
The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of ε-Toxin
Clostridium perfringens epsilon-toxin is a potent pore-forming toxin responsible for a devastating central nervous system disease in livestock and has been suggested as a possible environmental trigger for Multiple Sclerosis. Epsilon-toxin binds with great specificity to a restricted number of host cell types and structures, for example gut epithelial cells, blood-brain barrier endothelial cells, and myelin. While most pore-forming toxins achieve binding through specific interaction with respective receptors on the cell membrane, the receptor for epsilon-toxin, however, is unknown. In this report we identify the Myelin and Lymphocyte protein, MAL, as being necessary for binding and cytotoxic effects of epsilon-toxin, and we show its second extracellular loop is critical in this novel function. At a physiological level, mice homozygous for a targeted deletion of the MAL gene lack sensitivity to epsilon-toxin whereas the toxin is lethal in wild-type mice. These observations lead to the possibility that MAL is a candidate receptor for epsilon-toxin. However, we have not demonstrated a physical interaction between epsilon-toxin and MAL.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004896
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004896
Summary
Clostridium perfringens epsilon-toxin is a potent pore-forming toxin responsible for a devastating central nervous system disease in livestock and has been suggested as a possible environmental trigger for Multiple Sclerosis. Epsilon-toxin binds with great specificity to a restricted number of host cell types and structures, for example gut epithelial cells, blood-brain barrier endothelial cells, and myelin. While most pore-forming toxins achieve binding through specific interaction with respective receptors on the cell membrane, the receptor for epsilon-toxin, however, is unknown. In this report we identify the Myelin and Lymphocyte protein, MAL, as being necessary for binding and cytotoxic effects of epsilon-toxin, and we show its second extracellular loop is critical in this novel function. At a physiological level, mice homozygous for a targeted deletion of the MAL gene lack sensitivity to epsilon-toxin whereas the toxin is lethal in wild-type mice. These observations lead to the possibility that MAL is a candidate receptor for epsilon-toxin. However, we have not demonstrated a physical interaction between epsilon-toxin and MAL.
Introduction
Clostridium perfringens is a gram-positive, spore-forming, anaerobic bacillus that is possibly the most widespread pathogenic bacterium in the world [1, 2]. Conventionally, the species is categorized into five toxinotypes, A-E, based on carriage of one or more of the major toxin genes (alpha, beta, epsilon, or iota). C. perfringens types B and D carry the epsilon toxin (ETX) gene [1, 2]. In all, the C. perfringens species produces a remarkable seventeen exotoxins, and of these, ETX is by far the most deadly, ranked the third most potent Clostridial toxin following botulinum and tetanus toxins [3]
The ETX-producing C. perfringens type B and D strains are less prevalent than the type A strain and are best studied in ruminant livestock where colonized animals are susceptible to a devastating enterotoxemia disease, which is characterized by loss of vision, paresis, ataxia and other signs of CNS dysfunction [4–7]. C. perfringens type B or type D can be readily isolated from cultures of animals with characteristic signs of ETX enterotoxemia [8–10].
In C. perfringens colonized animals, ETX is typically secreted into the intestinal lumen as a 32.9 kDa protoxin (pro-ETX) and enzymatically activated into a 29 kDa toxin by extracellular trypsin, chymotrypsin, or a C. perfringens encoded lambda protease [11–15]. However, some strains may be able to activate ETX intracellularly [16]. Activated ETX is then absorbed into the bloodstream through partially understood mechanisms [17]. ETX demonstrates a unique tropism for kidney [18], CNS endothelial cells, and brain [19, 20]. Within the bloodstream, ETX encounters the CNS endothelium, causes disruption of BBB, and allows entry of toxin into the brain [21–25]. Once in the brain, ETX binds with high affinity to white matter and this is presumably responsible for its well-described effect of causing white matter lesions in sheep, goats and mice[6, 19, 26–30]. In light of these observations, it has been hypothesized that there may be a connection between ETX and MS [31]. Having obtained the first evidence supporting this notion, we recently proposed that ETX is an environmental factor responsible for initiating lesion formation in MS [32].
Despite its potency and unique tropism for CNS tissues, the mammalian receptor for ETX remains unidentified. Based on prior investigation, it has been postulated that the ETX receptor is a glycoprotein residing in detergent resistant membranes or lipid rafts [33–36]. ETX has also been reported to bind to the extracellular domains of Hepatitis A Virus Receptor 1 (HAVCR1) immobilized on paramagnetic beads [37]. However, although knockdown of HAVCR1 reduces ETX toxicity in previously susceptible cells [37], transfection of ETX-resistant cells with HAVCR1 fails to confer ETX binding and/or susceptibility. These experiments suggest HAVCR1 contributes to ETX toxicity but is insufficient to confer ETX toxicity. Since ETX specifically binds to distal collecting duct epithelium of the kidney [18, 38] and to myelin in the CNS [19, 23, 39, 40], and since the putative ETX receptor is thought to localize to lipid rafts, we searched the National Library of Medicine databases for proteins residing within lipid rafts of distal renal epithelium and myelin. The tetraspan proteolipid MAL [41, 42] uniquely fits these specific requirements. Therefore, we sought to test the hypothesis that MAL is the ETX receptor.
The crystal structure of active ETX reveals an elongated structure with three principle domains sharing structural and conformational homology with the aerolysin family of pore-forming toxins[43]. Domain 1 contains the putative receptor-binding site and includes a cluster of aromatic residues[44] within the N-terminus. An amphipathic beta-hairpin within domain 2 contributes to ETX insertion into the plasma membrane and channel architecture. Domain 3 is thought to be involved in protein-protein interactions between monomers that stabilize oligomers. The receptor-binding domain is distinct from the domain(s) required for oligomerization. Pro-ETX binds to receptive cells with the same affinity as active toxin but does not integrate into the membrane to generate pore-forming oligomers. Formation of the heptameric pore requires cleavage of the carboxy-terminal 22 amino acids [45, 46]. Upon binding to susceptible mammalian cells, ETX monomers form a heptameric prepore [47], which inserts into the plasma membrane generating a stable asymmetric pore of ~500 Da exclusion [48].
MAL is a highly hydrophobic, tetraspan membrane proteolipid with two extracellular loops. It is found in particular polarized epithelial cells [49], oligodendrocytes [42, 50, 51] and human T lymphocytes [52]. In human T cells, MAL is expressed on the plasma membrane and is implicated in the formation of immunological synapses [53, 54]. In the nervous system, MAL is expressed by oligodendrocytes and Schwann cells and localizes to compact as well as non-compact myelin membranes, and is thought to play a role in myelin biogenesis and function [55]. In polarized epithelial cells, MAL localizes to the apical membrane and is thought to shuttle between the Trans-Golgi-Network (TGN) apparatus and the plasma membrane, sorting apically associated membrane proteins to the surface [56, 57]. Moreover, MAL plays critical roles in the formation, stabilization and maintenance of lipid rafts [54, 55, 57–64]. Through a combination of gain and loss of function experiments we show that MAL is required for binding and activity of ETX.
Results
Specific binding of active ETX to its receptor is necessary for prepore assembly since resistant cells fail to form heptamers even at high concentrations of ETX [47]. If MAL is required for binding of ETX to cells, then expression of MAL in ETX-resistant cells should confer binding of the toxin and render the cells susceptible to ETX-mediated cell death. To test this hypothesis, we stably expressed a green fluorescent protein (GFP)-rat MAL fusion protein (GFP-rMAL) construct in ETX resistant Chinese Hamster Ovary (CHO) cells (CHOGFP-rMAL), and identified that these cells were able to bind Alexa-594 labeled ETX, while CHO cells transfected with GFP alone (CHOGFP) did not bind the toxin (Fig 1A). Alexa-594 labeled ETX retained normal function in cytotoxicity assays (S1 Fig). To confirm and quantify binding of ETX to MAL expressing cells, we tested these two stably transfected cell lines by flow cytometry using Alexa-647 labeled ETX (laser 640nm/detector 675 nm). ETX showed robust binding to CHOGFP-rMAL cells, while failing to bind to CHOGFP controls (Fig 1B). To assess the effects of ETX on cell viability in CHOGFP-rMAL and CHOGFP cells, we designed an assay using PrestoBlue as a cell viability indicator. PrestoBlue is a resazurin-based cell permeable compound converted to the fluorescently active resorufin in the reducing environment of metabolically active cells [65–67]. ETX killed CHOGFP-rMAL cells in a dose - and time-dependent fashion (Fig 1C). In contrast, CHOGFP cells remained completely viable at all doses of ETX tested. In addition, we assessed cell death using propidium iodide (PI) exclusion as an independent assay for cell death [68, 69]. CHOGFP or CHOGFP-rMAL cells were treated with 500 pM ETX 500pM and monitored for PI uptake over a 24-hour period. By the 4-hour time point, we observed robust PI uptake in the CHOGFP-rMAL but not the CHOGFP control cells (S2 Fig). By contrast, after 24 hours, PI uptake remained undetectable in the CHOGFP controls. Taken together, these experiments support the conclusion that MAL expression in ETX resistant cells is required to impart both binding and sensitivity to toxin.

We next sought to determine the temporal relationship between ETX pore formation and morphologic indicators of cell death. To accomplish this, we incubated CHOGFP-rMAL cells with 50 nM ETX for increasing times and assessed both PI uptake and nuclear morphology. PI has an estimated van der Waals volume of 447 cubic Å. The size of the propidium cation limits passage of PI through membrane pores, and the van der Waals dimensions of propidium requires a minimum diameter of 1.5 nm for a propidium-permeable pore [70]. Studies examining the size and physical properties of the pores formed by ETX suggest an asymmetrical shape of the pore with its larger radius at 1.0 nm [69, 71]. Therefore, the ETX heptameric pore is likely permeable to PI. It has been previously shown that ETX induces permeability of MDCK cells to PI, and that small molecule antagonists that do not block heptamerization but do block the pore, inhibit PI entry into cells [66, 68, 72]. In addition to time-course, we examined the change of localization patterns of MAL in response to ETX exposure. In untreated CHOGFP-rMAL cells and for those treated with ETX for 5 minutes, MAL is localized primarily in the plasma membrane with some perinuclear staining (Fig 2A and 2B). As shown, the complete exclusion of PI indicates preserved membrane integrity in these cells. At 15 minutes of ETX treatment some cells retain normal appearing plasma membrane morphology while others reveal disruption of the plasma membrane and internalization of MAL into vesicles. Interestingly, those cells with normal MAL localization on the plasma membrane completely exclude PI whereas cells with disrupted membrane architecture and internalization of MAL are PI-positive (Fig 2C). Cells that have incorporated PI show intense localization of PI in nucleoli and faint staining of morphologically normal nuclei. By 30 minutes of treatment, the majority of CHOGFP-rMAL cells shows disruption of the integrity of the plasma membrane and incorporation of PI (Fig 2D). However, incorporation of PI at 30 minutes of treatment falls into one of two morphologic categories: intense staining of nucleoli with faint staining of morphologically normal nuclei, or intense staining of condensed pyknotic nuclei characteristic of apoptosis. By 60 minutes of ETX treatment, all the cells show disrupted plasma membranes and all of the PI staining is in pyknotic condensed nuclei (Fig 2E).

If MAL is the receptor or a direct biding partner for ETX, then the protein-protein interaction is most likely to occur between the receptor binding domain of ETX and the extracellular domain(s) of MAL. The primary structure of full-length MAL predicts a tetraspan integral membrane protein with two extracellular loops, ECL1 and ECL2 [41, 51]. MAL shares 87–89% of amino acid residue identity among human, rat, and dog (Fig 3). Sequence alignment highlighting ECL1 and ECL2 for rat and human MAL reveals a single amino acid substitution in each of the two domains (Fig 3). In contrast, zebrafish MAL shares significantly less amino acid identity with either human or rat MAL, especially within ECL2. To determine if sequence differences in MAL orthologues influence ETX binding and cytotoxicity, we first transiently transfected CHO cells with vectors expressing human, rat, or zebrafish MAL and analyzed binding by flow cytometry. ETX fails to bind to cells expressing either zebrafish MAL or GFP control (Fig 4A). ETX binds to both human and rat MAL expressing cells but displays an approximate ten-fold increase in binding affinity for rat MAL when compared to the human orthologue (Fig 4A). We then generated stable CHO cells lines expressing rMAL, hMAL, or the GFP vector alone and tested these cells for sensitivity to ETX. Cells were treated with escalating concentrations of ETX for 6 hours followed by live-imaging in the presence of PI to assess pore formation and cell death. As shown, rMAL expressing CHO cells are markedly more sensitive to ETX than those expressing hMAL (Fig 4B). The IC50 for ETX-induced cell death was 0.6 nM for rMAL and 40 nM for hMAL. Notably, after 6 hours of treatment, rMAL-expressing CHO cells show evidence of cell death at 0.5 and 5.0 nM ETX, whereas those expressing hMAL are still PI - negative (Fig 4C).


To determine if disruption of the primary structure within ECL1 or ECL2 impacts ETX binding to MAL, we generated stable cell lines expressing mutants of the human MAL harboring the FLAG epitope in ECL1 or ECL2 [56], respectively designated hMAL-L1F and hMAL-L2F (Fig 5A). We assessed targeting and orientation of the mutant proteins by fluorescence microscopy for both GFP and the FLAG epitope. Stable clones of hMAL-L1F-expressing CHO cells were generated but these failed to reliably target to the plasma membrane and were therefore not used in subsequent analyses. hMAL and hMAL-L2F are targeted to the plasma membrane as well as show some peri-nuclear localization in both live (Fig 5B) and fixed cells (Fig 5C). In live cultures the anti-FLAG antibody binds to hMAL-L2F expressing cells (Fig 5B). This indicates extracellular localization of the FLAG epitope and thus of ECL2, which is consistent with previous description [56, 60]. We then assessed binding and localization of Alexa-594 labeled ETX to cells expressing rMAL, hMAL, hMAL-L2F, and GFP control. Expression levels and subcellular localization patterns for all of three fusion proteins (rMAL, hMAL, and hMAL-L2F) are similar, as shown by GFP fluorescence intensity and distribution (Fig 5C). ETX binding is greater in magnitude for rMAL than for hMAL (Fig 5D). ETX completely fails to bind to cells expressing hMAL-L2F, underscoring the potentially important role of this domain in ligand interaction.
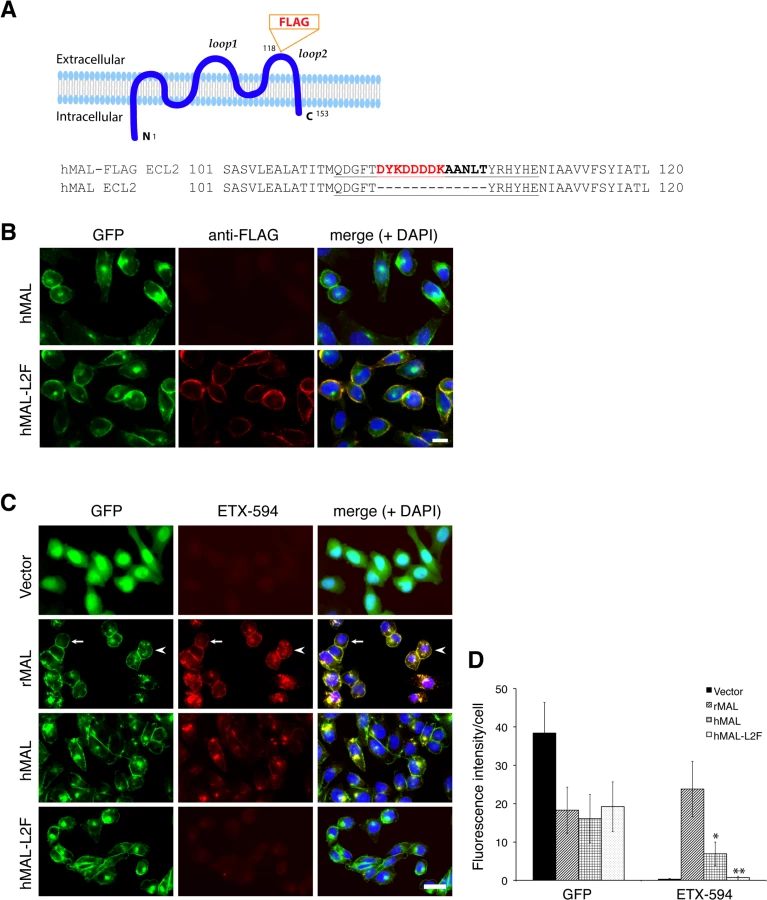
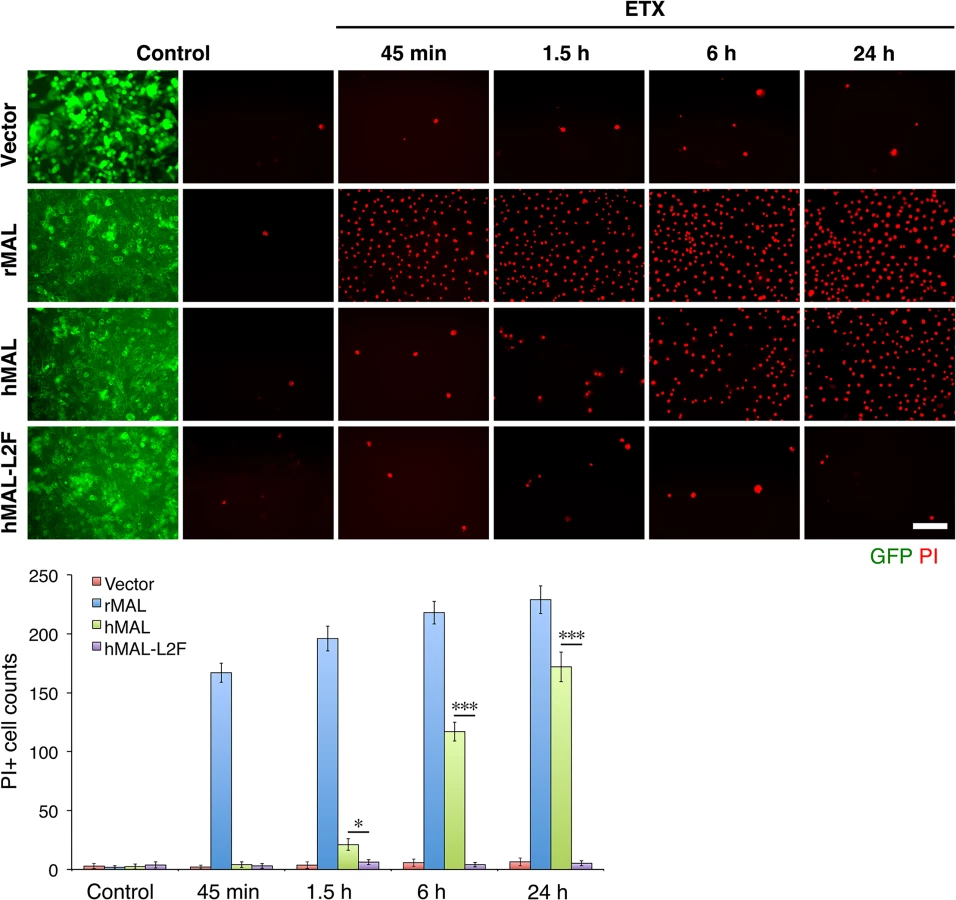
Having shown that the second extracellular loop of MAL is required for ETX binding, we next tested the sensitivity of the cells expressing hMAL-L2F using PI exclusion as an assessment of cell death. As previously noted, cells expressing rMAL were more sensitive to ETX in cell death assays than those expressing hMAL. Cells expressing hMAL-L2F are completely insensitive to ETX and thus behave like GFP vector control cells (Fig 6). Even after 24 hours of exposure to 50 nM ETX, hMAL-L2F expressing CHO cells show no evidence of cell death (Fig 6). By contrast, the wild-type hMAL-expressing cells exhibit significant cell death after 6 hours of ETX exposure. Since the hMAL-L2F mutant protein is correctly targeted and oriented within the plasma membrane, these results support a specific requirement for ECL2 in ETX binding and cytotoxicity.
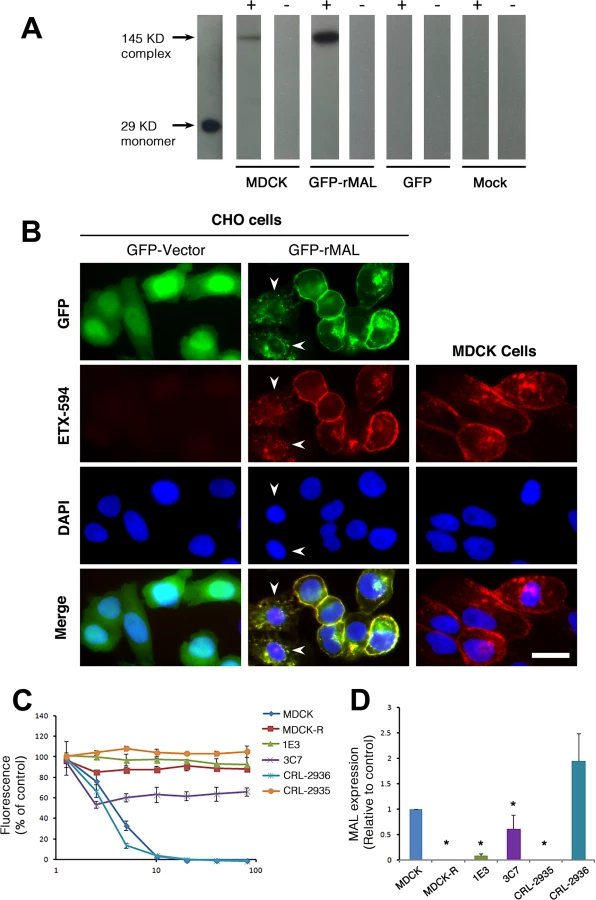
MDCK cells are possibly the most thoroughly studied ETX-sensitive cell line, and we therefore wanted to compare the binding and sensitivity of ETX between MDCK and MAL expressing CHO cells. ETX is a 29 KDa protein that heptamerizes [46, 48, 73] after binding to its cell surface specific receptor, generating an asymmetric pore leading to ATP depletion and loss of ion and water homeostasis [74]. To determine if MAL expression in CHO cells is required for ETX heptamerization, we treated CHOGFP, CHOGFP-rMAL and MDCK cells with ETX and performed Western-blot analysis of cell lysates for oligomerized ETX. As anticipated, treatment of MDCK and CHOGFP-rMAL cells with ETX result in formation of an approximate 145 kDa complex (Fig 7A) consistent with a heptameric pore [46, 73]. Both MDCK and CHOGFP-rMAL cells show similar localization patterns of ETX binding to the cell surface (Fig 7B). We next determined the relationship between MAL expression level and ETX sensitivity by analyzing various MDCK cell lines either sensitive or resistant to ETX (Fig 7C and 7D). MDCK (CCL-34) cells were treated with a dose of ETX sufficient to kill >95% of the monolayer and individual surviving cells were recovered as a pool (MDCK-R) or as individual clonal populations (1E3 and 3C7). In addition, two additional MDCK cell derivatives (CRL-2935 and CRL-2936) were acquired from ATCC. Sensitivity to ETX was determined by incubating cell monolayers with serial dilutions of purified ETX (Fig 7C). In all cases ETX-sensitive MDCK cells express MAL and ETX-resistant MDCK cells show no expression or negligible levels of MAL (Fig 7D).
Previous studies have shown that a group of closely spaced aromatic amino acid residues within domain 1 of ETX are necessary for binding and toxicity to MDCK cells [44, 75, 76]. Specifically, glutamate substitutions at Y29, Y30, Y36 or F199 each result in loss of ETX binding, pore formation and cytotoxicity [44]. At Y196, also within domain 1, glutamate substitution result in significantly reduced ETX activity [44]. Here, we tested whether these ETX mutations similarly impact ETX toxicity in MAL-expressing CHO cells. Wild-type ETX (wt-Etx), mutant toxin containing the Y30E substitution (Etx-Y30E), and mutant toxin containing the Y196E substitution (Etx-Y196E) were examined, and rMAL - or hMAL-expressing CHO cells were compared to MDCK cells in cytotoxicity assays (Fig 8). As controls, we included CHO cells expressing the irrelevant tetraspan PLLP [77] as well as GFP vector alone. As expected, wild-type toxin induces robust cell death in MDCK cells and in CHO cells expressing rMAL or hMAL. rMAL-expressing CHO cells are more sensitive than MDCK cells, which exhibit greater sensitivity than hMAL-expressing CHO cells. Consistent with published reports [44, 75, 76], Y30 mutation (Etx-Y30E) completely abolishes ETX-induced cell death while Y196 mutation (Etx-Y196E) significantly reduces ETX-toxicity in MDCK cells. Remarkably, the effects of these ETX mutations on MDCK cells are essentially reproduced in MAL-expressing CHO cells. Y30 mutation (Etx-Y30E) renders ETX ineffective in both rMAL - and hMAL-expressing CHO cells. Intriguingly, while Etx-Y196E attenuates ETX effect approximately 10 folds in rMAL-expressing CHO cells compared to Wt-Etx, this mutant toxin slightly increases ETX toxicity in hMAL-expressing CHO cells. PLLP-expressing CHO cells are completely insensitive to wild-type ETX, indicating the specificity of MAL for toxin activity. These findings demonstrate that MAL expression alone is not only necessary to impart ETX toxicity in insensitive naïve CHO cells but is also able to confer differential response to ETX mutants. Moreover, our data suggest that ETX-MAL interaction involves the aromatic side chains in domain 1 of ETX and the second extracellular domain of MAL.

We have so far provided evidence that introduction of MAL into ETX-resistant cells renders them capable of binding to toxin and sensitive to its effects. We further showed that insertion of the FLAG epitope into ECL2 results in loss of ETX toxicity. To elaborate on the loss-of-function strategy, we made use of the mice deficient in MAL (MAL-/-) [55], which exhibit no overt physical or behavioral abnormalities but do have a defect in the maintenance of CNS paranodal myelin loop structure [55]. We first investigated the behavioral responses of animals injected with ETX using a detailed scoring system. Adult mice were injected via the tail vein with 5 ng of ETX per gram of body weight and then placed under continuous observation for an hour. The mean time for onset of symptoms following ETX administration is 11.8 minutes for wild-type mice (Table 1), and all of the wild-type mice undergone ETX injection progressed rapidly to a moribund state requiring euthanasia. None of the MAL-/ - mice injected with ETX developed symptoms and they remain healthy throughout the next 48 hours after which they were routinely sacrificed. Neither wild-type nor MAL-/ - mice in the untreated control groups developed behavioral symptoms. In a second experiment, mice were injected with 50 ng of ETX and videotaped. MAL-/-mice were completely resistant to ETX, whereas wild-type mice rapidly became obtunded (S1 Movie).

2–5 month-old wild-type (MAL+/+) or MAL-Knockout (MAL-/-) mice were untreated (control) or injected via the tail vein with ETX at the dose of 5 ng/gram of body weight. Time until symptoms was recorded for the next hour. Data are means and SD from a single experiment and are representative of two independent experiments
We next examined binding of ETX to tissues from wild-type and MAL-/ - mice [55]. In wild-type mouse tissues examined, Alexa-594-labeled ETX stains gut microvasculature, retinal microvessels, renal tubules, CNS myelin, and squamous epithelium of the eye sclera (Fig 9). However, ETX fails to stain these same structures from MAL-/ - mice (Fig 9). Staining of markers for CNS myelin (PLP), microvasculature and eye sclera epithelium (BSL1), and renal tubules (Shiga toxin 1 beta) show no difference between wild-type and knockout mice. These results indicate that MAL loss-of-function abrogates specific ETX binding in vivo.

Discussion
Is MAL the ETX receptor?
We have shown that introduction of the MAL gene imparts ETX sensitivity to previously insensitive CHO cells, and that the characteristic responses of MDCK cells to ETX are shared by MAL-expressing CHO cells. These cellular responses include cell surface binding of ETX, ETX oligomerization and pore assembly, differential response to ETX mutant proteins according to the nature of the mutations [43, 44, 76, 78], and cell death. Such remarkable consistencies led us to speculate that MAL is either 1) the bona fide ETX receptor and interacts directly with ETX via protein-protein interactions or 2) part of the ETX receptor complex and plays a role in the sorting, chaperoning, and/or recruitment of a novel direct binding partner of ETX.
We employed several different experimental approaches to determine if ETX interacts directly with MAL, however, the results of these experiments were inconclusive. Numerous variations on traditional co-immunoprecipitation (co-IP) and far-Western blotting experiments altering antibodies, salt concentrations and detergents yielded inconsistent results. One possible explanation for these inconsistences is that transmembrane proteins containing multiple hydrophobic domains, as is the case of MAL, often possess different tertiary structures and binding affinities when in solution relative to those occurring within a lipid bilayer [79]. Such changes might confound solution-based analysis such as co-IP and far-Western detection or even render physical interaction between ETX and MAL impossible in these conditions. We also obtained negative results from far-Western blotting and fluorescence resonance energy transfer (FRET) experiments. While FRET analysis was performed under physiologic conditions with MAL in its native conformation within a lipid bilayer, molecular orientation and physical distance between the relevant fluorophores on ETX and MAL could easily preclude FRET detection. Investigation using other advanced methods for detecting protein-protein interactions, including co-IP/protein mass fingerprinting and surface plasmon resonance biosensor assays are currently underway. Our results thus far do not distinguish between a function of MAL in direct binding of ETX versus a role of MAL in recruiting or organizing the ETX receptor complex.
Although we have been unable to determine if MAL interacts directly with ETX, theoretical modeling suggests MAL may have the structural requirements to facilitate direct protein-to-protein interaction. The primary structure of the extracellular loop 2 (ECL2) for MAL reveals a Q to F substitution between human and rat respectively at position 114, a change from the highly polar side chain of glutamine to the non-polar side chain of phenylalanine. Considering the functional significance of aromatic side chains in the receptor-binding domain of ETX [44], the aromatic residue in ECL2 for rMAL may contribute to additional aromatic stacking interactions with residues in the receptor-binding domain of ETX [44]. If MAL is the binding receptor for ETX, then the second ECL appears to play an essential role. Insertion of the FLAG epitope into ECL2 leads to abrogation of both ETX binding and cytotoxicity while retaining normal localization of MAL in the plasma membrane. Computer-aided analysis predicts that the ECL2 FLAG sequence insertion changes structural dynamics of ECL2 - particularly in the aspect of protein surface accessibility. This, if experimentally proved, would suggest a central role for ECL2 structural integrity in ETX binding and toxicity. Interestingly, previous studies using the same construct show FLAG insertion in ECL2 neither alters the incorporation of MAL into glycolipid-enriched membrane microdomains and lipid rafts [80], nor influences MAL trafficking between the cell membrane and the trans-Golgi network [56]. That the FLAG insertion in ECL2 results in loss of ETX binding and toxicity thus favors, but does not prove, a direct role of MAL in transmitting ETX activity into target cells.
Despite MAL’s structural possibilities as the ETX receptor and the requirement of MAL for both ETX binding and toxicity, it is possible that MAL is not the direct binding receptor for ETX. MAL could be functioning in ETX binding either as a co-receptor, part of a multmeric protein complex, or as a membrane lipid micro-domain organizer necessary for assembly of the receptor complex, without directly binding to ETX. The lethal effects of C. perfringens ETX require first, binding of ETX monomers to a specific cell surface receptor; second, oligomerization into a prepore complex; and third insertion of the stable pore into the plasma membrane culminating in the free passage of ions and water and thus loss of electrochemical gradients essential for cell viability [46–48, 73]. MAL could play a role in any of these required steps without directly interacting with ETX. Indeed, MAL is known to localize to the apical surface or within specialized microdomains of particular polarized cells [49, 53, 56–60, 63, 80–83]. Localization of MAL on cell surface and MAL’s role in protein sorting provide the ideal localization, microenvironment and cellular machinery for ETX to target and damage cells [54, 55, 58, 64, 82, 84].
Previous studies show that HAVCR1 is required, at least partially, for ETX-mediated cytotoxicity [37, 44]. HAVCR1 increases the sensitivity of cells to ETX if they are already sensitive to the toxin. However, HAVCR1 expression alone is insufficient to confer ETX sensitivity upon cell lines resistant to the ETX effect. Also, both ETX binding and ETX-mediated cell death occur in the absence of HAVCR1. Moreover, ETX binding with HAVCR1 on cell surface has not been demonstrated [37]. In contrast, cells insensitive to ETX become completely sensitive when MAL expression is induced. In addition, ETX binds to the MAL-expressing cell membrane surface and becomes internalized in a time-dependent fashion. Thus, MAL seems to play a more central role than HAVCR1 in the context of ETX binding and toxicity. It would be interesting to determine whether MAL is physically and/or functionally linked to HAVCR1 in this context.
Taken together, we have identified, for the first time, a protein that is required for both binding and activity of ETX to cells. These results provide novel insights on how this toxin interacts with host cells. Since ETX is responsible for a devastating disease in livestock and may be a possible environmental factor contributing to the initiation of new MS lesion formation, understanding the molecular basis of toxin binding to cells and cytotoxicity opens investigation into new treatment strategies.
Does MAL structure and function make sense for enhancing ETX pore formation?
The normal physiological function of MAL in apical sorting of proteins to specialized lipid rafts affords the ideal context for a pore-forming toxin. ETX oligomerization requires close proximity of monomers to facilitate interaction between amphipathic residues within domain 3 of the active toxin [43]. Highly concentrated MAL within lipid rafts favors these necessary protein-protein interactions by bringing MAL-bound toxin monomers within binding range of one another [54, 55, 58, 64, 82, 84]. Intriguingly, MAL itself has been shown to self-oligomerize [64], which may further support ETX oligomerization and pore formation. The same membrane biophysical characteristics achieved within the lipid raft microenvironment that enhance endosome formation also appear to enhance pore formation. Lipids that favor spontaneous negative curvature in bilayers such as cholesterol, diacylglcerol, long-chain unsaturated fatty acids, glycosphingolipids and phosphatidylethanolamine are enriched in lipid rafts and have been shown to enhance ETX pore formation [34, 36]. Caveolins 1 and 2, essential scaffolding proteins localized to lipid rafts, promote formation of caveolae and are necessary for oligomerization of ETX and pore formation [85]. In essence, the MAL microenvironment is well suited for a pore-forming toxin that requires oligomerization.
What accounts for the variability in ETX pathogenicity between species?
Why are the pathological effects of ETX so variable between species? In part, this occurs due to known differences in the kinetics of bacterial growth within the gastrointestinal tracts of species with fermenting 4-chamber stomachs (ruminant) and the linear gut of the rodent or human [3, 7, 86–98]. A second level of difference explaining the distinct pathologic effects of ETX between species is receptor affinity. The binding receptor for ETX remains undefined. In this report we have shown that MAL is required for both ETX binding to cells and for cellular toxicity. Interestingly, the binding affinity and the susceptibility of cells to cytotoxic effects of ETX vary dramatically between rodent and human. Cells expressing human MAL bind ETX with approximately 1/10 the affinity of cells expressing rat MAL, and cells expressing zebrafish MAL do not bind ETX at all. The differences in affinity and cytotoxicity of ETX for cells expressing different MAL orthologues offers an additional level of complexity for the remarkable differences in toxin mediated pathologies between species. It will be of interest to test the relative binding affinity of ETX to MAL from a wide range of species.
Materials and Methods
Ethics statement
All animal work was conducted according to federal guidelines and approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee.
Generation of MAL constructs
The pβ-actin-EGFP-rat MAL (rMAL) and human MAL (hMAL) constructs and hMAL harboring a FLAG sequence insertion in its second extracellular loop (hMAL-L2F) have been previously published [56, 58]. hMAL and hMAL-L2F were subcloned and inserted into the N-terminal of GFP gene in the pβ-actin-EGFP vector. hMAL with a FLAG insertion in the first extracellular loop (hMAL-L1F) was synthesized with RsrII/XbaI restriction sites, and purchased from Genewiz, Inc. A plasmid encoding zebrafish MAL was purchased from ATCC, subcloned and inserted into the pβ-actin-EGFP vector. The pEGFP-C1 vector was purchased from Addgene and used as the GFP control. All plasmids were amplified and purified (Qiagen) in One Shot Top 10 Escherichia coli (Life Technologies). Sequence analysis and verification was performed (Macrogen USA).
ETX
ETX and pro-ETX are provided by BEI at a minimum > 95% purity. Each lot was provided with a certificate of analysis including a Western-blot, which showed a dominant band at ~30 kDa. In some lots, a faint low molecular weight band was also observed. Importantly, use of an ETX specific neutralizing antibody completely abrogated the cell death-inducing activity of the reagent underscoring its purity.
Fluorophore labeling of ETX/pro-ETX
His-tagged ε-prototoxin was procured from BEI Resources. One mg was fluorescently labeled using Alexa Fluor 594 Protein Labeling Kit (Life Technologies) and 1 mg was labeled using Alexa Fluor 647 Protein Labeling Kit (Life Technologies) as per manufacturer's instructions. His tagged Shiga toxin 1β was procured for BEI Resources and fluorescently labeled using Alexa Fluor 488 Protein Labeling Kit (Life Technologies).
Expression and purification of recombinant ε -prototoxin
Expression and purification of recombinant wild-type and mutant ε-prototoxins were performed as described previously [44, 72]. Purified ε-prototoxins were treated with trypsin-coated agarose beads and conversion of the prototoxin to active toxin was assessed based on SDS-PAGE and immunoblotting with anti-ETX antibodies [37].
Cell culture and transfection
Chinese hamster ovary (CHO) cells (ATCC Cat#CCL-61) were grown in Dulbecco's Modified Eagle's Medium/Ham's F12 medium (Life Technologies) with 10% heat-inactivated fetal bovine serum, 50 units/ml penicillin and 50 μg/ml streptomycin. CHO cells were transfected with the indicated expression plasmids using Turbofect (Fermentas) according to the manufacturer’s instructions. For stable cells, clumped cells were collected after transfection and growth in media containing G418 400 μg per ml for three weeks. Cells were then analyzed under a fluorescence microscope for GFP expression. Homogenous GFP expressing cells were selected using a Becton-Dickinson Vantage cell sorter. Colonies derived from single cells were viable and expanded to generate frozen stocks. MDCK cells were routinely cultured in Minimum Essential Medium (MEM) supplemented with glutamax (Life Technologies) and 10% FetalClone III serum (HyClone) and incubated at 37°C in 5% CO2.
Cell viability assays
PrestoBlue assay
Stably transfected rat GFP-MAL (CHOGFP-rMAL) and GFP (CHOGFP) cells were grown in 384-well plates and incubated with ETX overnight. PrestoBlue Reagent (Life Technologies), which is quickly reduced by metabolically active cells to a red fluorescent metabolite, was added to each well using a multi-drop liquid dispenser. Fluorescence intensity was measured with a microtiter plate reader (Perkin-Elmer Envision). For cytotoxicity analyses of MDCK cells, cells were plated in Leibovitz L-15 medium supplemented with 10% fetal bovine serum at a density of 5×103 cells per well in 384-well plates [66]. ETX was added and the cells were incubated at 37°C overnight. Cytotoxicity was determined by treating cells with resazurin (CellTiter Blue, Promega) at 37°C for 4 hours. Fluorescence was measured at 590 nm following excitation at 560 nm using a BioTek FLx800 plate reader. Results were normalized to the fluorescent signal from cells incubated in the absence of toxin (100%) and in 0.1% Triton X-100 (0%).
Propidium iodide (PI) uptake assay
Cell membrane permeability was assessed by the ability of cells to exclude the DNA-binding fluorescent dye PI. Specifically, CHO cells stably expressing GFP, GFP-rMAL (rMAL), GFP-hMAL (hMAL), or GFP-hMAL-L2f were treated in triplicate wells for each condition indicated in the experiments. Live images of randomly chosen fields in each well were acquired under an inverted fluorescence microscope (Nikon) equipped with a CCD camera (Carl Zeiss), and were then imported into ImageJ64 (NIH) in 8-bit grey format. For quantification of PI-positive cells, the images were converted into binary images by applying the same threshold value to all images collected from the same experiment. Analyze Particles function was selected to automatically count the particle numbers and to analyze particle properties, such as size shape, and distribution patterns. Data were exported into Excel (Microsoft) and statistical significance was assessed by two-tailed student’s t-test. In the experiment indicated, the toxin dose required to kill 50% of the cells (IC50) was determined by nonlinear regression analysis in Prism (GraphPad Software).
Flow cytometry
Transfected cells were removed from the tissue culture substrate by incubating with citric saline (135μM potassium chloride, 15μM sodium citrate dissolved in sterile water) for 5 minutes at 37°C. Cells were washed off the plate and then triturated into single cell suspensions. Cells were washed 3 times with DMEM without phenol red (Life Technologies), containing 10% fetal calf serum, 50 U/ml penicillin, and 50mg/ml streptomycin. Cells were diluted to 5x105/ml and incubated with 20 nM Alexa 647-labeled ETX for 1 hour at 37°C. Labeled cells were washed 3 times in DMEM lacking phenol red, and read on a AccuriC6 flow cytometer (BD Biosciences).
Fluorescence microscopy
Cells stably transfected with rat MAL (CHO-MAL) or GFP (CHO-GFP) were plated on glass bottom dishes (Mattek) at a density of 6x104 cells/ml, and allowed to grow overnight. Cells were subsequently washed and fixed with 4% PFA for 10 minutes at room temperature (RT). Cells were washed and incubated with Alexa 594 labeled ETX (50nM) for 1 hour at RT. After incubation, the cells were washed 3 times in PBS and Hoechst (1μg/ml) was added for visualization of nuclei. Stained cells were visualized by confocal microscopy at the Rockefeller University Bio-Imaging Facility.
Analyses of ETX binding and ETX-MAL co-localization
CHO cells stably expressing GFP, GFP-rMAL, GFP-hMAL or GFP-hMAL-L2F cultured in triplicated wells were incubated with ALexa 594-labeled ETX (ETX-594) for an hour. In some experiments, MDCK cells were incubated with ETX-594 for 30 minutes. Cells were fixed then with 4% PFA for 10 minutes at RT and were subsequently mounted onto slides in VECTASHIELD mounting medium containing DAPI (Vector Laboratories). Digital images were acquired under Axiskop2 fluorescence microscope (Carl Zeiss) coupled with a SPOT cooled camera (Diagnostic Instruments) and were imported into ImageJ64 (NIH) in 8-bit grey format. Images for GFP-expression and ETX-binding intensity analyses were processed in separate channels and converted into binary images by applying the same respective threshold values. Measurement was automatically performed on the binary images for mean and integrated fluorescent density. Data were exported into Excel (Microsoft) for statistical analysis.
FLAG expression analysis
CHO cells stably expressing GFP-hMAL or GF-hMAL-L2F were incubated with the anti-FLAG antibody (Sigma) at 37°C for 10 minutes, followed by two quick washes with Hank's Balanced Salt Solution (HBSS, Life Technologies). Cells were fixed with 4% PFA at RT for 10 minutes and then mounted onto slides in VECTASHIELD mounting medium with DAPI (Vector Laboratories). Images were acquired under Axiskop2 fluorescence microscope (Carl Zeiss) coupled with a SPOT cooled camera (Diagnostic Instruments) and processed in Photoshop (Adobe).
MAL expression in cell lines
Total RNA was isolated from cells with TRIZol reagent (Life Technologies). Complementary DNA (cDNA) was prepared from 2 μg total RNA using iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR was performed using a MAL-specific primer set from Applied Biosystems and a StepOne Plus instrument (Applied Biosystems). A primer set specific to 18S rRNA was used as control. Results were analyzed by comparing CT values [99].
Western-blot analysis of the ETX complex
MDCK, CHOGFP-rMAL, CHOGFP and mock transfected CHO cells (CHOmock) cells were grown in 6 - well plates (Costar) until confluence. Activated ETX (60nM) or vehicle control was administered to cells and allowed to incubate for 1 hour at 37 C°. Cells were detached from plate following trypsin digestion. Cells were collected, washed 3 times in PBS and re-suspended in PBS containing 0.2% SDS. Harvested cells were then triturated with a serological pipette followed by trituration with an insulin syringe until DNA was sufficiently sheared. ETX complex containing detergent resistant membranes was isolated by centrifugation at 14, 000G for 5 minutes and collecting the pellet. The pellet was washed 3 times in PBS and re-suspended in 100 ul PBS. An equal volume of 2 X Lamelli sample buffer (Bio-rad) was added to each sample for SDS page electrophoresis. Proteints were transferred to an Immobilon-P membrane (Millipore) and probed with a custom rabbit anti-ETX antibody generated against the N terminal of ETX sequence KASYDNVDTLIEKGR (Pacific Immunology). HRP conjugated donkey anti-Rabbit IgG (Jackson Immunoresearch) was used to visualize the ETX complex.
Immunofluorescence analysis with mouse tissues
Brain
Fresh frozen tissue sections were fixed in 4% PFA for 10 minutes at RT, permeabilized in a 1% sodium cholate, 1% BSA, 10% donkey serum, PBST solution overnight at 4°C16. Sections were then incubated with rabbit anti-PLP (ThermoScientific) at 1 : 1000 overnight at 4 C°. Following three washes with PBS, sections were then incubated with donkey anti-rabbit Alexa 488 (Jackson ImmunoResearch) at 1 : 1000, and Alexa 594 labeled His-tagged protoxin (50nM) for 2 hours at RT.
Retina/eye
Fresh frozen tissue sections were incubated with BSLI (Vector Labs) 1 : 200, and Alexa 594 labeled His-tagged protoxin (50 nM) for 1 hr at RT. After three 5-minute washes in PBS, stained sections were post fixed in 4% PFA for 10 minutes at RT.
Kidney
Fresh frozen tissue sections were fixed in 4% PFA for 10 minutes at room temperature, washed and incubated with Alexa 488 labeled His-tagged Shiga toxin 1β (200 nM) and Alexa 594 labeled His-tagged protoxin (50 nM) for 1 hour RT.
The above stained tissues were washed 3 times in PBS, mounted and imaged under Axiskop2 fluorescence microscope (Carl Zeiss) coupled with a SPOT cooled camera (Diagnostic Instruments.
Intestines
Anesthetized wild-type and MAL-/ - mice were injected intravenously via tail vein injection with 1ug Alexaflour 594 labeled active ETX and 1.6 μl FITC-conjugated BLS1 (Vector Laboratories) per gram of mouse. After ten minutes, mice were euthanized by cardiac perfusion with PBS. Tissue was removed and fresh frozen in OTC.
Mouse lines
MAL-deficient mice (MAL-/-) were generated by replacing the first exon of the MAL gene with the lacZ gene sequence [55]. MAL-/ - mice were backcrossed with C57 / Bl6 mice for more than six generations. MAL-deficient mice bred normally and had a normal life span.
ETX toxicity in mice
Animal handling
All animal work was conducted under an approved WCMC animal protocol. Each mouse received ETX via intraperitoneal injection. Animals were injected once IP with the dose of ETX indicated or with vehicle. Behavior was monitored every 15 minutes for the first 2 hours and every half hour for the next six hours. If animals showed signs of severe neurological toxicity (see below) then they were sacrificed by a lethal injection of Ketamine/Xylazine (450 mg/kg Ketamine IP: 45 Xylazine mg/kg IP).
Scoring system and actions for humane care of animals
For all animals a medical record and a neurological scoring chart was kept. For neurological signs the scoring system used was the same as for experimental allergic encephalomyelitis experiments. The scoring system was as follows: 0: No abnormality. Action: Continued monitoring. 1: Initial signs but no paraparesis. This included clumsiness, incontinence, flaccid tail. Action: Moistened food and hydration packs were added to the cage bottom. Continued monitoring. 2: Mild paraparesis: Characterized by trouble initiating movement, but walking well once started. Action: Affected mice were fed with moistened food and gel hydration packs. Continued monitoring. 3: Moderate paraparesis: Characterized by an inability to move one or both hindlegs, noticeable gait disturbance, and possible atonic bladder. Action: Moistened food and hydration packs were placed on floor of cage. Expressed urinary bladder if necessary. Supplemental heat if necessary. 4: Moderate quadraparesis: Characterized by weakness in all limbs, associated gait disturbance and possible atonic bladder. Animals could still move and feed from moistened food or hydration packs. Action: Moistened food and hydration packs were placed on the cage floor. Animals in this state were monitored every 15–30 minutes. The urinary bladder was manually expressed if needed. Animals were provided an external means of heat, if needed (heat lamp, Safe and Warm, nestlets with additional bedding). If animals were unable to move or unable to feed, then they were euthanized immediately. 5: Moribund: Characterized by an inability to move even with prodding, inability to feed, or labored respirations. Action: Euthanized immediately.
Data analysis and statistics
In most experiments, data were analyzed with Excel (Microsoft) and statistical significance was assessed by two-tailed student’s t-test. * p < 0.05; ** p < 0.01; *** p < 0.001. In MAL expression analysis, statistical different between samples were performed by ANOVA followed by Dunnett’s post hoc test. * P<0.05. In experiments determining ETX IC50 on vector-,rMAL - or, hMAL-expressing CHO cells, nonlinear regression analysis was performed in Prism (GraphPad Software).
Accession numbers
Rat (Rattus norvegicus) myelin and lymphocyte mRMA/Mal NM_012798, Rat Mal protein NP_036930, Human (Homo sapiens) MAL mRNA NM_002371, Human MAL protein NP_002362, Dog (Canis lupus familiaris) Mal mRNA NM_001003253, Dog Mal protein NP_001003253, Zebrafish (Danio rerio) MAL-like protein mRMA NM_001017686, Zebrafish Mal-like protein NP_001017686, epsilon-toxin (Clostridium perfringens B strain ATCC 3626) gene ABDV01000020.1, epsilon-toxin protein EDT23265.1
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Hatheway CL. Toxigenic clostridia. Clin Microbiol Rev. 1990;3(1):66–98. Epub 1990/01/01. 2404569
2. Popoff MR. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 2011;278(23):4602–15. doi: 10.1111/j.1742-4658.2011.08145.x 21535407
3. Alves GG, Machado de Avila RA, Chavez-Olortegui CD, Lobato FC. Clostridium perfringens epsilon toxin: The third most potent bacterial toxin known. Anaerobe. 2014;30C:102–7.
4. Blackwell TE, Butler DG, Bell JA. Enterotoxemia in the goat: the humoral response and local tissue reaction following vaccination with two different bacterin-toxoids. Can J Comp Med. 1983;47(2):127–32. 6309346
5. Finnie JW. Neurological disorders produced by Clostridium perfringens type D epsilon toxin. Anaerobe. 2004;10(2):145–50. 16701511
6. Uzal FA, Kelly WR, Morris WE, Bermudez J, Baison M. The pathology of peracute experimental Clostridium perfringens type D enterotoxemia in sheep. J Vet Diagn Invest. 2004;16(5):403–11. 15460322
7. Uzal FA, Songer JG. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J Vet Diagn Invest. 2008;20(3):253–65. 18460610
8. Gkiourtzidis K, Frey J, Bourtzi-Hatzopoulou E, Iliadis N, Sarris K. PCR detection and prevalence of alpha-, beta-, beta 2-, epsilon-, iota - and enterotoxin genes in Clostridium perfringens isolated from lambs with clostridial dysentery. Vet Microbiol. 2001;82(1):39–43. 11423193
9. Goncalves LA, Lobato ZI, Silva RO, Salvarani FM, Pires PS, Assis RA, et al. Selection of a Clostridium perfringens type D epsilon toxin producer via dot-blot test. Arch Microbiol. 2009;191(11):847–51. doi: 10.1007/s00203-009-0510-y 19779698
10. Uzal FA, Kelly WR, Thomas R, Hornitzky M, Galea F. Comparison of four techniques for the detection of Clostridium perfringens type D epsilon toxin in intestinal contents and other body fluids of sheep and goats. J Vet Diagn Invest. 2003;15(2):94–9. 12661718
11. Bhown AS, Habeerb AF. Structural studies on epsilon-prototoxin of Clostridium perfringens type D. Localization of the site of tryptic scission necessary for activation to epsilon-toxin. Biochem Biophys Res Commun. 1977;78(3):889–96. 199192
12. Freedman JC, Li J, Uzal FA, McClane BA. Proteolytic Processing and Activation of Clostridium perfringens Epsilon Toxin by Caprine Small Intestinal Contents. MBio. 2014;5(5).
13. Habeeb AF, Lee CL, Atassi MZ. Conformational studies on modified proteins and peptides. VII. Conformation of epsilon-prototoxin and epsilon-toxin from Clostridium perfringens. Conformational changes associated with toxicity. Biochim Biophys Acta. 1973;322(2):245–50. 4358084
14. Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N - and C-terminal peptides. Microbiol Immunol. 1997;41(7):527–35. 9272698
15. Worthington RW, Mulders MS. Physical changes in the epsilon prototoxin molecule of Clostridium perfringens during enzymatic activation. Infect Immun. 1977;18(2):549–51. 200566
16. Harkness JM, Li J, McClane BA. Identification of a lambda toxin-negative Clostridium perfringens strain that processes and activates epsilon prototoxin intracellularly. Anaerobe. 2012;18(5):546–52. Epub 2012/09/18. doi: 10.1016/j.anaerobe.2012.09.001 22982043
17. Adamson RH, Ly JC, Fernandez-Miyakawa M, Ochi S, Sakurai J, Uzal F, et al. Clostridium perfringens epsilon-toxin increases permeability of single perfused microvessels of rat mesentery. Infect Immun. 2005;73(8):4879–87. 16041001
18. Tamai E, Ishida T, Miyata S, Matsushita O, Suda H, Kobayashi S, et al. Accumulation of Clostridium perfringens epsilon-toxin in the mouse kidney and its possible biological significance. Infect Immun. 2003;71(9):5371–5. 12933886
19. Finnie JW. Ultrastructural changes in the brain of mice given Clostridium perfringens type D epsilon toxin. J Comp Pathol. 1984;94(3):445–52. 6088599
20. Finnie JW. Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: a review. Aust Vet J. 2003;81(4):219–21. 15080445
21. Finnie JW, Manavis J, Casson RJ, Chidlow G. Retinal microvascular damage and vasogenic edema produced by Clostridium perfringens type D epsilon toxin in rats. J Vet Diagn Invest. 2014;26(3):470–2. 24741023
22. Finnie JW, Manavis J, Chidlow G. Loss of endothelial barrier antigen immunoreactivity as a marker of Clostridium perfringens type D epsilon toxin-induced microvascular damage in rat brain. J Comp Pathol. 2014;151(2–3):153–6. doi: 10.1016/j.jcpa.2014.07.002 25172052
23. Soler-Jover A, Dorca J, Popoff MR, Gibert M, Saura J, Tusell JM, et al. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon. 2007;50(4):530–40. 17572464
24. Worthington RW, Mulders MS. Effect of Clostridium perfringens epsilon toxin on the blood brain barrier of mice. Onderstepoort J Vet Res. 1975;42(1):25–7. 171606
25. Zhu C, Ghabriel MN, Blumbergs PC, Reilly PL, Manavis J, Youssef J, et al. Clostridium perfringens prototoxin-induced alteration of endothelial barrier antigen (EBA) immunoreactivity at the blood-brain barrier (BBB). Exp Neurol. 2001;169(1):72–82. 11312560
26. Uzal FA, Kelly WR. Effects of the intravenous administration of Clostridium perfringens type D epsilon toxin on young goats and lambs. J Comp Pathol. 1997;116(1):63–71. 9076601
27. Finnie JW, Blumbergs PC, Manavis J. Neuronal damage produced in rat brains by Clostridium perfringens type D epsilon toxin. J Comp Pathol. 1999;120(4):415–20. 10208737
28. Lonchamp E, Dupont JL, Wioland L, Courjaret R, Mbebi-Liegeois C, Jover E, et al. Clostridium perfringens epsilon toxin targets granule cells in the mouse cerebellum and stimulates glutamate release. PLoS One. 2010;5(9).
29. Wioland L, Dupont JL, Bossu JL, Popoff MR, Poulain B. Attack of the nervous system by Clostridium perfringens Epsilon toxin: from disease to mode of action on neural cells. Toxicon. 2013;75 : 122–35. doi: 10.1016/j.toxicon.2013.04.003 23632158
30. Garcia JP, Giannitti F, Finnie JW, Manavis J, Beingesser J, Adams V, et al. Comparative Neuropathology of Ovine Enterotoxemia Produced by Clostridium perfringens Type D Wild-Type Strain CN1020 and Its Genetically Modified Derivatives. Vet Pathol. 2015;52(3):465–75. doi: 10.1177/0300985814540543 24964921
31. Murrell TG, O'Donoghue PJ, Ellis T. A review of the sheep-multiple sclerosis connection. Med Hypotheses. 1986;19(1):27–39. 2871478
32. Rumah KR, Linden J, Fischetti VA, Vartanian T. Isolation of Clostridium perfringens Type B in an Individual at First Clinical Presentation of Multiple Sclerosis Provides Clues for Environmental Triggers of the Disease. PLoS One. 2013;8(10):e76359. doi: 10.1371/journal.pone.0076359 24146858
33. Nagahama M, Hara H, Fernandez-Miyakawa M, Itohayashi Y, Sakurai J. Oligomerization of Clostridium perfringens epsilon-toxin is dependent upon membrane fluidity in liposomes. Biochemistry. 2006;45(1):296–302. 16388606
34. Nagahama M, Itohayashi Y, Hara H, Higashihara M, Fukatani Y, Takagishi T, et al. Cellular vacuolation induced by Clostridium perfringens epsilon-toxin. FEBS J. 2011;278(18):3395–407. doi: 10.1111/j.1742-4658.2011.08263.x 21781280
35. Turkcan S, Alexandrou A, Masson JB. A Bayesian inference scheme to extract diffusivity and potential fields from confined single-molecule trajectories. Biophys J. 2012;102(10):2288–98. doi: 10.1016/j.bpj.2012.01.063 22677382
36. Turkcan S, Masson JB, Casanova D, Mialon G, Gacoin T, Boilot JP, et al. Observing the confinement potential of bacterial pore-forming toxin receptors inside rafts with nonblinking Eu(3+)-doped oxide nanoparticles. Biophys J. 2012;102(10):2299–308. doi: 10.1016/j.bpj.2012.03.072 22677383
37. Ivie SE, Fennessey CM, Sheng J, Rubin DH, McClain MS. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens epsilon-toxin. PLoS One. 2011;6(3):e17787. doi: 10.1371/journal.pone.0017787 21412435
38. Soler-Jover A, Blasi J, Gomez de Aranda I, Navarro P, Gibert M, Popoff MR, et al. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J Histochem Cytochem. 2004;52(7):931–42. 15208360
39. Dorca-Arevalo J, Soler-Jover A, Gibert M, Popoff MR, Martin-Satue M, Blasi J. Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet Microbiol. 2008;131(1–2):14–25.
40. Finnie JW. Histopathological changes in the brain of mice given Clostridium perfringens type D epsilon toxin. J Comp Pathol. 1984;94(3):363–70. 6088597
41. Alonso MA, Weissman SM. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc Natl Acad Sci U S A. 1987;84(7):1997–2001. 3494249
42. Schaeren-Wiemers N, Valenzuela DM, Frank M, Schwab ME. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J Neurosci. 1995;15(8):5753–64. 7643216
43. Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, et al. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J Biol Chem. 2011;286(22):19549–55. doi: 10.1074/jbc.M111.228478 21489981
44. Ivie SE, McClain MS. Identification of amino acids important for binding of Clostridium perfringens epsilon toxin to host cells and to HAVCR1. Biochemistry. 2012;51(38):7588–95. doi: 10.1021/bi300690a 22938730
45. Knapp O, Maier E, Benz R, Geny B, Popoff MR. Identification of the channel-forming domain of Clostridium perfringens Epsilon-toxin (ETX). Biochim Biophys Acta. 2009;1788(12):2584–93. doi: 10.1016/j.bbamem.2009.09.020 19835840
46. Miyata S, Matsushita O, Minami J, Katayama S, Shimamoto S, Okabe A. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J Biol Chem. 2001;276(17):13778–83. 11278924
47. Robertson SL, Li J, Uzal FA, McClane BA. Evidence for a prepore stage in the action of Clostridium perfringens epsilon toxin. PLoS One. 2011;6(7):e22053. doi: 10.1371/journal.pone.0022053 21814565
48. Miyata S, Minami J, Tamai E, Matsushita O, Shimamoto S, Okabe A. Clostridium perfringens epsilon-toxin forms a heptameric pore within the detergent-insoluble microdomains of Madin-Darby canine kidney cells and rat synaptosomes. J Biol Chem. 2002;277(42):39463–8. 12177068
49. Zacchetti D, Peranen J, Murata M, Fiedler K, Simons K. VIP17/MAL, a proteolipid in apical transport vesicles. FEBS Lett. 1995;377(3):465–9. 8549777
50. Kim T, Fiedler K, Madison DL, Krueger WH, Pfeiffer SE. Cloning and characterization of MVP17: a developmentally regulated myelin protein in oligodendrocytes. J Neurosci Res. 1995;42(3):413–22. 8583510
51. Schaeren-Wiemers N, Schaefer C, Valenzuela DM, Yancopoulos GD, Schwab ME. Identification of new oligodendrocyte - and myelin-specific genes by a differential screening approach. J Neurochem. 1995;65(1):10–22. 7790852
52. Millan J, Puertollano R, Fan L, Rancano C, Alonso MA. The MAL proteolipid is a component of the detergent-insoluble membrane subdomains of human T-lymphocytes. Biochem J. 1997;321 (Pt 1):247–52.
53. Anton O, Batista A, Millan J, Andres-Delgado L, Puertollano R, Correas I, et al. An essential role for the MAL protein in targeting Lck to the plasma membrane of human T lymphocytes. J Exp Med. 2008;205(13):3201–13. doi: 10.1084/jem.20080552 19064697
54. Anton OM, Andres-Delgado L, Reglero-Real N, Batista A, Alonso MA. MAL protein controls protein sorting at the supramolecular activation cluster of human T lymphocytes. J Immunol. 2011;186(11):6345–56. doi: 10.4049/jimmunol.1003771 21508261
55. Schaeren-Wiemers N, Bonnet A, Erb M, Erne B, Bartsch U, Kern F, et al. The raft-associated protein MAL is required for maintenance of proper axon—glia interactions in the central nervous system. J Cell Biol. 2004;166(5):731–42. 15337780
56. Puertollano R, Alonso MA. MAL, an integral element of the apical sorting machinery, is an itinerant protein that cycles between the trans-Golgi network and the plasma membrane. Mol Biol Cell. 1999;10(10):3435–47. 10512878
57. Zhou G, Liang FX, Romih R, Wang Z, Liao Y, Ghiso J, et al. MAL facilitates the incorporation of exocytic uroplakin-delivering vesicles into the apical membrane of urothelial umbrella cells. Mol Biol Cell. 2012;23(7):1354–66. doi: 10.1091/mbc.E11-09-0823 22323295
58. Caduff J, Sansano S, Bonnet A, Suter U, Schaeren-Wiemers N. Characterization of GFP-MAL expression and incorporation in rafts. Microsc Res Tech. 2001;52(6):645–55. 11276117
59. Erne B, Sansano S, Frank M, Schaeren-Wiemers N. Rafts in adult peripheral nerve myelin contain major structural myelin proteins and myelin and lymphocyte protein (MAL) and CD59 as specific markers. J Neurochem. 2002;82(3):550–62. 12153479
60. Magal LG, Yaffe Y, Shepshelovich J, Aranda JF, de Marco Mdel C, Gaus K, et al. Clustering and lateral concentration of raft lipids by the MAL protein. Mol Biol Cell. 2009;20(16):3751–62. doi: 10.1091/mbc.E09-02-0142 19553470
61. Martin-Belmonte F, Arvan P, Alonso MA. MAL mediates apical transport of secretory proteins in polarized epithelial Madin-Darby canine kidney cells. J Biol Chem. 2001;276(52):49337–42. 11673461
62. Martin-Belmonte F, Puertollano R, Millan J, Alonso MA. The MAL proteolipid is necessary for the overall apical delivery of membrane proteins in the polarized epithelial Madin-Darby canine kidney and fischer rat thyroid cell lines. Mol Biol Cell. 2000;11(6):2033–45. 10848627
63. Puertollano R, Martinez-Menarguez JA, Batista A, Ballesta J, Alonso MA. An intact dilysine-like motif in the carboxyl terminus of MAL is required for normal apical transport of the influenza virus hemagglutinin cargo protein in epithelial Madin-Darby canine kidney cells. Mol Biol Cell. 2001;12(6):1869–83. 11408592
64. Ramnarayanan SP, Tuma PL. MAL, but not MAL2, expression promotes the formation of cholesterol-dependent membrane domains that recruit apical proteins. Biochem J. 2011;439(3):497–504. doi: 10.1042/BJ20110803 21732912
65. Lall N, Henley-Smith CJ, De Canha MN, Oosthuizen CB, Berrington D. Viability Reagent, PrestoBlue, in Comparison with Other Available Reagents, Utilized in Cytotoxicity and Antimicrobial Assays. Int J Microbiol. 2013;2013 : 420601. doi: 10.1155/2013/420601 23653650
66. Lewis M, Weaver CD, McClain MS. Identification of Small Molecule Inhibitors of Clostridium perfringens epsilon-Toxin Cytotoxicity Using a Cell-Based High-Throughput Screen. Toxins (Basel). 2010;2(7):1825–47. 20721308
67. Riss TL, Moravec RA, Niles AL, Benink HA, Worzella TJ, Minor L. Cell Viability Assays. In: Sittampalam GS, Gal-Edd N, Arkin M, Auld D, Austin C, Bejcek B, et al., editors. Assay Guidance Manual. Bethesda (MD)2004.
68. Petit L, Gibert M, Gourch A, Bens M, Vandewalle A, Popoff MR. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell Microbiol. 2003;5(3):155–64. 12614459
69. Petit L, Maier E, Gibert M, Popoff MR, Benz R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J Biol Chem. 2001;276(19):15736–40. 11278669
70. Bowman AM, Nesin OM, Pakhomova ON, Pakhomov AG. Analysis of plasma membrane integrity by fluorescent detection of Tl(+) uptake. J Membr Biol. 2010;236(1):15–26. Epub 2010/07/14. doi: 10.1007/s00232-010-9269-y 20623351
71. Nestorovich EM, Karginov VA, Bezrukov SM. Polymer partitioning and ion selectivity suggest asymmetrical shape for the membrane pore formed by epsilon toxin. Biophys J. 2010;99(3):782–9. Epub 2010/08/05. doi: 10.1016/j.bpj.2010.05.014 20682255
72. Pelish TM, McClain MS. Dominant-negative inhibitors of the Clostridium perfringens epsilon-toxin. J Biol Chem. 2009;284(43):29446–53. doi: 10.1074/jbc.M109.021782 19720828
73. Petit L, Gibert M, Gillet D, Laurent-Winter C, Boquet P, Popoff MR. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J Bacteriol. 1997;179(20):6480–7. 9335299
74. Chassin C, Bens M, de Barry J, Courjaret R, Bossu JL, Cluzeaud F, et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am J Physiol Renal Physiol. 2007;293(3):F927–37. 17567938
75. Bokori-Brown M, Hall CA, Vance C, Fernandes da Costa SP, Savva CG, Naylor CE, et al. Clostridium perfringens epsilon toxin mutant Y30A-Y196A as a recombinant vaccine candidate against enterotoxemia. Vaccine. 2014;32(23):2682–7. doi: 10.1016/j.vaccine.2014.03.079 24709588
76. Bokori-Brown M, Kokkinidou MC, Savva CG, Fernandes da Costa S, Naylor CE, Cole AR, et al. Clostridium perfringens epsilon toxin H149A mutant as a platform for receptor binding studies. Protein Sci. 2013;22(5):650–9. doi: 10.1002/pro.2250 23504825
77. Magyar JP, Ebensperger C, Schaeren-Wiemers N, Suter U. Myelin and lymphocyte protein (MAL/MVP17/VIP17) and plasmolipin are members of an extended gene family. Gene. 1997;189(2):269–75. 9168137
78. Bokori-Brown M, Savva CG, Fernandes da Costa SP, Naylor CE, Basak AK, Titball RW. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011;278(23):4589–601. doi: 10.1111/j.1742-4658.2011.08140.x 21518257
79. Cooper MA. Advances in membrane receptor screening and analysis. J Mol Recognit. 2004;17(4):286–315. Epub 2004/07/01. 15227637
80. Puertollano R, Alonso MA. Targeting of MAL, a putative element of the apical sorting machinery, to glycolipid-enriched membranes requires a pre-golgi sorting event. Biochem Biophys Res Commun. 1999;254(3):689–92. 9920802
81. Marazuela M, Acevedo A, Adrados M, Garcia-Lopez MA, Alonso MA. Expression of MAL, an integral protein component of the machinery for raft-mediated pical transport, in human epithelia. J Histochem Cytochem. 2003;51(5):665–74. 12704214
82. Cheong KH, Zacchetti D, Schneeberger EE, Simons K. VIP17/MAL, a lipid raft-associated protein, is involved in apical transport in MDCK cells. Proc Natl Acad Sci U S A. 1999;96(11):6241–8. 10339572
83. Puertollano R, Martin-Belmonte F, Millan J, de Marco MC, Albar JP, Kremer L, et al. The MAL proteolipid is necessary for normal apical transport and accurate sorting of the influenza virus hemagglutinin in Madin-Darby canine kidney cells. J Cell Biol. 1999;145(1):141–51. 10189374
84. Alonso MA, Millan J. The role of lipid rafts in signalling and membrane trafficking in T lymphocytes. J Cell Sci. 2001;114(Pt 22):3957–65. 11739628
85. Fennessey CM, Sheng J, Rubin DH, McClain MS. Oligomerization of Clostridium perfringens epsilon toxin is dependent upon caveolins 1 and 2. PLoS One. 2012;7(10):e46866. doi: 10.1371/journal.pone.0046866 23056496
86. Batty I, Bullen JJ. The effect of Clostridium welchii type D culture filtrates on the permeability of the mouse intestine. J Pathol Bacteriol. 1956;71(2):311–23. 13398877
87. Chen J, Ma M, Uzal FA, McClane BA. Host cell-induced signaling causes Clostridium perfringens to upregulate production of toxins important for intestinal infections. Gut Microbes. 2014;5(1):96–107. doi: 10.4161/gmic.26419 24061146
88. Fernandez Miyakawa ME, Uzal FA. The early effects of Clostridium perfringens type D epsilon toxin in ligated intestinal loops of goats and sheep. Vet Res Commun. 2003;27(3):231–41. 12777097
89. Garcia JP, Adams V, Beingesser J, Hughes ML, Poon R, Lyras D, et al. Epsilon toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect Immun. 2013;81(7):2405–14. doi: 10.1128/IAI.00238-13 23630957
90. Goldstein J, Morris WE, Loidl CF, Tironi-Farinati C, McClane BA, Uzal FA, et al. Clostridium perfringens epsilon toxin increases the small intestinal permeability in mice and rats. PLoS One. 2009;4(9):e7065. doi: 10.1371/journal.pone.0007065 19763257
91. Layana JE, Fernandez Miyakawa ME, Uzal FA. Evaluation of different fluids for detection of Clostridium perfringens type D epsilon toxin in sheep with experimental enterotoxemia. Anaerobe. 2006;12(4):204–6. 16857397
92. Li J, Sayeed S, Robertson S, Chen J, McClane BA. Sialidases affect the host cell adherence and epsilon toxin-induced cytotoxicity of Clostridium perfringens type D strain CN3718. PLoS Pathog. 2011;7(12):e1002429. doi: 10.1371/journal.ppat.1002429 22174687
93. Martin PK, Naylor RD, Sharpe RT. Detection of Clostridium perfringens beta toxin by enzyme-linked immunosorbent assay. Res Vet Sci. 1988;44(2):270–1. 3387684
94. Miserez R, Frey J, Buogo C, Capaul S, Tontis A, Burnens A, et al. Detection of alpha - and epsilon-toxigenic Clostridium perfringens type D in sheep and goats using a DNA amplification technique (PCR). Lett Appl Microbiol. 1998;26(5):382–6. 9674169
95. Pyakural S, Singh NB. Initial studies on "six months disease" in sheep. Vet Rec. 1976;98(3):49–50. 176766
96. Raibaud P. Experimental models for studying the microbial ecology in the intestinal tract. Acta Gastroenterol Latinoam. 1989;19(4):219–26. 2561802
97. Smedley JG 3rd, Fisher DJ, Sayeed S, Chakrabarti G, McClane BA. The enteric toxins of Clostridium perfringens. Rev Physiol Biochem Pharmacol. 2004;152 : 183–204. 15517462
98. Uzal FA. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. Anaerobe. 2004;10(2):135–43. 16701510
99. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. Epub 2002/02/16. 11846609
100. McWilliam H LW, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res 2013 Jul;41(Web Server issue):W597–600. http://www.ebi.ac.uk/Tools/msa/clustalw2/2013. doi: 10.1093/nar/gkt376 23671338
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance