
Hepatitis C Virus RNA Replication Depends on Specific and -Acting Activities of Viral Nonstructural Proteins
RNA viruses encode one or more proteins that form an RNA replicase, which serves to propagate the viral RNA genome. Some replication genes can function in trans, meaning that proteins expressed by one viral genome can complement and assist in the replication of a defective mutant virus within the same cell. Other viral genes work in cis, meaning that they are unable to complement defective mutants; these cis-acting genes therefore link the replication of a viral RNA genome to its translation. Because genetic complementation reflects the formation of molecular interactions that were previously absent or nonfunctional, understanding the cis - and trans-activities of viral replication proteins can provide insights into the structure and function of viral replicases. Here we develop a novel methodology to study interactions between components of the viral replicase to define essential cis - and trans-activities for all five hepatitis C virus (HCV) replication gene products. Our studies show that representative defects in each HCV replication protein can be complemented in trans. However, specific features of NS3 and NS5B were required in cis, indicating that these proteins specifically function to replicate the genome that encodes them. These data define important activities of the HCV replication proteins and provide new insight into how these proteins function together as an enzyme complex.
Published in the journal:
. PLoS Pathog 11(4): e32767. doi:10.1371/journal.ppat.1004817
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004817
Summary
RNA viruses encode one or more proteins that form an RNA replicase, which serves to propagate the viral RNA genome. Some replication genes can function in trans, meaning that proteins expressed by one viral genome can complement and assist in the replication of a defective mutant virus within the same cell. Other viral genes work in cis, meaning that they are unable to complement defective mutants; these cis-acting genes therefore link the replication of a viral RNA genome to its translation. Because genetic complementation reflects the formation of molecular interactions that were previously absent or nonfunctional, understanding the cis - and trans-activities of viral replication proteins can provide insights into the structure and function of viral replicases. Here we develop a novel methodology to study interactions between components of the viral replicase to define essential cis - and trans-activities for all five hepatitis C virus (HCV) replication gene products. Our studies show that representative defects in each HCV replication protein can be complemented in trans. However, specific features of NS3 and NS5B were required in cis, indicating that these proteins specifically function to replicate the genome that encodes them. These data define important activities of the HCV replication proteins and provide new insight into how these proteins function together as an enzyme complex.
Introduction
The low-fidelity of RNA virus replication gives rise to swarms of mutant variants [1], which can genetically interact with one another. Mutant viruses may complement or inhibit one another in trans, with important consequences on virus evolution, pathogenesis, and the emergence of drug-resistance [2–5]. For instance, subgenomic (i.e. less than genome length) defective-interfering (DI) viruses are dependent on, and compete with, helper viruses with intact genomes [6]. Because of this competition, many positive-strand RNA viruses have evolved genome quality control mechanisms that require specific viral genes in cis [7–14]. In some cases, essential cis-acting RNA sequences or secondary structures are embedded within the viral coding region [15–19]. In other cases, RNA replication requires ribosomal translation through a specific gene region, although the gene product is not necessarily required in cis [7,11,20]. Finally, some cis-acting viral proteins recruit the RNA from which they were translated into RNA replication complexes [9–11,21].
One group of viruses exhibiting marked cis-preferences is the Flaviviridae, a large family of enveloped, positive-strand RNA viruses that includes several globally important human and animal pathogens [22]. Members of this family are genetically diverse but share similarities in genome organization, mechanisms of gene expression, and replication strategies. As a model of RNA replication, the best-characterized member of this family is hepatitis C virus (HCV), a human pathogen that causes chronic liver disease, cirrhosis, and liver cancer, and is a major target of antiviral design [23]. The 9.6-kb HCV genome lacks a 5' cap and 3' polyadenylated tail but has highly structured 5' and 3' non-coding regions, as well as an internal RNA cis-replication element (CRE) within the coding region (Fig 1A). These RNA structures are important cis-acting sequences that direct translation and replication of the viral genome, including a 5' internal ribosome entry site (IRES) to initiate cap-independent translation.

As for all positive-strand RNA viruses, the HCV genome is translated directly, producing a polyprotein that is cleaved by cellular and viral proteinases into structural proteins—core, E1, and E2—and nonstructural (NS) proteins—p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Fig 1A and 1B). After multiple rounds of translation, a subset of NS proteins then recruits the genome out of translation and into a membrane-bound replication complex (replicase) that contains multiple copies of NS3–5B (Fig 1B and 1C). The core–NS2 genes are involved in virus particle assembly but are dispensable for RNA replication [24]; in contrast, NS3–5B are all required for RNA replication and is the minimal set of HCV genes necessary to produce a self-replicating subgenomic RNA replicon (SGR) [25]. RNA replication is thought to occur via de novo (i.e., unprimed) initiation at or near the 3' end of the template genome, production of a complementary negative strand, and iterative use of the negative strand as a secondary template to make many new positive-strand genomes [26,27].
Much is known about the structure and function of individual NS3–5B proteins, although the molecular architecture of the replicase is not yet known. NS3 is a bifunctional enzyme, encoding an N-terminal serine protease domain and a C-terminal helicase domain. The serine protease requires interaction with NS4A for optimal activity and is responsible for cleaving the downstream polyprotein [28]; it also proteolytically inactivates specific cellular substrates including MAVS [29,30], TRIF [31], DDB1 [32], and GPx8 [33]. The C-terminal RNA helicase/NTPase activities of NS3 are essential for RNA replication [34,35], although the specific roles of these activities are unknown. For instance, this domain can unwind double-stranded RNA and DNA in vitro [36], but direct evidence is lacking that it binds to or unwinds viral RNA during the replication cycle in vivo. The small NS4A protein binds to NS3, acts as a cofactor for both NS3 activities [28,37], and anchors the NS3-4A heterodimer in the membrane [38]. NS4B is a polytopic membrane protein that can oligomerize and may serve as a scaffold for replicase assembly [39,40]. NS5A is an RNA-binding phosphoprotein that contains an N-terminal amphipathic membrane anchor, a structured zinc-binding domain that directs NS5A homodimerization in either of two alternate conformations, and a large, natively unfolded C-terminal domain [41]. The precise role of NS5A during RNA replication is not yet clear, but it is known to recruit cellular factors essential for replicase assembly and function, including cyclophilin A [42–46], phosphatidylinositol 4-kinase IIIα [47], and the vesicle trafficking protein VAP-A [48–50]. NS5B is the viral RNA-dependent RNA polymerase and has a C-terminal membrane tail anchor [51,52]. The NS3 serine protease, NS5B polymerase, and NS5A proteins are the major targets of HCV-specific antiviral compounds [23].
Mutations in the HCV core-NS2 genes, including large in-frame deletions, can be complemented in trans [53–56]. However, among the genes required for RNA replication, only NS4B and NS5A have been shown to be trans-complemented [57–62]. These observations have led to the suggestion that the other NS genes work only in cis, although the underlying basis of these cis requirements remains unclear. Here, we describe newly developed quantitative tools to study the cis - and trans-requirements of HCV RNA replication and identify loss-of-function mutations in NS3, NS4A, and NS5B that can be complemented in trans. Whereas NS5B RNA binding and polymerase activities could be supplied in trans, NS5B expression was required in cis, suggesting that NS5B has a structural role in replicase assembly. A subset of NS3 activities was required in cis, including the helicase RNA binding and NTPase active site residues, implicating these helicase activities in the recruitment of an RNA template for replication. Complementation group analysis indicated that NS3-4A and NS4B activities are coordinated, whereas NS5B activity requires an intact set of upstream NS3-5A genes. We followed up on this latter observation by showing that NS5B segregates with a daclatasvir-sensitive activity of NS5A. These results reveal new aspects of HCV replicase structure-function, provide insights into combination anti-HCV therapy, and establish an experimental platform to study the genetic and functional interactions of gene products from positive-strand RNA viruses.
Results
NS5B can be supplied in trans
HCV SGRs encoding reporter genes provide a quantitative readout of HCV RNA replication [63,64]. To monitor HCV replication levels over time, we used SGR-Gluc (Fig 1A), an HCV genotype 2a (strain JFH-1) SGR that autonomously replicates and expresses the Gaussia princeps luciferase (Gluc) [65]. At early times post-transfection of Huh-7.5 hepatoma cells with SGR-Gluc RNA transcripts, Gluc expression increased and reached maximal expression by 48 hours (Fig 1D); the decline in Gluc activity at later times corresponded with the onset of cytopathic effects caused by JFH-1 replication [55,64,66]. In contrast, SGR-Gluc(5Bm1), a mutant replicon containing inactivating point mutations of the Mg++-coordinating polymerase active site residues (Table 1), expressed Gluc only at early time points post-transfection (Fig 1D), consistent with translation of the input RNA followed by RNA turnover [65].

We tested whether the replication defect of SGR-Gluc(5Bm1) could be trans-complemented in Huh-7.5[SGR-Neo] cells, which stably harbor a functional JFH-1 SGR that confers resistance to G418 [66]. SGR-Gluc efficiently replicated in these cells, whereas SGR-Gluc(5Bm1) did not (Fig 1E). Thus, SGR-Gluc(5Bm1) was not trans-complemented in Huh-7.5[SGR-Neo] cells, consistent with prior results indicating that NS5B is not supplied in trans by an active replicon [57,61,83]. We hypothesized that active RNA replication competed with complementation, such that NS5B expressed by a replicon might be unavailable to function in trans. To test this idea, we expressed NS3–5B outside the context of a functional HCV replicon by using a recoded 6015-nt NS3–5B gene region containing 1379 silent mutations (Fig 1A, 1F, and S1 Text). The rationale for recoding HCV NS3–5B was to: 1) ensure efficient expression by using preferred human codons; 2) minimize RNA secondary structures, including known and potential cis-acting replication elements within the NS3–5B coding region; and 3) allow for selective amplification of HCV replicons vs. synthetic NS3–5B genes during downstream analysis (e.g. RT-PCR). The recoded NS3–5B region was expressed in Huh-7.5 cells by using a noncytopathic SGR derived from Venezuelan equine encephalitis virus (VEEV), a member of the Alphavirus genus of the Togaviridae family of positive-strand RNA viruses. This expression vector was chosen because noncytopathic alphavirus vectors: 1) stably and abundantly express foreign genes [84,85]; 2) accommodate large insertions [86]; and 3) have been used successfully in trans-complementation studies with members of the Flaviviridae, including HCV [86–89]. After stable transduction of Huh-7.5 cells with the VEEV/NS3–5B vector to produce Huh-7.5[VEEV/NS3–5B] cells, the HCV NS3–5B polyprotein was expressed at physiologically relevant levels and properly processed through proteolytic cleavage, as evidenced by the patterns of NS3 and NS5A expression (Fig 1G). NS3–5B expression was stable for many weeks of passage. As a negative control, the green fluorescent protein (GFP) was expressed as an irrelevant protein via the VEEV vector (VEEV/GFP).
We next investigated whether SGR-Gluc(5Bm1) could be trans-complemented in Huh-7.5[VEEV/NS3–5B] cells. Indeed, SGR-Gluc(5Bm1) replicated and expressed Gluc in Huh-7.5[VEEV/NS3–5B] cells but not in Huh-7.5[VEEV/GFP] cells, although the kinetics of Gluc expression was slower and less robust than from wild-type SGR-Gluc (Fig 1H). In agreement with the Gluc expression data, SGR-Gluc(5Bm1) RNA levels were maintained in Huh-7.5[VEEV/NS3–5B] cells, albeit at reduced levels compared to SGR-Gluc RNA (Fig 1I). In contrast, SGR-Gluc(5Bm1) RNA levels fell below the limit of detection in Huh-7.5[VEEV/GFP] cells (Fig 1I). These data demonstrate that an HCV mutant containing an inactivating mutation of the NS5B polymerase active site can be trans-complemented.
To confirm that SGR-Gluc(5Bm1) replicated in Huh-7.5[VEEV/NS3–5B] cells, transfected cells were treated with 1 nM daclatasvir (DCV), a potent inhibitor of HCV RNA replication complex assembly [90–92]. DCV inhibits JFH-1 replication with an effective concentration (EC50) of 71 pM and cytostatic concentrations (CC50) of >10 μM [90,93]. Treatment with 1 nM DCV abolished replication of SGR-Gluc(5Bm1) in Huh-7.5[VEEV/NS3–5B] cells (Fig 1J), confirming that the observed trans-complementation of SGR-Gluc(5Bm1) requires HCV replicase activity.
We next examined the essential features of NS5B supplied in trans. As expected, complementation of SGR-Gluc(5Bm1) required an intact polymerase active site in the recoded NS3–5B polyprotein (S1A Fig). Furthermore, expression of NS5B alone, either by introducing an initiator Met codon (Met-NS5B) or by fusion with ubiquitin to ensure an authentic NS5B N-terminal Ser residue (Ubi-NS5B), failed to trans-complement SGR-Gluc(5Bm1) (S1B Fig). Processing of Ubi-NS5B into NS5B was confirmed by western blot (S1C Fig), consistent with efficient cleavage of N-terminal Ubi fusions by cellular ubiquitin C-terminal hydrolases [94,95]. These data indicate that expression of NS5B alone is insufficient to trans-complement a defect in NS5B polymerase activity, suggesting that NS5B may need to be expressed as part of the NS3–5B polyprotein.
We also examined the trans-complementation of a full-length HCV reporter virus, Jc1/Gluc2A [96], containing the 5Bm1 mutation. As expected, this mutant was unable to replicate in Huh-7.5, Huh-7.5[VEEV/GFP], or Huh-7.5[SGR-Neo] cells, but replicated in Huh-7.5[VEEV/NS3–5B] cells (S1D Fig). However, the dynamic range of trans-complementation (i.e., the ratio between the highest and lowest Gluc signals) was lower for Jc1/Gluc2(5Bm1) than for SGR-Gluc(5Bm1), consistent with the higher replication efficiency of SGRs vs. full-length HCV genomes [55,97]. As a result, only low titers (≈2.3 x 103 TCID50 units/ml) of infectious virus (as assayed on Huh-7.5[VEEV/NS3–5B] cells) were obtained from the full-length 5Bm1 mutant. Since our focus was to characterize the cis - and trans - activities of NS genes during RNA replication, SGRs were used in further studies because of their superior dynamic range in replication-dependent Gluc expression.
To determine the long-term stability of NS5B trans-complementation, we tested the ability of SGR-Neo(5Bm1) to replicate and transduce Neo-expression in Huh-7.5[VEEV/NS3–5B] cells. As shown in Fig 1K, SGR-Neo(5Bm1) formed G418-resistant colonies in Huh-7.5[VEEV/NS3–5B] cells with an efficiency similar to the wild-type SGR-Neo replicon. Following selection with G418, SGR-Neo(5Bm1) replicon-bearing Huh-7.5[VEEV/NS3–5B] cells could be maintained under selection indefinitely. The 5Bm1 mutation (“GNN”) was maintained stably in G418-selected SGR-Neo(5Bm1) replicon-bearing cells, as seen by direct sequencing of the resulting reverse-transcribed and PCR-amplified amplicon (S1E Fig) and by cloning the PCR product and sequencing twenty independent clones. Although reversion is possible, it was not observed in these experiments. Thus, stable replication of the SGR-Neo(5Bm1) replicon was not due to reversion or recombination with the recoded NS5B gene.
We also tested the trans-complementation of an NS5B mutant containing inactivating mutations in the polymerase RNA binding groove, SGR-Gluc(5Bm2), as well as a third mutant, SGR-Gluc(5Bm3), which contained both RNA binding and polymerase active site mutations (Table 1). These mutants were trans-complemented with efficiencies similar to SGR-Gluc(5Bm1) (Fig 1L). In contrast to prior results [57,61,83], these data establish that defects in NS5B can be complemented in trans by expressing NS3–5B outside the context of an actively replicating SGR.
NS5B protein expression is required in cis for RNA replication
We next examined whether the efficiency of NS5B complementation could be improved by preventing expression of the defective NS5B protein. However, the NS5B gene cannot simply be deleted because it contains an RNA structural element, the CRE, required for RNA replication. We therefore inserted a stop codon just downstream of NS5A to create SGR-Gluc(5A*5B) (Table 1 and Fig 2A). This mutant was unable to replicate, but surprisingly, was not complemented in trans (Fig 2B). We considered three explanations for these observations. First, the premature stop codon destabilized the SGR-Gluc(5A*5B) RNA. However, SGR-Gluc(5Bm1) and SGR-Gluc(5A*5B) expressed similar levels of residual Gluc (Fig 2B); given that nascent Gluc was collected at each time point (Materials and Methods), these data suggest that non-replicating SGR-Gluc(5Bm1) and SGR-Gluc(5A*5B) RNAs were turned over at similar rates. Second, RNA replication required ribosomal transit through the NS5B coding region, as has been observed for the 2A–3D coding region of poliovirus [11]. Third, the NS5B protein was required in cis. To distinguish between these possibilities, we designed a strategy to permit ribosomal translation through the NS5B coding region, without NS5B expression, by using an alternate reading frame. First, we designed the SGR-Gluc(5Brc) mutant containing 22 silent mutations in the NS5B coding region that: 1) removed stop codons from the -1 reading frame; 2) maintained the CRE structure; and 3) maintained a stop at codon 592 (Fig 2A and S2 Text). Thus, this mutant expresses a wild-type NS3–5B polyprotein but uses altered NS5B codon usage to create an untranslated -1 open reading frame. SGR-Gluc(5Brc) autonomously and efficiently replicated, although with slightly delayed kinetics compared to SGR-Gluc (Fig 2C), indicating that these silent mutations in NS5B had only a minor effect on RNA replication.

We then introduced a -1 frame-shift at codon 39 of the SGR-Gluc(5Brc) NS5B gene, to create SGR-Gluc(5Bfs), a mutant predicted to express a 65.3-kDa, 591 amino acid protein, 5Bfs, of unknown structure instead of the 66.7-kDa, 591 amino acid NS5B protein (S2 Text). The SGR-Gluc(5Bfs) mutant did not replicate, nor was it trans-complemented (Fig 2D). One possible explanation was that the 5Bfs protein non-specifically interfered with HCV replication. However defects in SGR-Gluc replication were not observed when a 5Bfs transgene was expressed in trans, arguing that the 5Bfs-encoded protein did not have a dominant negative effect on replication. We also considered the possibility that the frame-shift could have caused defects in processing of the NS3–5Bfs polyprotein. Because 5Bfs includes the N-terminal 39 amino acids of NS5B, it should allow proper processing of the NS5A/5B junction. Consistent with this, SGR-Gluc(5Bfs) expressed normal levels of NS3 and NS5A but not NS5B (Fig 2E), indicating that proper cleavage of the polyprotein occurred and that normal levels of NS3 and NS5A were expressed. Together, these data show that the recoded NS3–5B transgene was unable to trans-complement SGR-Gluc(NS5Bfs), a mutant that: 1) lacks a premature stop codon; 2) allows ribosomes to translate through the NS5B coding region; but 3) does not express NS5B. Thus, two mutants lacking NS5B expression, SGR-Gluc(5A*5B) and SGR-Gluc(5Bfs), were not complemented in trans. The simplest interpretation of these data is that RNA replication requires NS5B protein translation in cis. Since defects in NS5B RNA binding and polymerase activities can be trans-complemented (Fig 1), this cis-requirement is therefore independent of the known enzymatic activities of NS5B.
NS3 serine protease and RNA helicase activities are required in cis
Given the success in trans-complementing NS5B, we examined whether defects in other HCV NS genes could be trans-complemented in Huh-7.5[VEEV/NS3–5B] cells. We initially tested mutants containing lethal, inactivating mutations of the NS3 serine protease active site residues (Table 1), SGR-Gluc(3m1) and SGR-Gluc(3m2). Neither of these protease mutants replicated, nor were they complemented in trans (Fig 3A). We also examined NS3 RNA helicase domain mutants SGR-Gluc(3m3) and SGR-Gluc(3m4), which contained loss-of-function mutations abrogating RNA binding and NTPase activity, respectively (Table 1). Neither of the helicase domain mutants replicated, nor were they complemented in trans (Fig 3B). In comparison, a third helicase domain mutant, SGR-Gluc(3m5) was trans-complemented at levels similar to the 5Bm1 mutant (Table 1 and Fig 3C). As detailed in the Discussion, SGR-Gluc(3m5) is a loss-of-function mutant that lacks RNA unwinding activity but retains NS3 helicase RNA binding and NTPase activities [75–77].
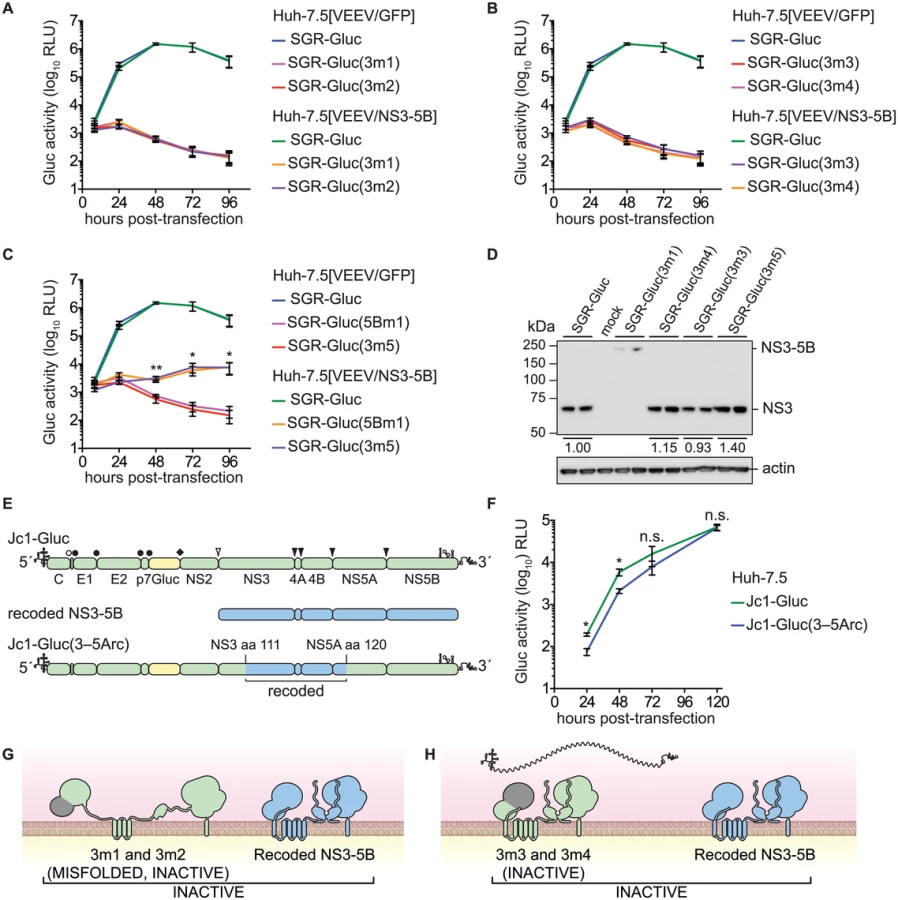
Although the data above suggested that HCV replication required the enzymatic activities of NS3 in cis, two alternative interpretations are that these specific NS3 mutations: 1) destabilized the inactive NS3 proteins, which could be required in cis; or 2) disrupted underlying RNA structures within the NS3 gene that are required in cis. Western blot analysis indicated that the 3m3 and 3m5 mutations caused slight (≤2-fold) increases in NS3 accumulation, and that the 3m4 mutation had no effect on NS3 accumulation (Fig 3D). As expected, the 3m1 mutation abrogated serine protease cleavage activity and caused accumulation of an unprocessed NS3–5B polyprotein (Fig 3D). These data show that the helicase mutations did not inhibit NS3 protein accumulation. To examine whether the NS3 mutations disrupted a putative underlying RNA structure required for replication, we replaced a large portion of a full-length HCV genome with the corresponding NS3-5A region from the recoded NS3–5B cassette, creating the Jc1-Gluc(3-5Arc) mutant (Fig 3E and Table 1). This mutant contained 524 silent mutations, including the NS3 RNA binding and NTPase active site codons T269, D290, and E291 that had been mutated in 3m3 and 3m4 (S3 Text). As expected, the recoded NS3 gene also exhibited pronounced differences in predicted RNA secondary structure (S2 Fig). The Jc1-Gluc(3-5Arc) mutant replicated efficiently with only a slight delay in kinetics (Fig 3F); it is therefore unlikely that the 3m3 and 3m4 mutations disrupted an important RNA element in the NS3 gene region. Given that the inactivating NS3 helicase mutations did not inhibit NS3 protein accumulation nor disrupt important RNA secondary structures, we conclude that HCV RNA replication requires the RNA binding and NTPase activities of the NS3 RNA helicase domain in cis. While the serine protease activity is required in cis because it is needed for polyprotein processing (Fig 3G), the RNA binding and NTPase activities of the helicase domain are likely needed for a post-translational step in replication, such as RNA template recruitment (Fig 3H).
trans-complementation of NS3–5B genes
We also examined the trans-complementation of additional NS3-4A mutants. The SGR-Gluc(3m6) mutant, which contains a loss-of-function, three-codon deletion in the NS3 linker region [78], replicated to high levels in Huh-7.5[VEEV/NS3–5B] cells (Fig 4A), indicating that it was trans-complemented. This mutant was also complemented in Huh-7.5[SGR-Neo] cells (Fig 4B), indicating that, unlike NS5B, NS3 can be complemented by an actively replicating replicon. Similarly, the SGR-Gluc(4Am1) mutant, which contains a lethal codon substitution in the NS4A acidic domain [65,79], replicated in both Huh-7.5[VEEV/NS3–5B] cells (Fig 4C) and Huh-7.5[SGR-Neo] cells (Fig 4D). Efficient trans-complementation was also observed for mutants SGR-Gluc(3m7) and SGR(4Am2), which contained lethal mutations in the NS3 linker and NS4A transmembrane domains, respectively (Table 1 and S2C Fig). Together, these data confirm that NS3-4A can be complemented in trans, either by expressing a recoded NS3–5B region or by an actively replicating SGR.

Prior work has shown that NS4B and NS5A can be supplied in trans [57–61]. Consistent with these results, SGR-Gluc(4Bm1) and SGR-Gluc(5Am1) (Table 1) had severe replication defects in Huh-7.5[VEEV/GFP] cells but were trans-complemented in Huh-7.5[VEEV/NS3–5B] cells (S3A and S3B Fig). Both mutants also were complemented in Huh-7.5[SGR-Neo] cells (S3C and S3D Fig), confirming that defects in NS4B and NS5A can be complemented in trans by expressing the wild-type gene either from an active replicon or from a synthetic mRNA encoding NS3–5B.
We then examined whether multiple defects could be complemented simultaneously by combining two (3m6 and 5Bm1), three (4Am1, 4Bm1, and 5Am1), or five inactivating mutations (3m6, 4Am1, 4Bm1, 5Am1, and 5Bm1) within the SGR-Gluc replicon (Table 1). All of the combined mutants replicated in Huh-7.5[VEEV/NS3–5B] cells but not in Huh-7.5[VEEV/GFP] cells (Fig 5A–5C). The efficiency of complementation inversely correlated with the number of mutations; nevertheless, replication levels were statistically significant and sensitive to DCV treatment, confirming that these signals were due to active HCV replication. None of the combined mutants replicated in Huh-7.5[SGR-Neo] cells (S4A–S4C Fig). To examine the long-term trans-complementation of a mutant containing multiple defects, we used G418 selection of a mutant SGR-Neo replicon. As shown in Fig 5D, SGR-Neo(3–5Bm) stably replicated and formed G418-resistant colonies in Huh-7.5[VEEV/NS3–5B] cells but not in Huh-7.5[VEEV/GFP] cells. RT-PCR and sequence analysis of twelve independent cDNA clones confirmed that the 3m6, 4Am1, 4Bm1, 5Am1, and 5Bm1 mutations were all retained in the G418-selected cell populations. These data demonstrate that HCV replicons with combined defects in all five NS genes required for RNA replication (NS3, NS4A, NS4B, NS5A, and NS5B) can be complemented in trans (Fig 5E).

NS3–5B complementation group analysis
The ability to complement defects in each NS3–5B gene enabled us to examine functional interactions between NS genes through complementation group analysis (Fig 6). To do so, we first created Huh-7.5[VEEV/NS3–5B] cell lines containing the 3m6, 4Am1, 4Bm1, 5Am1, and 5Bm1 mutations within the recoded NS3–5B transgene. Wild-type SGR-Gluc replicated similarly in all cell lines (Fig 6A), indicating that the mutant NS3–5B transgenes did not have dominant negative effects on HCV replication. A panel of SGR-Gluc NS3–5B mutants was then tested for complementation in these cell lines. As expected, each SGR-Gluc mutant failed to replicate in cells expressing the NS3–5B transgene containing a defect in the same gene (Fig 6), thus defining distinct complementation groups. Nonetheless, the efficiency of trans-complementation varied among mutant genes, suggesting that functional interactions exist between the corresponding gene products. Notably, the 3m6 mutant replicated less efficiently in Huh-7.5[VEEV/NS3–5B(4Bm1)] cells than in Huh-7.5[VEEV/NS3–5B] cells (Fig 6B); the 4Am1 mutant replicated less efficiently in Huh-7.5[VEEV/NS3–5B(3m6)] and Huh-7.5[VEEV/NS3–5B(4Bm1)] cells than in Huh-7.5[VEEV/NS3–5B] cells (Fig 6C); and the 4Bm1 mutant replicated less efficiently in Huh-7.5[VEEV/NS3–5B(3m6)] cells than in Huh-7.5[VEEV/NS3–5B] cells (Fig 6D). These data indicate that the NS3-4A and NS4B activities were enhanced by co-expression of these genes from the same polyprotein (Fig 6G). However, the most notable effect was that SGR-Gluc(5Bm1) failed to replicate in Huh-7.5[VEEV/NS3–5B(3m6)], Huh-7.5[VEEV/NS3–5B(4Am1)], and Huh-7.5[VEEV/NS3–5B(4Bm1)] cells and was poorly complemented in Huh-7.5[VEEV/NS3–5B(5Am1)] cells (Fig 6F). These data indicate that trans-complementing activity of NS5B depends on upstream NS3-5A activities within the same polyprotein (Fig 6H), consistent with our observation that the 5Bm1 mutant was not trans-complemented by expression of NS5B alone (S1A Fig). In contrast, the efficient replication of SGR-Gluc(3m6), SGR-Gluc(4Am1), SGR-Gluc(4Bm1), and SGR-Gluc(5Am1) on Huh-7.5[VEEV/NS3–5B(5Bm1)] cells (last column of Fig 6B–6F) indicates that the activity of NS5B in cis does not depend on upstream NS3–5A activities (Fig 6I).

NS5B activity segregates with a DCV-sensitive NS5A activity
Trans-complementation can impact the emergence of drug-resistance [2–4]. Given that trans-complementation of NS5B is linked to the function of upstream NS3-5A genes, we hypothesized that NS5B activity would segregate with sensitivity to an HCV-specific direct acting antiviral compound targeting an upstream gene product. To explore this hypothesis, we examined the complementation of the lethal 5Bm1 mutation in combination with DCVR, a point mutation in NS5A (Table 1) that shifts the EC50 of DCV by >7,500-fold and renders HCV resistant to DCV [59]. As expected, replication of the SGR-Gluc(DCVR) mutant was resistant to DCV (Fig 7A). When the DCVR mutation was introduced into VEEV/NS3–5B or VEEV/NS3–5B(5Bm1), SGR-Gluc replication was resistant to DCV but showed a kinetic lag (Fig 7B and 7E), again confirming that NS5A can be supplied in trans. Surprisingly, trans-complementation of the SGR-Gluc(5Bm1,DCVR) double mutant was sensitive to DCV (Fig 7C and 7F). However, trans-complementation of SGR-Gluc(5Bm1) was resistant to DCV when the DCVR mutation was introduced into VEEV/NS3–5B (Fig 7D and 7G). These data show that NS5A, and in particular, the DCV-sensitive function of NS5A, can function in trans, independent of NS5B. However, the trans-complementation activity of NS5B segregated with an NS5A activity that was sensitive to DCV, again confirming that NS5B is linked functionally to upstream NS3-5A activities within the same polyprotein.

Discussion
In this study we define essential cis - and trans-replication activities for all five HCV replicase genes. Previous studies showed that defects in HCV NS4B and NS5A were complemented in trans, whereas defects in NS3 and NS5B were not, leading to the suggestion that NS3-4A and NS5B can only function in cis [57–61]. In contrast, we found that specific genetic defects in HCV NS3, NS4A, NS4B, NS5A, and NS5B can be complemented in trans, yet other NS3 and NS5B activities are required in cis.
How do viral genes and their gene products work in cis? In the simplest case, a viral coding region can contain an important RNA sequence or secondary structure, such as the CREs present within the poliovirus 2C gene [15,16] or HCV NS5B gene [17–19]. In other cases, the process of RNA translation is required although the protein product may be irrelevant, such as the ORF encoded by DIs of mouse hepatitis virus [7,20]. As previously noted by Novak and Kirkegaard [11], such cis-translational requirements can be explained by ribosome-induced structural changes within the RNA template (Fig 8A), or by recruitment of ribosome-associated factors to the template. Finally, cis-acting viral proteins can recruit the RNA from which they were translated into a replication complex. For instance, the nascent N-terminal polypeptide of bovine coronavirus nsp1 targets its RNA template to sites of replication [9]; a similar mechanism has been observed for brome mosaic virus 2a protein [98]. In these cases, the nascent polypeptides are linked physically to the RNA through the actively translating ribosome (Fig 8B). Alternatively, cis-acting proteins that have completed synthesis may bind preferentially to their RNA template (Fig 8C) due to spatial limitations, such as physical barriers to protein diffusion, or temporal restrictions, such rapid protein turnover [11]. Similar mechanisms have been put forth to explain cis-acting proteins encoded by DNA viruses and other mobile genetic elements [99–104].

The cis and trans activities of NS5B
When expressed by an HCV replicon, NS5B exhibited a marked cis-preference and was unable to trans-complement replicons with defects in NS5B. These data agree with prior studies, which used replicons as the source of NS5B [57,61,83]. However, NS5B could function in trans when it was expressed as part of a synthetic NS3–5B polyprotein. We envision two non-exclusive mechanisms by which this could occur within a replication complex: 1) by intermingling of NS proteins and exchange of NS5B (Fig 8D); and/or 2) by transfer of template RNA (Fig 8E). In favor of the RNA transfer model, SGR-Gluc(5Bm1) was not complemented when NS5B was expressed alone (Fig 8F), nor was it complemented by NS3–5B polyproteins containing defects in NS3, NS4A, NS4B, or NS5A (Fig 6H). Thus, trans-complementation of NS5B may utilize RNA transfer; at the molecular level, this could reflect a propensity of NS5B to remain associated with the NS proteins with which it was translated. Nevertheless, protein exchange likely occurs during complementation of other viral proteins. For instance NS5A activity segregates promiscuously, independent of other NS protein activities.
While replicons expressing enzymatically inactive NS5B were complemented in trans, replicons lacking NS5B expression were not. We excluded nonspecific effects caused by a premature stop codon and a potential requirement for ribosomal transit through the NS5B coding region. These data indicate that NS5B has a cis-acting role in replicase assembly that is independent of its RNA binding and polymerase activities. Recent work has shown that the RNA polymerase of poliovirus plays an important, non-enzymatic role in RNA replication by assembling two-dimensional, membrane-bound oligomeric lattices that enhance RNA binding and synthesis through cooperativity [105–107]. Intriguingly, HCV NS5B also forms oligomers that show cooperativity in RNA synthesis rates [108]. Together, these data lend support to a conserved, structural role for viral polymerases during positive-strand RNA virus replicase assembly.
Complementation of HCV NS5B in trans only occurred when NS5B was expressed by an intact NS3-5B polyprotein. Analogously, trans-complementation of the NS5 polymerase from the related West Nile virus (subtype Kunjin; genus Flavivirus) also requires co-expression of upstream NS proteins [86], suggesting this may be common within the Flaviviridae. We found that HCV NS5B was functionally linked to NS5A, and more specifically, to the DCV-sensitive function of NS5A translated from the same polyprotein. Fridell and colleagues used SGR trans-complementation to define three NS5A complementation groups and showed that the DCV-sensitive function of NS5A works preferentially in cis [59,60]. In contrast, we found that the DCVR allele of NS5A can function in trans when it was expressed from a synthetic NS3–5B mRNA. Thus, the cis-preferences of NS5A and NS5B can be overcome by expressing NS3–5B outside the context of a functional replicon.
Complementation of NS5A also requires expression of an intact NS3-5A polyprotein [57,62] or an active replicon [57–61]. Similar results were observed for NS5A complementation with bovine viral diarrhea virus-1 (BVDV-1), a related member of the Flaviviridae [13]. These observations may be related to the influence of upstream proteins on the phosphorylation status and interactome of NS5A [109–112]. Thus, there appears to be multiple layers of cis-linkage, whereby NS5B activity is dependent on upstream NS5A activity (this study), which in turn is linked to the activities of NS3-4A and NS4B. This network of linkages could serve as a quality control mechanism to reduce the emergence of DI genomes despite the ability of NS3-5A to function in trans. In this regard, deletions in the NS3–5B region have not been observed in naturally occurring HCV deletion mutants [55,113–115].
Given that both NS5A and NS5B are targets of HCV-specific antivirals [23,116], our findings may have implications for the emergence of drug resistance. We predict that NS5A inhibitors like DCV may have synergistic effects when combined with inhibitors of the viral polymerase. Furthermore, the barrier for resistance to drugs targeting NS5A and NS5B may be high because replicase-expressed NS5B does not complement in trans and is linked to the daclatasvir-sensitive activity of upstream NS5A; thus resistant alleles would need to arise within the same genome. It is notable that DCV and sofosbuvir combination therapy shows high rates of sustained virological response in HCV-infected patients [117].
The cis and trans activities of NS3-4A
Defects in the essential NS3 serine protease, RNA binding, and NTPase active sites were not trans-complemented. This phenotype was not unexpected for the serine protease mutants, which express uncleaved, inactive NS3–5B polyproteins (Fig 3G). These data suggest that a protease inhibitor-sensitive virus would not be trans-complemented by an inhibitor-resistant virus in vivo. Defects in the NS3 RNA binding and NTPase activities also were not complemented in trans. We excluded nonspecific effects caused by instability of the mutant NS3 proteins or disruption of underlying cis-acting RNA structures within this region of the viral genome. Our data are most consistent with a requirement for the HCV NS3 RNA binding and NTPase activities in cis. Similarly, for other Flaviviridae, the BVDV-1 NS3 NTPase active site residues are required in cis [13,118], whereas a West Nile virus NS3 helicase mutant was trans-complemented with very low efficiency [14].
Surprisingly the 3m5 mutant, which contained a lethal W501A mutation of a helicase “bookend” residue, was complemented in trans. The NS3 W501 residue has a key role in RNA unwinding. Structurally, W501 stacks against the 3'-most base of RNA and DNA template strands and may keep the template in register for up to three single-nucleotide translocation steps [72,74,76]. In vitro, NS3 proteins containing the W501A mutation retain RNA binding and NTPase activities [73,75,119]; such mutants have defects in RNA unwinding but can unwind DNA [75,77]. Thus, the NS3 W501A mutant, SGR/Gluc(3m5), has basal helicase activity but likely has a defect in processivity on RNA templates. While it has been established that HCV helicase activity is required for RNA replication [34,35], the function of the helicase has been unclear, including whether it binds to or unwinds viral RNA during replication. Given that post-translational cis-preferences rely on protein-RNA interaction (Fig 8C), our data suggest that the RNA binding and NTPase activities, but not RNA unwinding activity, contribute to RNA template recruitment (Fig 3H). Remarkably, the RNA helicase/NTPase of brome mosaic virus also recruits viral RNA into membrane-bound replication complexes [120]. Thus, RNA template recruitment may be a conserved function of positive-strand RNA virus-encoded helicases.
Defects in the NS3 linker domain and NS4A C-terminal acidic domain also were complemented, both by recoded NS3–5B and by replicons. The NS3 linker domain, which is conserved and essential for RNA replication, coordinates interaction between the protease and helicase domains and may mediate protein-protein interaction via a putative SH3-binding motif [78]. The NS4A acidic domain contributes to helicase activity by promoting RNA-coupled ATP hydrolysis [37] and affects interactions with downstream NS4B and NS5A proteins [65,79]. The helicase activity of recombinant NS3-4A protein containing the 4Am1 mutation (NS4A Y45A) has efficient basal RNA-binding activity, but severely reduced ATP-coupled RNA binding, RNA-stimulated ATPase activity, and RNA unwinding [37]. Thus, the 4Am1 mutant independently confirms that RNA unwinding activity of the NS3-4A helicase can be supplied in trans.
Based on the ability to complement representative defects in NS3, NS4A, NS4B, NS5A, and NS5B, we conducted a detailed complementation group analysis for the HCV NS3–5B genes. These experiments revealed functional interactions between NS3-4A and NS4B: 3m6 - and 4Am1-mutant replicons were trans-complemented less efficiently in cells expressing the 4Bm1-mutant form of NS3–5B, whereas 4Am1 - and 4Bm1-mutant replicons were trans-complemented less efficiently in cells expressing the 3m6-mutant form of NS3–5B. One interpretation of these data is that NS3-4A and NS4B preferentially interact with each other from the same polyprotein (Fig 6G). Given that the NS3-4A-4B complementation grouping was reciprocal, it was surprising that 3m6 - and 4Bm1-mutant replicons replicated well on cells expressing the 4Am1-mutant form of NS3–5B. Furthermore, it was surprising that NS3 and NS4A activities segregated independently given that proteolytic processing of the NS3/NS4A junction is autocatalytic [121] and is predicted to lead to cis-preferential interaction between NS3 and NS4A [122]. One possible explanation is that NS3-4A dimerizes, such that one monomer provides NS3 activity while the other provides NS4A activity (Fig 8H). Indeed, we recently demonstrated that the NS4A transmembrane domain mediates homodimerization that is essential for RNA replication [80]. Alternatively, a second explanation might be that NS3-4A heterodimers exchange partners to form a single fully active NS3-4A heterodimer (Fig 8H).
Complete replication in trans
We were able to complement an HCV SGR containing loss-of-function mutations in every replication gene (Fig 5E). It is notable that a few HCV “minigenome” systems have been developed [123–127]. HCV minigenomes are RNAs encoding a reporter gene flanked by the HCV 5' and 3' noncoding regions; because they lack NS protein expression, minigenomes are presumably replicated in trans by HCV replicons or NS3–5B transgene expression. Given the cis-requirements of NS3 and NS5B described here, it is surprising that minigenomes can be replicated in trans. However, minigenome replication levels have not been compared directly to replicon or infectious virus replication levels, nor has their sensitivity to HCV-specific antiviral compounds been demonstrated. A key feature of these systems is that minigenome RNAs are produced continuously through transcription of transfected cDNA; hence they may represent a single round of RNA synthesis rather than bona fide RNA replication and amplification. In this regard, NS5B is capable of nonspecific RNA synthesis, in the absence of other HCV NS proteins, when overexpressed in cell culture [128,129]. In contrast, the trans-replication system we describe here supports authentic, long-term replication of defective HCV replicons initiated by RNA transfection.
Conclusions
In summary, we have identified several cis - and trans-acting activities of the HCV NS3–5B genes that are essential for RNA replication, including a non-enzymatic, cis-acting role of NS5B in replicase assembly, and that NS3 RNA binding and NTPase activities are likely involved in template recruitment. NS3-4A and NS4B activities co-segregated in complementation group analysis, whereas NS5B activity was dependent on the function of upstream NS3-5A genes. Together, these studies reveal a network of functional interactions that could serve as a quality control mechanism for the HCV replicase genes and can be exploited by combination therapies targeting multiple NS protein activities.
Materials and Methods
cDNA constructs and RNA transcription
SGR-Gluc was previously described as pYSGR-JFH/Gluc [65]. The noncytopathic VEEV vector 5'VEEVrep/L/GFP/Pac [84], a kind gift of Dr. Ilya Frolov (University of Alabama, Birmingham), was modified to replace the GFP insert with a short multi-cloning site. Briefly, oligos YO-0779 and YO-0780 (S1 Table) were annealed and ligated into the XbaI/BsrGI sites of p5'VEEVrep/L/GFP/Pac to yield pVEEV/MCS.
The JFH-1 NS3–5B region was recoded by GeneArt (Regensburg, Germany) to optimize human codon usage, minimize RNA structure, and remove repeat sequences, RNA instability motifs, and AT-rich or GC-rich sequences (S1 Text). The recoded NS3–5B region was subcloned as a 6066-bp PacI/Ecl136II fragment into pVEEV/MCS cut with PacI and PmeI. pJc1-Gluc(3–5Arc) was created by subcloning the SpeI/BstEII fragment of recoded NS3–5B into pJc1-Gluc2A [96]. To make pSGR-Gluc(5Brc), a recoded NS5B–HCV 3' end (S3 Text) was synthesized by Blue Heron Biotech (Bothell, WA) and cloned into pSGR-Gluc as a BsrGI/XbaI fragment. pSGR-Gluc(5Bfs) was constructed by cleaving pSGR-Gluc(NS5Brc) with BsrGI, fill-in with dNTPs and Klenow enzyme, followed by religation. pJc1-Gluc(3–5Arc) was constructed by subcloning a SpeI/BstEII fragment from the recoded NS3–5B region into pJc1/Gluc2A [96].
Mutations were introduced into SGR-Gluc or VEEV/NS3–5B by site-directed mutagenesis with appropriate primer pairs (S1 Table). SGR-Gluc(5Bm1) was described previously as pYSGR-JFH/Gluc(GNN) [65]. SGR-Gluc(4Am1) was constructed by subcloning the NS4A Y45A mutation from Jc1/Gluc2A (NS4A Y45A) [65]. The recoded Met-NS5B gene was created via PCR by using primers YO-0913 and YO-0908. The recoded Ubi-NS5B gene was constructed by overlap extension PCR with primers YO-0905, YO-0906, YO-0907, and YO-0908.
VEEV/NS3-5B is a large (16.6-kb) high copy number plasmid, and cloning efficiency was improved by using high capacity E. coli strains such as XL10-Gold (Agilent). In contrast, we were unable to obtain a VEEV vector expressing an unrecoded NS3-5B cassette (to test whether the recoding process itself enabled trans-complementation) despite numerous cloning attempts with several bacterial host strains. Given that the recoded NS3-5B cassette was successfully subcloned into the VEEV vector several times, we suspect that the authentic JFH-1 NS3-5B sequence adversely impacted stability of the pVEEV plasmid. Nevertheless, the recoded NS3–5B region functioned in trans and was used for our studies.
All constructs were confirmed by restriction digestion and/or Sanger sequencing. The sequence of all DNA segments that were amplified in vitro were confirmed and subcloned. HCV RNA and VEEV transcripts were produced as previously described [87,130]. RNA transcripts were treated with DNase I (Ambion) and purified with the Qiagen RNeasy kit with on-column DNase digestion [130]. RNA concentrations were checked by spectrophotometry, adjusted to 100 ng/μl, RNA integrity was checked by agarose gel electrophoresis, and 10 μl aliquots were stored at—80°C.
Cell culture and RNA transfection
Huh-7.5 human hepatoma cells were maintained in complete growth medium (Dulbecco’s modified Eagle medium (DMEM; Invitrogen) containing 10% heat-inactivated fetal calf serum (Omega Scientific) and 0.1 mM non-essential amino acids (Invitrogen)). Cells were transfected with VEEV RNAs by electroporation as described [130]. Two days after electroporation, transfected cells were selected in complete growth medium containing 5 μg/ml puromycin (Invivogen), passaged 3 to 4 times, then frozen. Thawed cells were maintained in puromycin-containing complete growth medium and used within 4 or 5 passages; cells were seeded for experiments in complete growth medium lacking puromycin. Cells were transfected with HCV replicons by using 1 μl RNA and the TransIT mRNA transfection reagent (Mirus) according to the manufacturer’s instructions. DCV was a kind gift of Mingjun Huang (Achillion Pharmaceuticals). For selection of stable replicon-bearing cells, the cells were transfected, as above, in 10 cm dishes. At two days post-transfection, the medium was changed to complete growth medium containing 1 mg/ml G418 (Invitrogen) and selected for 3 weeks with triweekly media changes. Colonies were fixed with methanol and stained with crystal violet [79].
Luciferase assays
At each time point, media were collected, cells were washed three times with phosphate buffered saline, and fresh media was added. The collected media were clarified by centrifugation (5 min at 21,130 x g), placed into fresh tubes, mixed with 1/4 volume 5x Lysis buffer (Promega or NEB), and stored frozen (-80°C). Samples were assayed for Gluc activity on a Berthold Centro LB 960 plate reader [96] or a Berthold Centro XS3 LB 960.
Protein analysis
Cell lysates were prepared and equal amounts of protein were electrophoresed by SDS-PAGE, transferred to polyvinylidene difluoride membranes and immunoblotted with NS3-specific monoclonal antibodies 9G2 (Virogen) or 4E11 (this study), NS5A-specific monoclonal antibody 9E10 [130], NS5B-specific monoclonal antibody 5D1 (BioFront), or ß-actin-specific monoclonal antibody AC-15 (Sigma) antibodies. We noticed that the NS5B-specific antibody 5D1 had large lot-to-lot variability in NS5B reactivity and non-specific background. Protein masses were estimated based on the migration of the SuperSignal Enhanced Molecular Weight protein ladder (Pierce, Rockford IL). To analyze polyprotein expression of defective replicons, the vaccinia-T7 RNA polymerase expression system was used [131]. Briefly, Huh-7.5 cells were seeded at 5 x 105/well into six-well plates the day before use. Cells were infected for 1 h at a multiplicity of infection (MOI) of 10 with vTF7-3 and transfected with pSGR-Gluc derivatives by using Mirus LT1 reagent according to the manufacturer’s recommendations. Lysates were collected at 24 h post-transfection. Relative NS3 expression (Fig 3D) was quantitated by using the gel analyzer densitometry feature of NIH ImageJ [132].
NS3 antibody production
Recombinant HCV NS3 protein was expressed and purified from E. coli as described [133]. BALB/c mice were primed and boosted at 3-week intervals with 25 μg recombinant NS3 protein complexed with adjuvant (RIBI Immunochemical). Approximately 1 month after the last boost, sera were harvested and tested for immunoreactivity against HCV-infected Huh-7.5 cells. Mice with high titers (specific signal at >1/1,000 dilution) were boosted intravenously with 15 μg purified NS3 protein in PBS. Splenocytes were harvested 3 d later and fused to P3X63Ag8.653 myeloma cells to generate hybridomas according to published procedures [134]. A total of 8 hybridomas were derived after repeated subcloning, and monoclonal antibodies were purified by standard protein A/protein G chromatography. Monoclonal antibody 4D11 was found to react with NS3 protein by western blot of HCV-infected cells vs. uninfected control cells. Other monoclonal antibodies developed in these fusions will be described elsewhere.
RNA analysis
RNA was harvested by using Trizol reagent (Invitrogen). For sequence analysis, HCV RNA was amplified from the selected cell populations by RT-PCR and amplicons were directly submitted for Sanger sequencing; PCR products were also cloned into pCR2.1TOPO (Invitrogen) and multiple independent cDNA clones were sequenced. HCV RNA was quantitated by qRT-PCR as previously described [96] and normalized to total RNA concentration. Briefly, 2 μl of each RNA were assayed in 20 μl reactions containing 200 nM of an HCV IRES-specific hydrolysis probe (6FAM-5'-TAT GAG TGT CGT GCA GCC TC-3'-MGBNFQ), 400 nM forward (5'-CTT CAC GCA GAA AGC GTC TA-3'), 400 nM reverse (5'-CAA GCA CCC TAT CAG GCA GT-3') primers, and the LightCycler RNA Amplification Kit (Roche). Reactions were analyzed in a Roche LightCycler 480 by using a 45 minute RT step at 56°C, a 5 minute denaturation step at 94°C, followed by 40 cycles of denaturation (10 seconds at 95°C), annealing (20 seconds at 60°C), and extension (3 seconds at 72°C). Quantitation curves were constructed by analyzing HCV RNA standards in parallel [96].
Statistical analysis
All transfection experiments were performed at least twice, with each experiment containing at least three replicate cell cultures that were independently plated, transfected, sampled, and analyzed. Mean relative light unit (RLU) values were calculated ± standard deviations (SD) based on replicate data and plotted against matched control samples collected in parallel. Because many samples could be processed in parallel, the same positive and negative controls are presented across multiple samples within large experiments, as explicitly noted in the Figure legends. Because the background sensitivity of luciferase assays was affected by the lysis buffer and luminometer hardware, data were normalized to untransfected cells by dividing the mean value of each sample by the mean value from matched, mock-transfected cells collected in parallel. To calculate SD values for the normalized data, the propagations of error were accounted for as described [135]. Statistical significance was calculated in GraphPad Prism by using two-tailed, paired Student’s t-tests of the normalized, replicate data for each time point.
Supporting Information
Zdroje
1. Eigen M, Schuster P (1979) The hypercycle. A principle of natural self-organization. Heidelberg: Springer-Verlag. 92 p.
2. Crowder S, Kirkegaard K (2005) Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet 37 : 701–709. 15965477
3. Li JP, Baltimore D (1988) Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J Virol 62 : 4016–4021. 2845120
4. Tanner EJ, Liu HM, Oberste MS, Pallansch M, Collett MS, et al. (2014) Dominant drug targets suppress the emergence of antiviral resistance. Elife 3.
5. Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R (2006) Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439 : 344–348. 16327776
6. Huang AS, Baltimore D (1970) Defective viral particles and viral disease processes. Nature 226 : 325–327. 5439728
7. van der Most RG, Luytjes W, Rutjes S, Spaan WJ (1995) Translation but not the encoded sequence is essential for the efficient propagation of the defective interfering RNAs of the coronavirus mouse hepatitis virus. J Virol 69 : 3744–3751. 7745722
8. Molenkamp R, Greve S, Spaan WJ, Snijder EJ (2000) Efficient homologous RNA recombination and requirement for an open reading frame during replication of equine arteritis virus defective interfering RNAs. J Virol 74 : 9062–9070. 10982351
9. Su YP, Fan YH, Brian DA (2014) Dependence of coronavirus RNA replication on an NH2-terminal partial nonstructural protein 1 in cis. J Virol 88 : 8868–8882. doi: 10.1128/JVI.00738-14 24872586
10. Yi G, Kao C (2008) cis - and trans-acting functions of brome mosaic virus protein 1a in genomic RNA1 replication. J Virol 82 : 3045–3053. 18160434
11. Novak JE, Kirkegaard K (1994) Coupling between genome translation and replication in an RNA virus. Genes Dev 8 : 1726–1737. 7958852
12. Collis PS, O'Donnell BJ, Barton DJ, Rogers JA, Flanegan JB (1992) Replication of poliovirus RNA and subgenomic RNA transcripts in transfected cells. J Virol 66 : 6480–6488. 1328676
13. Grassmann CW, Isken O, Tautz N, Behrens SE (2001) Genetic analysis of the pestivirus nonstructural coding region: defects in the NS5A unit can be complemented in trans. J Virol 75 : 7791–7802. 11483722
14. Khromykh AA, Sedlak PL, Westaway EG (2000) cis - and trans-acting elements in flavivirus RNA replication. J Virol 74 : 3253–3263. 10708442
15. Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E (2000) Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol 74 : 10359–10370. 11044080
16. Goodfellow I, Chaudhry Y, Richardson A, Meredith J, Almond JW, et al. (2000) Identification of a cis-acting replication element within the poliovirus coding region. J Virol 74 : 4590–4600. 10775595
17. Friebe P, Boudet J, Simorre JP, Bartenschlager R (2005) Kissing-loop interaction in the 3' end of the hepatitis C virus genome essential for RNA replication. J Virol 79 : 380–392. 15596831
18. Lee H, Shin H, Wimmer E, Paul AV (2004) cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J Virol 78 : 10865–10877. 15452207
19. You S, Stump DD, Branch AD, Rice CM (2004) A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J Virol 78 : 1352–1366. 14722290
20. Liao CL, Lai MM (1995) A cis-acting viral protein is not required for the replication of a coronavirus defective-interfering RNA. Virology 209 : 428–436. 7778278
21. Lin J, Guo J, Finer J, Dorrance AE, Redinbaugh MG, et al. (2014) The bean pod mottle virus RNA2-encoded 58-kilodalton protein P58 is required in cis for RNA2 accumulation. J Virol 88 : 3213–3222. doi: 10.1128/JVI.03301-13 24390330
22. Lindenbach BD, Murray CL, Thiel HJ, Rice CM (2013) Flaviviridae. In: Knipe DM, Howley PM, editors. Fields Virology. Sixth ed. Philadelphia: Lippincott Williams & Wilkins. pp. 712–746.
23. Bartenschlager R, Lohmann V, Penin F (2013) The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11 : 482–496. doi: 10.1038/nrmicro3046 23748342
24. Lindenbach BD, Rice CM (2013) The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11 : 688–700. doi: 10.1038/nrmicro3098 24018384
25. Lohmann V, Korner F, Koch JO, Herian U, Theilmann L, et al. (1999) Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285 : 110–113. 10390360
26. Bartenschlager R, Frese M, Pietschmann T (2004) Novel insights into hepatitis C virus replication and persistence. Adv Virus Res 63 : 71–180. 15530561
27. Ranjith-Kumar CT, Kao CC (2006) Biochemical Activities of the HCV NS5B RNA-Dependent RNA Polymerase. In: Tan SL, editor. Hepatitis C Viruses: Genomes and Molecular Biology. Norfolk (UK).
28. Steinkühler C (2004) Hepacivirin. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of proteolytic enzymes. 2nd ed. London: Elsevier. pp. 1773–1779.
29. Li XD, Sun L, Seth RB, Pineda G, Chen ZJ (2005) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102 : 17717–17722. 16301520
30. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, et al. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437 : 1167–1172. 16177806
31. Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, et al. (2005) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102 : 2992–2997. 15710891
32. Kang X, Chen X, He Y, Guo D, Guo L, et al. (2013) DDB1 is a cellular substrate of NS3/4A protease and required for hepatitis C virus replication. Virology 435 : 385–394. doi: 10.1016/j.virol.2012.10.025 23137809
33. Morikawa K, Gouttenoire J, Hernandez C, Dao Thi VL, Tran HT, et al. (2014) Quantitative proteomics identifies the membrane-associated peroxidase GPx8 as a cellular substrate of the hepatitis C virus NS3-4A protease. Hepatology 59 : 423–433. doi: 10.1002/hep.26671 23929719
34. Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM (2000) Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3' nontranslated region are essential for virus replication in vivo. J Virol 74 : 2046–2051. 10644379
35. Lam AM, Frick DN (2006) Hepatitis C virus subgenomic replicon requires an active NS3 RNA helicase. J Virol 80 : 404–411. 16352565
36. Pyle AM (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37 : 317–336. doi: 10.1146/annurev.biophys.37.032807.125908 18573084
37. Beran RK, Lindenbach BD, Pyle AM (2009) The NS4A protein of hepatitis C virus promotes RNA-coupled ATP hydrolysis by the NS3 helicase. J Virol 83 : 3268–3275. doi: 10.1128/JVI.01849-08 19153239
38. Tanji Y, Hijikata M, Satoh S, Kaneko T, Shimotohno K (1995) Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J Virol 69 : 1575–1581. 7853491
39. Gouttenoire J, Penin F, Moradpour D (2010) Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20 : 117–129. doi: 10.1002/rmv.640 20069613
40. Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, et al. (2011) NS4B Self-Interaction through Conserved C-Terminal Elements Is Required for the Establishment of Functional Hepatitis C Virus Replication Complexes. J Virol 85 : 6963–6976. doi: 10.1128/JVI.00502-11 21543474
41. Ross-Thriepland D, Harris M (2014) Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol.
42. Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, et al. (2011) Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem 286 : 20441–20454. doi: 10.1074/jbc.M110.182436 21489988
43. Grise H, Frausto S, Logan T, Tang H (2012) A conserved tandem cyclophilin-binding site in hepatitis C virus nonstructural protein 5A regulates Alisporivir susceptibility. J Virol 86 : 4811–4822. doi: 10.1128/JVI.06641-11 22345441
44. Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, et al. (2009) Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem 284 : 13589–13601. doi: 10.1074/jbc.M809244200 19297321
45. Fernandes F, Ansari IU, Striker R (2010) Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5: e9815. doi: 10.1371/journal.pone.0009815 20352119
46. Foster TL, Gallay P, Stonehouse NJ, Harris M (2011) Cyclophilin A Interacts with Domain II of Hepatitis C Virus NS5A and Stimulates RNA Binding in an Isomerase-Dependent Manner. J Virol 85 : 7460–7464. doi: 10.1128/JVI.00393-11 21593166
47. Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, et al. (2011) Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9 : 32–45. doi: 10.1016/j.chom.2010.12.002 21238945
48. Evans MJ, Rice CM, Goff SP (2004) Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci U S A 101 : 13038–13043. 15326295
49. Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, et al. (2005) Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol 79 : 13473–13482. 16227268
50. Tu H, Gao L, Shi ST, Taylor DR, Yang T, et al. (1999) Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 263 : 30–41. 10544080
51. Brass V, Gouttenoire J, Wahl A, Pal Z, Blum HE, et al. (2010) Hepatitis C Virus RNA Replication Requires a Conserved Structural Motif within the Transmembrane Domain of the NS5B RNA-Dependent RNA Polymerase. J Virol 84 : 11580–11584. doi: 10.1128/JVI.01519-10 20739529
52. Moradpour D, Brass V, Bieck E, Friebe P, Gosert R, et al. (2004) Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J Virol 78 : 13278–13284. 15542678
53. Adair R, Patel AH, Corless L, Griffin S, Rowlands DJ, et al. (2009) Expression of hepatitis C virus (HCV) structural proteins in trans facilitates encapsidation and transmission of HCV subgenomic RNA. J Gen Virol 90 : 833–842. doi: 10.1099/vir.2008.006049-0 19223490
54. Ishii K, Murakami K, Hmwe SS, Zhang B, Li J, et al. (2008) Trans-encapsidation of hepatitis C virus subgenomic replicon RNA with viral structure proteins. Biochem Biophys Res Commun 371 : 446–450. doi: 10.1016/j.bbrc.2008.04.110 18445476
55. Pacini L, Graziani R, Bartholomew L, De Francesco R, Paonessa G (2009) Naturally occurring hepatitis C virus subgenomic deletion mutants replicate efficiently in Huh-7 cells and are trans-packaged in vitro to generate infectious defective particles. J Virol 83 : 9079–9093. doi: 10.1128/JVI.00308-09 19587042
56. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T (2008) Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol 82 : 7034–7046. doi: 10.1128/JVI.00118-08 18480457
57. Appel N, Herian U, Bartenschlager R (2005) Efficient rescue of hepatitis C virus RNA replication by trans-complementation with nonstructural protein 5A. J Virol 79 : 896–909. 15613318
58. Jones DM, Patel AH, Targett-Adams P, McLauchlan J (2009) The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J Virol 83 : 2163–2177. doi: 10.1128/JVI.01885-08 19073716
59. Fridell RA, Qiu D, Valera L, Wang C, Rose RE, et al. (2011) Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J Virol 85 : 7312–7320. doi: 10.1128/JVI.00253-11 21593143
60. Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, et al. (2013) Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J Virol 87 : 2320–2329. doi: 10.1128/JVI.02861-12 23236071
61. Tong X, Malcolm BA (2006) Trans-complementation of HCV replication by non-structural protein 5A. Virus Res 115 : 122–130. 16146661
62. Herod MR, Schregel V, Hinds C, Liu M, McLauchlan J, et al. (2014) Genetic complementation of hepatitis C virus nonstructural protein functions associated with replication exhibits requirements that differ from those for virion assembly. J Virol 88 : 2748–2762. doi: 10.1128/JVI.03588-13 24352463
63. Krieger N, Lohmann V, Bartenschlager R (2001) Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol 75 : 4614–4624. 11312331
64. Lohmann V (2009) HCV replicons: overview and basic protocols. Methods Mol Biol 510 : 145–163. doi: 10.1007/978-1-59745-394-3_11 19009259
65. Phan T, Kohlway A, Dimberu P, Pyle AM, Lindenbach BD (2011) The acidic domain of hepatitis C virus NS4A contributes to RNA replication and virus particle assembly. J Virol 85 : 1193–1204. doi: 10.1128/JVI.01889-10 21047963
66. Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, et al. (2003) Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125 : 1808–1817. 14724833
67. Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, et al. (1999) Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol 6 : 937–943. 10504728
68. Kim YC, Russell WK, Ranjith-Kumar CT, Thomson M, Russell DH, et al. (2005) Functional analysis of RNA binding by the hepatitis C virus RNA-dependent RNA polymerase. J Biol Chem 280 : 38011–38019. 16166071
69. Mosley RT, Edwards TE, Murakami E, Lam AM, Grice RL, et al. (2012) Structure of hepatitis C virus polymerase in complex with primer-template RNA. J Virol 86 : 6503–6511. doi: 10.1128/JVI.00386-12 22496223
70. Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM (1993) Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol 67 : 2832–2843. 8386278
71. Tomei L, Failla C, Santolini E, De Francesco R, La Monica N (1993) NS3 is a serine protease required for processing of hepatitis C virus polyprotein. Journal of virology 67 : 4017–4026. 7685406
72. Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, et al. (1998) Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6 : 89–100. 9493270
73. Lin C, Kim JL (1999) Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J Virol 73 : 8798–8807. 10482634
74. Appleby TC, Anderson R, Fedorova O, Pyle AM, Wang R, et al. (2011) Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J Mol Biol 405 : 1139–1153. doi: 10.1016/j.jmb.2010.11.034 21145896
75. Preugschat F, Danger DP, Carter LH 3rd, Davis RG, Porter DJ (2000) Kinetic analysis of the effects of mutagenesis of W501 and V432 of the hepatitis C virus NS3 helicase domain on ATPase and strand-separating activity. Biochemistry 39 : 5174–5183. 10819985
76. Gu M, Rice CM (2010) Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc Natl Acad Sci U S A 107 : 521–528. doi: 10.1073/pnas.0913380107 20080715
77. Kim JW, Seo MY, Shelat A, Kim CS, Kwon TW, et al. (2003) Structurally conserved amino Acid W501 is required for RNA helicase activity but is not essential for DNA helicase activity of hepatitis C virus NS3 protein. J Virol 77 : 571–582. 12477861
78. Kohlway A, Pirakitikulr N, Ding SC, Yang F, Luo D, et al. (2014) The linker region of NS3 plays a critical role in the replication and infectivity of hepatitis C virus. J Virol 88 : 10970–10974. doi: 10.1128/JVI.00745-14 24965468
79. Lindenbach BD, Prágai BM, Montserret R, Beran RK, Pyle AM, et al. (2007) The C terminus of hepatitis C virus NS4A encodes an electrostatic switch that regulates NS5A hyperphosphorylation and viral replication. J Virol 81 : 8905–8918. 17581983
80. Kohlway A, Pirakitikulr N, Barrera FN, Potapova O, Engelman DM, et al. (2014) Hepatitis C Virus RNA Replication and Virus Particle Assembly Require Specific Dimerization of the NS4A Protein Transmembrane Domain. Journal of Virology 88 : 628–642. doi: 10.1128/JVI.02052-13 24173222
81. Appel N, Pietschmann T, Bartenschlager R (2005) Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79 : 3187–3194. 15709040
82. Blight KJ, Kolykhalov AA, Rice CM (2000) Efficient initiation of HCV RNA replication in cell culture. Science 290 : 1972–1974. 11110665
83. Evans MJ, Rice CM, Goff SP (2004) Genetic interactions between hepatitis C virus replicons. J Virol 78 : 12085–12089. 15479852
84. Petrakova O, Volkova E, Gorchakov R, Paessler S, Kinney RM, et al. (2005) Noncytopathic replication of Venezuelan equine encephalitis virus and eastern equine encephalitis virus replicons in Mammalian cells. J Virol 79 : 7597–7608. 15919912
85. Agapov EV, Frolov I, Lindenbach BD, Prägai BM, Schlesinger S, et al. (1998) Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc Natl Acad Sci USA 95 : 12989–12994. 9789028
86. Khromykh AA, Sedlak PL, Guyatt KJ, Hall RA, Westaway EG (1999) Efficient trans-complementation of the flavivirus kunjin NS5 protein but not of the NS1 protein requires its coexpression with other components of the viral replicase. J Virol 73 : 10272–10280. 10559344
87. Lindenbach BD, Rice CM (1997) trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol 71 : 9608–9617. 9371625
88. Lindenbach BD, Rice CM (1999) Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol 73 : 4611–4621. 10233920
89. Yi M, Ma Y, Yates J, Lemon SM (2009) Trans-complementation of an NS2 defect in a late step in hepatitis C virus (HCV) particle assembly and maturation. PLoS Pathog 5: e1000403. doi: 10.1371/journal.ppat.1000403 19412343
90. Gao M (2013) Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr Opin Virol.
91. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, et al. (2010) Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465 : 96–100. doi: 10.1038/nature08960 20410884
92. Berger C, Romero-Brey I, Radujkovic D, Terreux R, Zayas M, et al. (2014) Daclatasvir-Like Inhibitors of NS5A Block Early Biogenesis of Hepatitis C Virus-Induced Membranous Replication Factories, Independent of RNA Replication. Gastroenterology 147 : 1094–1105 e1025. doi: 10.1053/j.gastro.2014.07.019 25046163
93. National Center for Biotechnology Information PubChem BioAssay Database; CID = 25154714, Source = Bristol Meyers Squibb Co.
94. Bachmair A, Finley D, Varshavasky A (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234 : 179–186. 3018930
95. Baker RT, Tobias JW, Varshavsky A (1992) Ubiquitin-specific proteases of Saccharomyces cerevisiae. Cloning of UBP2 and UBP3, and functional analysis of the UBP gene family. J Biol Chem 267 : 23364–23375. 1429680
96. Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD (2009) Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol 83 : 8379–8395. doi: 10.1128/JVI.00891-09 19515772
97. Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, et al. (2002) Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J Virol 76 : 4008–4021. 11907240
98. Chen J, Noueiry A, Ahlquist P (2003) An alternate pathway for recruiting template RNA to the brome mosaic virus RNA replication complex. J Virol 77 : 2568–2577. 12551995
99. Derbyshire KM, Grindley ND (1996) Cis preference of the IS903 transposase is mediated by a combination of transposase instability and inefficient translation. Mol Microbiol 21 : 1261–1272. 8898394
100. Duval-Valentin G, Chandler M (2011) Cotranslational control of DNA transposition: a window of opportunity. Mol Cell 44 : 989–996. doi: 10.1016/j.molcel.2011.09.027 22195971
101. Burt DW, Brammar WJ (1982) The cis-specificity of the Q-gene product of bacteriophage lambda. Mol Gen Genet 185 : 468–472. 6212756
102. Francke B, Ray DS (1972) Cis-limited action of the gene-A product of bacteriophage phiX174 and the essential bacterial site (E. coli-electron microscopy-cis-acting protein-specifically-nicked RF). Proc Natl Acad Sci U S A 69 : 475–479. 4551145
103. Kulpa DA, Moran JV (2006) Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat Struct Mol Biol 13 : 655–660. 16783376
104. Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, et al. (2001) Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21 : 1429–1439. 11158327
105. Spagnolo JF, Rossignol E, Bullitt E, Kirkegaard K (2010) Enzymatic and nonenzymatic functions of viral RNA-dependent RNA polymerases within oligomeric arrays. RNA 16 : 382–393. doi: 10.1261/rna.1955410 20051491
106. Pata JD, Schultz SC, Kirkegaard K (1995) Functional oligomerization of poliovirus RNA-dependent RNA polymerase. Rna 1 : 466–477. 7489508
107. Hobson SD, Rosenblum ES, Richards OC, Richmond K, Kirkegaard K, et al. (2001) Oligomeric structures of poliovirus polymerase are important for function. EMBO J 20 : 1153–1163. 11230138
108. Wang QM, Hockman MA, Staschke K, Johnson RB, Case KA, et al. (2002) Oligomerization and cooperative RNA synthesis activity of hepatitis C virus RNA-dependent RNA polymerase. J Virol 76 : 3865–3872. 11907226
109. Asabe SI, Tanji Y, Satoh S, Kaneko T, Kimura K, et al. (1997) The N-terminal region of hepatitis C virus-encoded NS5A is important for NS4A-dependent phosphorylation. J Virol 71 : 790–796. 8985418
110. Kaneko T, Tanji Y, Satoh S, Hijikata M, Asabe S, et al. (1994) Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem Biophys Res Commun 205 : 320–326. 7999043
111. Koch JO, Bartenschlager R (1999) Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J Virol 73 : 7138–7146. 10438800
112. Neddermann P, Clementi A, De Francesco R (1999) Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J Virol 73 : 9984–9991. 10559312
113. Bernardin F, Stramer SL, Rehermann B, Page-Shafer K, Cooper S, et al. (2007) High levels of subgenomic HCV plasma RNA in immunosilent infections. Virology 365 : 446–456. 17493654
114. Noppornpanth S, Smits SL, Lien TX, Poovorawan Y, Osterhaus AD, et al. (2007) Characterization of hepatitis C virus deletion mutants circulating in chronically infected patients. J Virol 81 : 12496–12503. 17728237
115. Sugiyama K, Suzuki K, Nakazawa T, Funami K, Hishiki T, et al. (2009) Genetic analysis of hepatitis C virus with defective genome and its infectivity in vitro. J Virol 83 : 6922–6928. doi: 10.1128/JVI.02674-08 19369330
116. Delang L, Neyts J, Vliegen I, Abrignani S, Neddermann P, et al. (2013) Hepatitis C virus-specific directly acting antiviral drugs. Curr Top Microbiol Immunol 369 : 289–320. doi: 10.1007/978-3-642-27340-7_12 23463206
117. Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, et al. (2014) Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med 370 : 211–221. doi: 10.1056/NEJMoa1306218 24428467
118. Grassmann CW, Isken O, Behrens SE (1999) Assignment of the multifunctional NS3 protein of bovine viral diarrhea virus during RNA replication: an in vivo and in vitro study. J Virol 73 : 9196–9205. 10516027
119. Tai CL, Pan WC, Liaw SH, Yang UC, Hwang LH, et al. (2001) Structure-based mutational analysis of the hepatitis C virus NS3 helicase. J Virol 75 : 8289–8297. 11483774
120. Wang X, Lee WM, Watanabe T, Schwartz M, Janda M, et al. (2005) Brome mosaic virus 1a nucleoside triphosphatase/helicase domain plays crucial roles in recruiting RNA replication templates. J Virol 79 : 13747–13758. 16227294
121. Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H (1993) Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J Virol 67 : 3835–3844. 8389908
122. Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC (1999) Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7 : 1353–1363. 10574797
123. Dumas E, Masante C, Astier-Gin T, Lapaillerie D, Ventura M (2007) The hepatitis C virus minigenome: a new cellular model for studying viral replication. J Virol Methods 142 : 59–66. 17320981
124. King RW, Zecher M, Jeffries MW, Carroll DR, Parisi JM, et al. (2002) A cell-based model of HCV-negative-strand RNA replication utilizing a chimeric hepatitis C virus/reporter RNA template. Antivir Chem Chemother 13 : 353–362. 12718407
125. Yang J, Lei YF, An QX, Yin W, Lu X, et al. (2010) Properties of hepatitis C virus minigenome containing mutated 5'UTR region and luciferase transgene. Acta Virol 54 : 105–112. 20545439
126. Zhang J, Yamada O, Sakamoto T, Yoshida H, Araki H, et al. (2005) Exploiting cis-acting replication elements to direct hepatitis C virus-dependent transgene expression. J Virol 79 : 5923–5932. 15857978
127. Zhang J, Yamada O, Yoshida H, Sakamoto T, Araki H, et al. (2007) Helper virus-independent trans-replication of hepatitis C virus-derived minigenome. Biochem Biophys Res Commun 352 : 170–176. 17112469
128. DeMarini DJ, Johnston VK, Konduri M, Gutshall LL, Sarisky RT (2003) Intracellular hepatitis C virus RNA-dependent RNA polymerase activity. J Virol Methods 113 : 65–68. 14500128
129. Ranjith-Kumar CT, Wen Y, Baxter N, Bhardwaj K, Cheng Kao C (2011) A cell-based assay for RNA synthesis by the HCV polymerase reveals new insights on mechanism of polymerase inhibitors and modulation by NS5A. PLoS One 6: e22575. doi: 10.1371/journal.pone.0022575 21799903
130. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, et al. (2005) Complete replication of hepatitis C virus in cell culture. Science 309 : 623–626. 15947137
131. Fuerst TR, Niles EG, Studier FW, Moss B (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83 : 8122–8126. 3095828
132. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9 : 671–675. 22930834
133. Beran RK, Pyle AM (2008) Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J Biol Chem 283 : 29929–29937. doi: 10.1074/jbc.M804065200 18723512
134. Harlow E, Lane DP (1988) Antibodies: a laboratory manual. In: Harlow E, Lane DP, editors. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory.
135. Lindberg V (2000) Uncertainties and Error Propagation: Part I of a manual on Uncertainties, Graphing, and the Vernier Caliper.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 4
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Toxin-Induced Necroptosis Is a Major Mechanism of Lung Damage
- Transgenic Fatal Familial Insomnia Mice Indicate Prion Infectivity-Independent Mechanisms of Pathogenesis and Phenotypic Expression of Disease
- Role of Hypoxia Inducible Factor-1α (HIF-1α) in Innate Defense against Uropathogenic Infection
- A Temporal Gate for Viral Enhancers to Co-opt Toll-Like-Receptor Transcriptional Activation Pathways upon Acute Infection