoblotting, immunoprecipitation, and antibodies
To prepare whole cell extracts for immunoblotting, media were removed after 24-h incubation and AGS cells were washed with ice-cold PBS 5 times to remove H. pylori cells. The whole cell lysates for western blotting were prepared with RIPA lysis buffer (Thermo Scientific Pierce, Rockford IL) with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific Pierce). Lysates were separated by SDS-PAGE (Expedeon Inc. San Diego CA) and transferred to Immobolin-P PVDF Transer Membrane (Fisher Scientific). Membranes were blocked in TBST with 3% BSA or 5% skim milk for 1 or 2 h at room temperature. Membranes were incubated with the following antibodies according to the instructions of the manufacturer. Immunodetection of CagA peptides and the various proteins of interest were performed using horseradish peroxidase–conjugated anti-mouse or anti-rabbit polyvalent sheep immunoglobulin secondary antibodies and using chemiluminescence reagents, West Femto Chemiluminescent Substrate (Thermo Scientific Pierce) or Amersham ECL Western Blotting Detection Reagents (GE Healthcare, Piscataway NJ) in accordance with the manufacturers’ instructions [83]. After exposure to X-ray film, the target band intensities were quantified using ImageJ software (NIH, Bethesda MD). For immunoprecipitation, the whole cell lysates were prepared with IP Lysis/Wash buffer (Thermo Scientific Pierce, Rockford IL) with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific Pierce). Immune complexes were prepared using Pierce Crosslink Immunoprecipitation kit (Thermo Scientific Pierce) in accordance with the manufacturers’ instructions. The immunoprecipitates were subjected to SDS-PAGE, as described above. Anti-actin, anti-AKT, anti-phospho-AKT (pThr308), and anti-phospho-AKT (pSer473) ePI3-kinase were obtained from Cell Signaling Technology (Danvers MA). Anti-HA, anti-Shp2, anti-Crk II, and anti-phospho-CrkII (pTyr221) were obtained from Thermo Scientific Pierce, anti-phosphotyrosine was obtained from EMD Millipore Inc. (Billerica MA), anti-Csk, anti-phospho-Csk (pSer364), anti-GAPDH and pan-phosphotyrosine antibody pY-99 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz CA), and anti-CagA was produced as described [49]. Anti-PI3-kinase (p85) antibodies were obtained from Cell Signaling Technology (Danvers MA) or Santa Cruz Biotechnology, Inc. (Santa Cruz CA). Rabbit polyclonal and mouse monoclonal α-CagA antibodies were from Austral Biologicals, or from Emd Millipore Corporation (Billerica MA).
The phosphorylation status of CagA and bound PI3-kinase also was verified by immunoprecipitation experiments as described [73,79]. Briefly, co-cultured or control AGS cells were washed with cold PBS and lysed for 30 min at 4°C in lysis buffer (20 mM Tris pH 7.2, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM Na3VO4, COMPLETE™ inhibitor mix from Roche). Lysates were pre-cleared with protein G-Sepharose (Pharmacia, Uppsala, Sweden) for 2 h at 4°C. Two micrograms of the monoclonal α-CagA antibody (Austral Biologicals, San Ramon CA) or polyclonal antibody against the p85 subunit of PI3-kinase (Santa Cruz Biotechnology, Dallas TX) were added to the supernatant and incubated overnight at 4°C on a shaker. Immune complexes were precipitated by addition of protein G-sepharose for 2 h, washed three times in 0.5× PBS and then mixed with equal amounts of 2× SDS-PAGE buffer. Precipitates were analyzed by SDS-PAGE and immunoblotting.
Quantification of Dotblot and Western blot signals
Spot or band intensities on blots probed with the different α-phosphotyrosine antibodies were quantitated with the Lumi-Imager F1 (Roche Diagnostics, Mannheim, Germany). Densitometric measurement of signal intensities revealed the percentage of phosphorylation per sample [84].
3D-Modeling
The CagA-PI3-kinase interaction was modeled using the crystal structure of the N-terminal PI3-kinase SH2-domain in complex with a C-kit phosphotyrosyl peptide (PDB: 2IUH) as a template [85]. Modeling of the Cag-A B-TPM motif was performed using SwissModel [86] and included the sequence stretches “EPIYTQVA” or “EPIYAQVA”. Structural analysis and visualization was performed using RasMol [87].
Assessment of CagA EPIYA motif polymorphisms
A total of 2561 H. pylori complete or partial CagA protein sequences available at GenBank on August 8th 2013 were collected. The cagA EPIYA A-, B-, C-, and D-TPM types were defined as described [46]. The numbers of each type of EPIYA TPMs and the polymorphisms within the five specified amino acids were tabulated independently three times. A Chi-square test was performed to evaluate the variance in the representation of each EPIYA TPM.
Supporting Information
Zdroje
1. Murata-Kamiya N (2011) Pathophysiological functions of the CagA oncoprotein during infection by Helicobacter pylori. Microbes Infect 13 : 799–807. doi: 10.1016/j.micinf.2011.03.011 21477660
2. Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1 : 1311–1315. 6145023
3. Linz B, Balloux F, Moodley Y, Manica A, Liu H, et al. (2007) An African origin for the intimate association between humans and Helicobacter pylori. Nature 445 : 915–918. doi: 10.1038/nature05562 17287725
4. Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, et al. (1999) Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol 32 : 459–470. 10320570
5. Wroblewski LE, Peek RM Jr., Wilson KT (2010) Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23 : 713–739. doi: 10.1128/CMR.00011-10 20930071
6. Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, et al. (1994) Helicobacter pylori infection and gastric lymphoma. N Engl J Med 330 : 1267–1271. 8145781
7. el-Serag HB, Sonnenberg A (1998) Opposing time trends of peptic ulcer and reflux disease. Gut 43 : 327–333. 9863476
8. Wiseman EF, Ang YS (2011) Risk factors for neoplastic progression in Barrett’s esophagus. World J Gastroenterol 17 : 3672–3683. doi: 10.3748/wjg.v17.i32.3672 21990948
9. Eucker TP, Samuelson DR, Hunzicker-Dunn M, Konkel ME (2014) The focal complex of epithelial cells provides a signaling platform for interleukin-8 induction in response to bacterial pathogens. Cell Microbiol.
10. Oertli M, Muller A (2012) Helicobacter pylori targets dendritic cells to induce immune tolerance, promote persistence and confer protection against allergic asthma. Gut Microbes 3 : 566–571. doi: 10.4161/gmic.21750 22895083
11. Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, et al. (1995) Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55 : 2111–2115. 7743510
12. Hatakeyama M (2014) Helicobacter pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 15 : 306–316. doi: 10.1016/j.chom.2014.02.008 24629337
13. Jurik A, Hausser E, Kutter S, Pattis I, Prassl S, et al. (2010) The coupling protein Cag{beta} and its interaction partner CagZ are required for type IV secretion of the Helicobacter pylori CagA protein. Infect Immun.
14. Tummuru MK, Cover TL, Blaser MJ (1993) Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun 61 : 1799–1809. 8478069
15. Tegtmeyer N, Wessler S, Backert S (2011) Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. Febs J.
16. Yamaoka Y, El-Zimaity HM, Gutierrez O, Figura N, Kim JG, et al. (1999) Relationship between the cagA 3’ repeat region of Helicobacter pylori, gastric histology, and susceptibility to low pH. Gastroenterology 117 : 342–349. 10419915
17. Azuma T, Yamakawa A, Yamazaki S, Fukuta K, Ohtani M, et al. (2002) Correlation between variation of the 3’ region of the cagA gene in Helicobacter pylori and disease outcome in Japan. J Infect Dis 186 : 1621–1630. 12447739
18. Furuta Y, Yahara K, Hatakeyama M, Kobayashi I (2011) Evolution of cagA Oncogene of Helicobacter pylori through Recombination. PLoS One 6: e23499. doi: 10.1371/journal.pone.0023499 21853141
19. Higashi H, Yokoyama K, Fujii Y, Ren S, Yuasa H, et al. (2005) EPIYA motif is a membrane-targeting signal of Helicobacter pylori virulence factor CagA in mammalian cells. J Biol Chem 280 : 23130–23137. 15831497
20. Nagase L, Murata-Kamiya N, Hatakeyama M (2011) Potentiation of Helicobacter pylori CagA virulence through homodimerization. J Biol Chem.
21. Backert S, Feller SM, Wessler S (2008) Emerging roles of Abl family tyrosine kinases in microbial pathogenesis. Trends Biochem Sci 33 : 80–90. doi: 10.1016/j.tibs.2007.10.006 18182299
22. Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, et al. (2002) c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol 43 : 971–980. 11929545
23. Tammer I, Brandt S, Hartig R, Konig W, Backert S (2007) Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 132 : 1309–1319. 17408661
24. Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, et al. (2009) Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe 5 : 23–34. doi: 10.1016/j.chom.2008.11.010 19154985
25. Muller A (2012) Multistep activation of the Helicobacter pylori effector CagA. J Clin Invest 122 : 1192–1195. doi: 10.1172/JCI61578 22378039
26. Backert S, Tegtmeyer N, Selbach M (2010) The Versatility of Helicobacter pylori CagA Effector Protein Functions: The Master Key Hypothesis. Helicobacter 15 : 163–176. doi: 10.1111/j.1523-5378.2010.00759.x 20557357
27. Hayashi T, Senda M, Morohashi H, Higashi H, Horio M, et al. (2012) Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe 12 : 20–33. doi: 10.1016/j.chom.2012.05.010 22817985
28. Selbach M, Paul FE, Brandt S, Guye P, Daumke O, et al. (2009) Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 5 : 397–403. doi: 10.1016/j.chom.2009.03.004 19380118
29. Ne Sbreve Ic D, Miller MC, Quinkert ZT, Stein M, Chait BT, et al. (2009) Helicobacter pylori CagA inhibits PAR1-MARK family kinases by mimicking host substrates. Nat Struct Mol Biol.
30. Nesic D, Buti L, Lu X, Stebbins CE (2014) Structure of the Helicobacter pylori CagA oncoprotein bound to the human tumor suppressor ASPP2. Proc Natl Acad Sci U S A 111 : 1562–1567. doi: 10.1073/pnas.1320631111 24474782
31. Lee IO, Kim JH, Choi YJ, Pillinger MH, Kim SY, et al. (2010) Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem 285 : 16042–16050. doi: 10.1074/jbc.M110.111054 20348091
32. Neel BG, Gu H, Pao L (2003) The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28 : 284–293. 12826400
33. Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, et al. (2007) Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447 : 330–333. 17507984
34. Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, et al. (2002) SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295 : 683–686. 11743164
35. Segal ED, Cha J, Lo J, Falkow S, Tompkins LS (1999) Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A 96 : 14559–14564. 10588744
36. Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, et al. (2004) Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 279 : 17205–17216. 14963045
37. Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, et al. (2009) Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis 199 : 641–651. doi: 10.1086/596660 19199544
38. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, et al. (2008) Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A 105 : 1003–1008. doi: 10.1073/pnas.0711183105 18192401
39. Ren S, Higashi H, Lu H, Azuma T, Hatakeyama M (2006) Structural basis and functional consequence of Helicobacter pylori CagA multimerization in cells. J Biol Chem 281 : 32344–32352. 16954210
40. Suzuki R, Shiota S, Yamaoka Y (2013) Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori. Infect Genet Evol 12 : 203–213. doi: 10.1016/j.meegid.2011.12.002 22197766
41. Logan RP, Berg DE (1996) Genetic diversity of Helicobacter pylori. Lancet 348 : 1462–1463. 8942769
42. Alm RA, Trust TJ (1999) Analysis of the genetic diversity of Helicobacter pylori: the tale of two genomes. J Mol Med 77 : 834–846. 10682319
43. Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, et al. (2003) Traces of human migrations in Helicobacter pylori populations. Science 299 : 1582–1585. 12624269
44. Aras RA, Lee Y, Kim SK, Israel D, Peek RM Jr., et al. (2003) Natural variation in populations of persistently colonizing bacteria affect human host cell phenotype. J Infect Dis 188 : 486–496. 12898434
45. Azuma T, Yamazaki S, Yamakawa A, Ohtani M, Muramatsu A, et al. (2004) Association between diversity in the Src homology 2 domain—containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J Infect Dis 189 : 820–827. 14976598
46. Hatakeyama M (2004) Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 4 : 688–694. 15343275
47. Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, et al. (2002) Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A 99 : 14428–14433. doi: 10.1073/pnas.222375399 12391297
48. Jones KR, Joo YM, Jang S, Yoo YJ, Lee HS, et al. (2009) Polymorphism in the CagA EPIYA Motif Impacts Development of Gastric Cancer. J Clin Microbiol.
49. Kim SY, Lee YC, Kim HK, Blaser MJ (2006) Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell Microbiol 8 : 97–106. 16367869
50. Huang H, Li L, Wu C, Schibli D, Colwill K, et al. (2008) Defining the specificity space of the human SRC homology 2 domain. Mol Cell Proteomics 7 : 768–784. 17956856
51. Suzuki M, Mimuro H, Suzuki T, Park M, Yamamoto T, et al. (2005) Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J Exp Med 202 : 1235–1247. doi: 10.1084/jem.20051027 16275761
52. Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Nichols PH, et al. (1994) Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol 47 : 61–66. 8132812
53. Beswick EJ, Reyes VE (2008) Macrophage migration inhibitory factor and interleukin-8 produced by gastric epithelial cells during Helicobacter pylori exposure induce expression and activation of the epidermal growth factor receptor. Infect Immun 76 : 3233–3240. doi: 10.1128/IAI.01534-07 18474653
54. Argent RH, Hale JL, El-Omar EM, Atherton JC (2008) Differences in Helicobacter pylori CagA tyrosine phosphorylation motif patterns between western and East Asian strains, and influences on interleukin-8 secretion. J Med Microbiol 57 : 1062–1067. doi: 10.1099/jmm.0.2008/001818-0 18719174
55. Takata S, Ito M, Wada Y, Yoshihara M, Tanaka S, et al. (2009) Pathogenetic role of the tyrosine-phosphorylated CagA EPIYA sequence of Helicobacter pylori in histological gastritis in Japanese patients. J Gastroenterol.
56. Matsunari O, Shiota S, Suzuki R, Watada M, Kinjo N, et al. (2012) Association between Helicobacter pylori virulence factors and gastroduodenal diseases in Okinawa, Japan. J Clin Microbiol 50 : 876–883. doi: 10.1128/JCM.05562-11 22189111
57. Lind J, Backert S, Pfleiderer K, Berg DE, Yamaoka Y, et al. (2014) Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of Western-type Helicobacter pylori strains. PLoS One 9: e96488. doi: 10.1371/journal.pone.0096488 24800748
58. Ubersax JA, Ferrell JE Jr. (2007) Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol 8 : 530–541. 17585314
59. Li SP, Chen XJ, Sun AH, Zhao JF, Yan J (2010) CagA(+)H.pylori Induces Akt1 Phosphorylation and Inhibits Transcription of p21(WAF1/CIP1) and p27(KIP1) via PI3K/Akt1 Pathway. Biomed Environ Sci 23 : 273–278. doi: 10.1016/S0895-3988(10)60063-3 20934114
60. Huang F-C, Li Q, Cherayil BJ (2005) A phosphatidyl-inositol-3-kinase-dependent anti-inflammatory pathway activated by Salmonella in epithelial cells. FEMS Microbiology Letters 243 : 265–270. 15668028
61. Schneider N, Krishna U, Romero-Gallo J, Israel DA, Piazuelo MB, et al. (2009) Role of Helicobacter pylori CagA Molecular Variations in Induction of Host Phenotypes with Carcinogenic Potential. J Infect Dis 199 : 1218–1221. doi: 10.1086/597416 19278338
62. Zhang Y, Takeuchi H, Nishioka M, Morimoto N, Kamioka M, et al. (2009) Relationship of IL-8 production and the CagA status in AGS cells infected with Helicobacter pylori exposed to low pH and activating transcription factor 3 (ATF3). Microbiol Res 164 : 180–190. 17449233
63. Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, et al. (2014) Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A 111 : 1455–1460. doi: 10.1073/pnas.1318093111 24474772
64. Ando T, Peek RM Jr., Lee YC, Krishna U, Kusugami K, et al. (2002) Host cell responses to genotypically similar Helicobacter pylori isolates from United States and Japan. Clin Diagn Lab Immunol 9 : 167–175. doi: 10.1128/CDLI.9.1.167-175.2002 11777849
65. Brandt S, Kwok T, Hartig R, Konig W, Backert S (2005) NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102 : 9300–9305. doi: 10.1073/pnas.0409873102 15972330
66. Zhu Y, Zhong X, Zheng S, Du Q, Xu W (2005) Transformed immortalized gastric epithelial cells by virulence factor CagA of Helicobacter pylori through Erk mitogen-activated protein kinase pathway. Oncogene 24 : 3886–3895. 15856031
67. Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13 : 140–156. doi: 10.1038/nrd4204 24481312
68. Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27 : 5497–5510. doi: 10.1038/onc.2008.245 18794884
69. Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M (2006) Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol 26 : 261–276. doi: 10.1128/MCB.26.1.261-276.2006 16354697
70. Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269 : 5241–5248. 8106507
71. Tsutsumi R, Higashi H, Higuchi M, Okada M, Hatakeyama M (2003) Attenuation of Helicobacter pylori CagA × SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem 278 : 3664–3670. 12446738
72. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, et al. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388 : 539–547. 9252185
73. Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, et al. (2012) c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 122 : 1553–1566. doi: 10.1172/JCI61143 22378042
74. Zhang XS, Blaser MJ (2012) DprB facilitates inter - and intragenomic recombination in Helicobacter pylori. J Bacteriol 194 : 3891–3903. doi: 10.1128/JB.00346-12 22609923
75. Tegtmeyer N, Rivas Traverso F, Rohde M, Oyarzabal OA, Lehn N, et al. (2013) Electron microscopic, genetic and protein expression analyses of Helicobacter acinonychis strains from a Bengal tiger. PLoS One 8: e71220. doi: 10.1371/journal.pone.0071220 23940723
76. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning, A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
77. Zhang XS, Blaser MJ (2012) Natural transformation of an engineered Helicobacter pylori strain deficient in type II restriction endonucleases. J Bacteriol 194 : 3407–3416. doi: 10.1128/JB.00113-12 22522893
78. Tegtmeyer N, Lind J, Schmid B, Backert S (2014) Helicobacter pylori CagL Y58/E59 mutation turns-off type IV secretion-dependent delivery of CagA into host cells. PLoS One 9: e97782. doi: 10.1371/journal.pone.0097782 24893039
79. Tegtmeyer N, Wittelsberger R, Hartig R, Wessler S, Martinez-Quiles N, et al. (2011) Serine Phosphorylation of Cortactin Controls Focal Adhesion Kinase Activity and Cell Scattering Induced by Helicobacter pylori. Cell Host Microbe 9 : 520–531. doi: 10.1016/j.chom.2011.05.007 21669400
80. Pillinger MH, Marjanovic N, Kim SY, Scher JU, Izmirly P, et al. (2005) Matrix metalloproteinase secretion by gastric epithelial cells is regulated by E prostaglandins and MAPKs. J Biol Chem 280 : 9973–9979. 15640153
81. Bourzac KM, Botham CM, Guillemin K (2007) Helicobacter pylori CagA induces AGS cell elongation through a cell retraction defect that is independent of Cdc42, Rac1, and Arp2/3. Infect Immun 75 : 1203–1213. doi: 10.1128/IAI.01702-06 17194805
82. Tegtmeyer N, Hartig R, Delahay RM, Rohde M, Brandt S, et al. (2010) A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J Biol Chem.
83. Hirsch C, Tegtmeyer N, Rohde M, Rowland M, Oyarzabal OA, et al. (2012) Live Helicobacter pylori in the root canal of endodontic-infected deciduous teeth. J Gastroenterol 47 : 936–940. doi: 10.1007/s00535-012-0618-8 22722905
84. Krause-Gruszczynska M, Boehm M, Rohde M, Tegtmeyer N, Takahashi S, et al. (2011) The signaling pathway of Campylobacter jejuni-induced Cdc42 activation: Role of fibronectin, integrin beta1, tyrosine kinases and guanine exchange factor Vav2. Cell Commun Signal 9 : 32. doi: 10.1186/1478-811X-9-32 22204307
85. Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, Harrison SC (1996) Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat Struct Biol 3 : 364–374. 8599763
86. Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18 : 2714–2723. 9504803
87. Sayle RA, Milner-White EJ (1995) RASMOL: biomolecular graphics for all. Trends Biochem Sci 20 : 374. 7482707
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- Dimorphism in Fungal Pathogens of Mammals, Plants, and Insects
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging