
Induces the Premature Death of Human Neutrophils through the Action of Its Lipopolysaccharide
The absence of obvious clinical symptoms during the early stages of brucellosis is linked to the Brucella stealthy strategy and its non-canonical PAMPs, which are low PRRs agonists. Still, there are clinical profiles that require explanation. For instance ‒despite the fact that neutrophils readily ingest Brucella during the onset of infection, brucellosis courses without neutrophilia, and just a low number of infected neutrophils are present in target organs. In the chronic phases, a significant proportion of the patients display absolute neutropenia and bone marrow pancytopenia linked to the myeloid cell linage. Examination of the Brucella infected bone marrow reveals granulomas and phagocytosis of myeloid cells. Based on these observations we explored the fate of native neutrophils during their interaction with Brucella. We found that the bacterium induces the premature cell death of neutrophils without inducing proinflammatory phenotypic changes. This event was reproduced by the lipid A of the Brucella LPS and depends on NADPH-oxidase activation and low ROS formation. We believe that this phenomenon explains ‒at least in part ‒ the hematological and histological profiles observed during brucellosis. In addition, it may be that dying Brucella-infected neutrophils serve as “Trojan horse” vehicles for infecting phagocytic cells without promoting activation.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004853
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004853
Summary
The absence of obvious clinical symptoms during the early stages of brucellosis is linked to the Brucella stealthy strategy and its non-canonical PAMPs, which are low PRRs agonists. Still, there are clinical profiles that require explanation. For instance ‒despite the fact that neutrophils readily ingest Brucella during the onset of infection, brucellosis courses without neutrophilia, and just a low number of infected neutrophils are present in target organs. In the chronic phases, a significant proportion of the patients display absolute neutropenia and bone marrow pancytopenia linked to the myeloid cell linage. Examination of the Brucella infected bone marrow reveals granulomas and phagocytosis of myeloid cells. Based on these observations we explored the fate of native neutrophils during their interaction with Brucella. We found that the bacterium induces the premature cell death of neutrophils without inducing proinflammatory phenotypic changes. This event was reproduced by the lipid A of the Brucella LPS and depends on NADPH-oxidase activation and low ROS formation. We believe that this phenomenon explains ‒at least in part ‒ the hematological and histological profiles observed during brucellosis. In addition, it may be that dying Brucella-infected neutrophils serve as “Trojan horse” vehicles for infecting phagocytic cells without promoting activation.
Introduction
Polymorphonuclear leukocytes (PMNs) represent a key cellular component of the host’s antibacterial arsenal. Once in the circulation, the average lifespan of PMNs is close to 5.4 days, period after which they undergo spontaneous apoptosis [1]. This is in frank contrast to the previously reported short lifespans of a few hours for these cells [2]. Then, these dead PMNs are removed by phagocytic cells laying in the reticuloendothelial system, such as monocytes (Mo), macrophages (Mϕ) and dendritic cells (DCs) [3]. This physiological phenomenon does not induce proinflammatory signals and is regarded as a constitutive mechanism to maintain leukocyte homeostasis [4].
Upon bacterial infection, PMNs are activated and migrate into tissues, where they may survive three to five days to perform their phagocytic, microbicidal and proinflammatory functions [1,5]. These events are part of the innate immune response commonly triggered by pathogen-associated molecular patterns (PAMPs) [6] or by danger signals that guide the PMNs response [7].
A variety of microbes have evolved strategies to influence the timing and mode of PMN cell death [8–10]. For instance, Shigella flexneri kills PMNs by necrosis, a process characterized by the release of tissue-injurious granular proteins. This contributes to disruption of the intestinal epithelial barrier, leading to the dysentery observed in shigellosis and allowing the bacterium to enter its colonic host cells [11]. Similarly, Pseudomonas aeruginosa infections may cause lysis or oncosis of PMNs, leading to persistent infections by depleting these cells and contributing to the pulmonary pathophysiology by facilitating bacterial extracellular replication [12,13]. Others, such as the obligate intracellular Anaplasma phagocytophilum and Chlamydia pneumoniae are able to inhibit PMN cell death to achieve intracellular replication within these leukocytes [14,15].
Brucella microorganisms are stealthy alpha-protobacterial intracellular pathogens of mammals, including humans [16,17]. In the early stages of infection, Brucella minimizes the host proinflammatory response, opening an immunological window that allows this bacterium to invade and reach sheltered intracellular niches before adaptive immunity becomes effective [16,18,19]. Once established, Brucella organisms survive and extensively replicate within the intracellular milieu of Mo, Mϕ, DCs and placental trophoblasts [20,21]. As part of its parasitic strategy, Brucella inhibits apoptosis and prolongs the life of these infected mononuclear phagocytic cells [16,22]. Although Brucella is readily internalized by PMNs [23,24], the bacterium survives inside the phagosomes of these cells resisting their killing action including oxidative components and isolated lysosomal extracts [16,25,26].
During the course of human and animal brucellosis, there are several clinical and pathological features related to PMNs which biological mechanisms remain unclear. Among the most striking signs are the neutropenia observed during chronic brucellosis, the absence of recruitment of PMNs at the site of infection and the low numbers of Brucella infected PMNs in the target organs [16,27–30]. Moreover, PMNs have an unexpected influence in dampening the immune response against intracellular Brucella infection and strengthen the notion that PMNs actively participate in regulatory circuits shaping both innate and adaptive immunity [19].
In an attempt to improve our understanding of the mechanisms underlying the fate of PMNs during brucellosis, we have explored the outcome of these leukocytes upon interaction with Brucella abortus. Our findings reveal a novel microbial-host cross-talk through which B. abortus is able to hinder and evade host innate PMN response and suggest a mechanism by which Brucella may hamper the presence of infected PMNs in the target organs and promote neutropenia during chronic brucellosis.
Results
B. abortus resists the killing action of PMNs
Confirming previous reports [16,18,31], Brucella is more resistant than other bacteria to the killing action of PMNs (Fig 1A). This resistance is not related to reduced bacterial internalization, since at multiplicity of infection (MOI) of 5, both B. abortus and Salmonella enterica, were phagocytized at similar rates. Due to the toxic effects mediated by Salmonella on PMNs, higher MOIs of this bacterium were precluded. Compared to latex beads, fluorescent B. abortus-GFP was internalized more efficiently by PMNs at different MOIs, suggesting an active PAMP receptor-mediated phagocytosis (Fig 1B). Early phagocytosis of B. abortus-GFP (MOI < 50) was not accompanied by obvious PMN shape changes such as nuclear rounding, chromatin condensation, cell fragmentation, degranulation (Fig 1C) or myeloperoxidase activity [16]. This observation is in agreement with previous reports [16,25,31,32]. Only when high loads of B. abortus-GFP were tested (MOI > 50) changes in nuclear morphology was detected in a proportion of PMNs containing more than 50 bacteria/cell (Fig 1C).

B. abortus infection induces PMN cell death in a dose-dependent manner
After infection with Brucella-GFP, PMN cell death was assessed by flow cytometry using Annexin V and AquaDead as markers. After two hours of incubation (MOI = 10), Brucella infected PMNs (whole blood or purified PMNs, see below) became positive for both markers, following a bacterial dose dependence (Fig 2). This phenomenon did not require live bacteria, since similar effects were observed in PMNs exposed to equivalent doses of live or heat killed B. abortus (HKBA) (Fig 3).


B. abortus releases Br-LPS inside vacuoles of PMNs
We have demonstrated that B. abortus sheds non-toxic Br-LPS inside cells and that this molecule traffics in vacuoles and influences antigen presentation to T cells [33–35]. Following this, we explored the shedding of Br-LPS inside PMNs by live B. abortus. For this purpose, we used a double labeling fluorescence method [36]. First PMNs were infected with B. abortus-RFP at a MOI of 5. After 1 h of incubation, PMNs were permeabilized and treated with anti-Br-LPS FITC-antibody and counterstained with DAPI. This approach revealed that significant amounts of Br-LPS molecules (green fluorescence) were released intracellularly by live (red fluorescent) Brucella in the proximity of bacteria-containing PMN phagosomes (Fig 4). Almost all B. abortus-RFP infected PMNs exhibited this pattern after 1 h infection, most strikingly evident by immunofluorescence in cells containing between 2–3 bacteria/PMN.

In order to determine if Br-LPS was released inside vacuoles or translocated to the cytosol, B. abortus infected PMNs were subjected to immunodetection of Br-LPS by electron microscopy. Regular osmium tetroxide staining of infected PMNs (1 hour) demonstrated that all phagocytized B. abortus reside inside phagosomes, and just a few of them within phagolysosomes, confirming previous results [37]. As expected, sensitive staining for detection of immunogold particles revealed the presence of Br-LPS in the bacterial cells. However, vesicles in the proximity of the ingested Brucella also contained gold particles, indicating the presence of Br-LPS within vacuoles (Fig 5). In some cells, immunogold stained Br-LPS molecules were detected within a phagosome containing partially digested bacteria or in the proximity of cell membrane ruffle-like structures (Fig 5E). Gold particles were practically absent in the cytosol and not detected in the extracellular milieu (Fig 5A).

Br-LPS specifically induces the cell death of PMNs
Intracellular Br-LPS influences the antigen presentation of Mϕ without affecting the survival of these cells [34,35]. Therefore, we assessed the effects of Br-LPS on PMNs cell survival. As demonstrated in Fig 6A, Br-LPS induced PMN cell death in a dose-dependent manner in blood or in purified (see the results presented in the next sections) PMNs. This effect was specific for PMNs since other cells, such as lymphocytes, treated and gated under the same conditions, did not display death cell markers (Fig 6B). Consistent with previous observations [22,38], Br-LPS did not induce cell death of Mϕ, Mo and DCs. In contrast, Escherichia coli LPS (Ec-LPS) did not induce cell death in blood (Fig 6A) or in purified PMNs under the same experimental conditions.

In view of the relatively high amounts of Br-LPS added to induce PMN cell death, a quantitative determination of the Br-LPS interacting with these cells was performed. For this purpose, purified PMNs were incubated with Br-LPS and the associated amounts determined by Western blotting (Fig 7). In order to have a saturating positive control, the assay was also performed in the presence of human antibodies against Br-LPS. The estimated quantities of associated Br-LPS in the absence of antibodies ranged between 5–25 ng/106 PMNs. Likewise, the amounts of associated Br-LPS in the presence of antibodies were between 10–50 ng/106 PMNs. This result indicates that the actual quantities of Br-LPS interacting with PMNs under these experimental conditions corresponded just 0.05–0.25% of the total Br-LPS added. As expected, antibodies increased close to 10 times the quantities of Br-LPS associated to PMNs through the concourse of Fc receptors. It should be noticed that the molecules associated to PMNs corresponded to the lower molecular weight (~30–40 MW) fraction of Br-LPS. This indicates that among all the Br-LPS molecules available, just specific classes are selected by PMNs.

The lipid A moiety of the Br-LPS is responsible for the induction of PMN cell death
The non-toxic Br-LPS is built of an O-chain constructed of N-formyl perosamine sugar homopolymer, a positively charged core oligosaccharide and a lipid A containing a diaminoglucose disaccharide backbone substituted with long chain hydroxylated, cyclic and non-hydroxylated fatty acids (S1 Fig). In an attempt to identify the moiety responsible for inducing the PMN cell death, we first tested the biological action of different LPSs that shared at least some of the Br-LPS structural features [18,39–45]: i) Yersinia enterocolitica O:9 LPS displays the same O-chain homopolymer as Br-LPS but has different lipid A and core oligosaccharide; ii) Ochrobactrum anthropi LPS shares the lipid A structural features with Br-LPS but possesses different O chain and core oligosaccharide; iii) B. abortus ∆WadC LPS displays the same lipid A and O chain as the Br-LPS, but has a different core oligosaccharide; finally, iv) the overall structure of Ec-LPS differs from that of Br-LPS, but it shares the lipid A and core features with Y. enterocolitica O:9 LPS. As shown in Fig 8A, LPSs from B. abortus ∆WadC and O. anthropi sharing the same lipid A structure as the Br-LPS were able to induce cell death more readily than other LPSs. A similar pattern of PMN cell death was observed when these cells were treated with increasing quantities of purified B. abortus 2308 lipid A (Fig 8B). Altogether, these results demonstrate that the lipid A of Br-LPS is the moiety responsible for inducing the premature cell death of human PMNs.

Blocking of CD14 molecule prevents the Br-LPS-induced PMN cell death
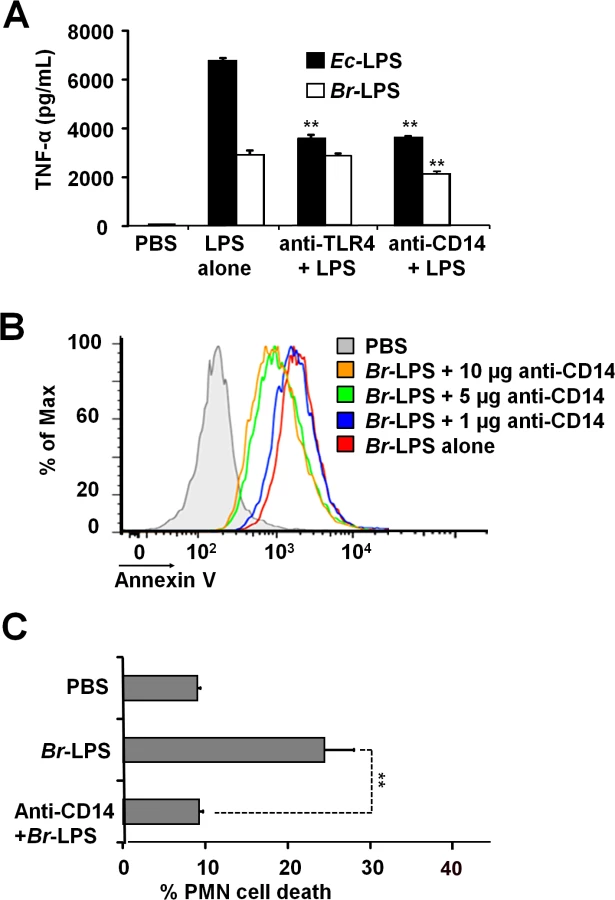
It is well known that the coordinated interaction of CD14, MD-2/TLR4 molecules mediate LPS recognition in mammalian cells [46] and that binding of these membrane molecules may promote cell survival or cell death depending on the context [47,48]. Therefore, we explored the roles of TLR4 and CD14 Br-LPS-induced the cell death in PMNs.
When TLR4 or CD14 receptors were blocked with specific antibodies prior to the exposure of blood with Ec-LPS, the secretion of TNF-α was significantly abrogated (Fig 9A), indicating that the amounts of antibodies used were suitable. Despite of the lower amounts of TNF-α induced by Br-LPS as compared to those stimulated by Ec-LPS, the blocking of TLR4 does not have any effect on the action of the former bacterial molecule on blood cells. This phenomenon is consistent with previous findings demonstrating that Br-LPS is a poor agonist of the MD-2/TLR4 pathway [16,43]. Likewise, when TLR4 was blocked, PMN cell death mediated by Br-LPS was not inhibited (S2 Fig). In contrast, anti-CD14 antibodies significantly inhibited the secretion of TNF-α (Fig 9A) in blood as well as PMN cell death induced by Br-LPS (Fig 9B). Since anti-CD14 treatment of blood could modulate other leukocytes and influence the dead of PMNs, we then performed the experiment using purified PMNs (Fig 9C) to confirm the blocking effect of anti-CD14. In preparations of purified human PMNs, blocking of CD14 totally abolished the Br-LPS induction of cell death after a short incubation (Fig 9C). Anti-CD14 alone or low amounts of this antibody (≤1μg) did not have any observable effect in PMN cell death.
Br-LPS-induced PMN cell death correlates with low ROS formation
The low and slow kinetics of ROS formation induced by Br-LPS correlates with the kinetics of the PMN cell death observed (Fig 10A). Although it is dose dependent, this profile is in clear contrast and opposite to the kinetics of ROS formation and cell death measured in Ec-LPS treated PMNs (Fig 10B). Brucella-infected PMNs did not undergo NETosis or display typical signs of apoptosis or necrosis (Fig 1C). Likewise, the doses of Br-LPS that promoted PMN cell death failed to induce NETosis (S4 Fig) or degranulation, as demonstrated before [49]. Therefore, this phenomenon seems to be specifically mediated by Br-LPS and its lipid A. This also agrees with previous data demonstrating that these bacterial molecules barely induce degranulation or activation of PMNs [49].

Br-LPS triggers PMN cell death through the action of NADPH-oxidase and ROS mediators
Many of the cell death features displayed by PMNs are unique for these leukocytes [50–52]. Since microscopically the Brucella-induced PMN cell death does not fit with any of the classical cell death types described for these phagocytes, then we investigated the action of several chemical inhibitors (Fig 11). Among the most conspicuous were the NADPH-oxidase inhibitor, acetovanillone (apocynin) [53] and the superoxide and hydrogen peroxide scavengers, tiron and catalase, respectively [54,55]. These chemicals almost completely abrogated the Br-LPS-induced PMN cell death.

Since inhibition of the check point kinase 1 (Chk1) significantly prevented the cell death of Br-LPS treated PMNs, then we explored the induction of DNA damage. One hour after B. abortus infection, the fragmentation of PMN DNA was already evident (S5A Fig). The DNA damage induced by B. abortus infection or by Br-LPS treatment was reversed by pan-caspase inhibition (S5B Fig), suggesting the participation of caspase-activated DNase (CAD) [56].
Blocking of caspase 5 and to minor extent of caspase 4, prevented cell death; however, specific inhibition of caspases 1 had very little effect. Although related to caspase 1, caspases 5 and 4 have different substrates than caspase 1, and the activation of the former caspases induce cell death independently from the later [57]; therefore not necessarily linked in function. This result, suggests the absence of inflammosome recruitment in the Brucella induced PMNs cell death. The modest action of BAPTA/AM and Necrostatin-5 indicates partial involvement of Ca++ and the RIP1 kinase/FADD cell death routes [58].
Caspase-8 and caspase-9 are important mediators of cell death through the extrinsic and intrinsic pathways [59,60]. As shown in Fig 12, both caspases became activated after treatment of PMNs with Br-LPS. In spite of this, specific inhibitors for these caspases had little effect in preventing the death of PMNs (Fig 11). This effect was specific for PMNs since caspase triggering was not observed in other blood cells, such as lymphocytes (S3 Fig). This suggests the downstream recruitment of caspases 8 and 9, after the initial cell death triggering mechanisms. Other inhibitors, such as those used for preventing necrosis, apoptosome formation or the activity of Ca++ dependent-ATPase or MAP-, tyrosine - or PI3-kinases did not have any effect in blocking the action of Br-LPS (Fig 11).

Brucella and Br-LPS induce low levels of proinflammatory cytokines in PMNs
Pro-inflammatory TNF-α, IL-1β and IL-6 cytokines and IL-8 chemokine, may influence the life of PMNs, either prolonging or inducing the death of these phagocytic leukocytes [61–64]. Therefore, we assessed the release of these cytokines and chemokines in whole blood cell preparations or purified PMNs after Brucella or Br-LPS treatment. S. enterica was included as a control. As shown in Fig 13, there were significant quantitative differences in cytokine production between blood and purified PMNs. Salmonella stimulates the release of cytokines by PMNs (Fig 13), induces degranulation and does not prematurely promote the cell death of these cells [65] Regardless whether blood or purified PMNs were tested, the levels of TNF-α, IL-1β and IL-6 were comparatively low after Brucella infection or Br-LPS treatment. This result is consistent with the low cytokine production by Brucella infected or Br-LPS treated macrophages [66–69], or by Brucella infected mice at early time points of infection [16].

The concentrations of IL-8 induced by Brucella or Br-LPS in purified PMNs were significantly higher than the levels of the other cytokines (Fig 13). This is striking since it has been established that the chemoattractant IL-8, rather than inducing cell death, promotes PMNs survival [61,70]. Transcription of IL-8 is constitutive in PMNs, making the synthesis of this chemokine readily available after simulation [71]. Still, the cell death remains evident in both PMN preparations, being more conspicuous in blood that in purified PMNs.
Discussion
The consensus in Gram negative bacterial infections is that the endotoxic LPS molecule and other PAMPs, engage PMNs into activation and prolongation of their life span [63]. This phenomenon is linked to the activation of other cells such as Mϕ, Mo and DCs. In purified PMNs, stimulation of TLR2 and TLR4 with agonists modestly inhibits apoptosis, while in the presence of Mϕ, Mo and DCs, the inhibition of PMN cell death is very potent [47,72]. PMNs are able to use this time delay to recruit other cells and to promote proinflammatory events to eliminate the invading bacteria [73] through actions that involve the respiratory burst [5]. It has been shown that high levels of ROS inhibit caspases activities, suggesting that reactive oxygen species may prevent these proteases from functioning optimally in PMNs [74]. During these processes, some PMNs degranulate, others undergo NETosis, while others may die by necrosis or oncosis, triggering proinflammatory signals [51,52].
In contrast, upon invasion Brucella resists the killing action of PMNs and prematurely induces the cell death of these phagocytes. The Brucella-induced PMN cell death occurs without bacterial replication [23,24] and without promoting those classical phenotypic changes associated with NETosis, degranulation, necrosis, oncosis or classical apoptosis. The cell death of Brucella-infected PMNs seems to be triggered after active phagocytosis of the microorganism followed by the intracellular release of the Br-LPS inside cell vacuoles, either by alive or death bacteria. Although the process by which B. abortus sheds Br-LPS inside the cells has not been elucidated, it is likely that it occurs through blebbing of outer membrane fragments enriched in Br-LPS, a phenomenon that is well known in Brucella [75]. This is significant, since Br-LPS is capable to circulate in the body and reside inside phagocytes for months without being destroyed [33], and consequently, capable to exert its biological action on PMNs in vivo.
There are reports claiming that B. abortus and Brucella lipoproteins activate PMNs [76]. However, in those experiments the ex vivo PMNs viability was less than expected and the assays were performed with heat killed bacteria and lipoproteins acylated in the E. coli background; thus preventing comparison with our results, as explained before [17]. The interaction between the lipid A of Br-LPS and PMNs mostly precludes TLR4, as well as other TLRs, as demonstrated before for Mϕ [18,43,67,77,78]. However, it does not exclude the CD14 molecule, since antibodies against the later co-receptor abrogates the PMN cell death and to less extent the release of TNF-α, suggesting some signaling through this co-receptor. The interaction of Br-LPS with intracellular CD14 molecules is feasible, since this lipoprotein is also found inside PMN vesicles [79] and in concordance with the transport of Br-LPS to CD14 containing lipid rafts in Mϕ membranes [80]. Moreover, in agreement with our results, it has been shown that in Mϕ, Brucella signals through CD14 for the production of low amounts of TNF-α [81]. Finally, the involvement of CD14 in the induction of cell death is not without precedent and it has been demonstrated that direct binding of LPS to CD14 ‒without the concourse of TLR4 ‒ prompts apoptosis in DCs [48].
We have demonstrated in murine Mϕ that Br-LPS follows the classical endocytic pathway used by protein antigens but with a slower kinetics [80]. Then, Br-LPS is transported to cellular compartments enriched in MHC-II and recycled to the cell surface, where it forms dense macrodomains. Once in the cell membrane, the Br-LPS macrodomains segregate several lipid-raft components and interfere with the MHC-II presentation of peptides to specific CD4+ T cells [80]. The initial release of Br-LPS inside PMN phagosomes and its subsequently transit within vacuoles seems to occur by a similar mechanism proposed for Mϕ [82]. Likewise, in some infected PMNs, Br-LPS was also observed in cell membrane ruffles-like structures. However, the biogenesis and life span between infected human PMNs and murine Mϕ is rather different: while in the former leukocytes Brucella induces premature cell death, in the later it prologues the life span and protects against apoptosis [16,22]. Moreover, the amounts of Br-LPS internalized by Mϕ are comparatively much higher than those ingested by PMNs [33]. This difference may be linked to the numbers of CD14 surface molecules present in Mo and Mϕ, which are from 30–40 times more abundant than in PMNs [83]. However, the amounts of intracellular Br-LPS available in PMNs at early times of cell infection may be considerably larger than in Mϕ; since the former leukocytes ingest larger number of Brucella organisms than the latter, which internalize just a few bacteria [84]. These and other differences make quite difficult to perform a detail experimentation of the intracellular trafficking of Br-LPS inside PMNs, and alternative methodological approaches would be required. For the moment, this is beyond of our possibilities.
The dose-dependent Br-LPS-induced PMN cell death correlates with a modest but steadily increase of ROS mediated by NADPH oxidase. This seems to be the main triggering mechanisms by which the lipid A of Br-LPS induces the premature cell death of human PMNs. It is worth noting that several of the molecular pathways causing PMN cell death are dependent on ROS generation. While large amounts of ROS may inhibit caspases, promote necrosis or cause NETosis [74,85,86], low amounts may induce PMN cell death [52].
DNA damage by oxygen radicals is a well-known phenomenon in a variety of cells, including PMNs [52]; and even small amounts of ROS may induce DNA alterations. The recorded DNA fragmentation of B. abortus and Br-LPS treated PMNs recruited Chk1, a protein that coordinates the DNA damage response at the initiation of cell cycle [87]. Although in other cells inhibition of Chk1 induces apoptosis [87], it is likely that in non-dividing cells ‒such as PMNs ‒ this protein has a terminal role and its function is not to arrest the cell cycle, but to promote cell death. The PMN DNA fragmentation was partially reversed by pan-caspase inhibitors; event that suggests the participation of CAD [56].
At first glance, the profile of inhibitory substances suggests that caspase-8 could be extrinsically activated through the RIP1 kinase/FADD route [88]. In addition, the mobilization of Ca++ may activate several death signals, including the calcium-activated cysteine protease calpain that cleaves and thereby activates a number of molecules that have important functions in the apoptosis processes [89].
As already recorded in human monocytes [68] the amounts of IL-1β induced by B. abortus and its Br-LPS in PMNs are rather low. In addition, inhibition of caspase 1 did not block the Br-LPS mediated PMN cell death. These two observations, together with the low cytokine induction by Brucella and its Br-LPS in PMNs, seem to preclude the role of the inflammasome pathway in the premature death of these leukocytes. Though this seems relevant, it has been reported that caspase-1 induced pyroptotic cell death does not function in PMNs [90]. Moreover, upon inflammasome activation the amounts of IL-1β produced by purified PMNs are rather low [91]. This may be linked to the fact that human neutrophils express key components of the inflammasome machinery at non-canonical intracellular sites [91]. A general proposal of the mechanisms for the induction of the premature PMN cell death generated during B. abortus infection is presented in S6 Fig.
It is well known that under certain circumstances, proinflammatory cytokines produced by leukocytes during Gram negative endotoxemia are capable of inducing programmed cell death [92,93]. Among these, the TNF-α is the most conspicuous cytokine generating apoptosis through binding to its cognate TNFR1 [94,95]. However, it is unlikely that TNF-α is the signal that promotes the Br-LPS-induced PMN cell death. First, the amounts of proinflammatory cytokines ‒including TNF-α ‒ produced upon exposure of PMNs to Brucella or Br-LPS were very low (Fig 13). Second, it is well known that Brucella and Br-LPS are low agonist of pattern recognition receptors and low activators of NF-κB [16,43,67]. Third, under similar experimental conditions the Ec-LPS ‒which induces the production of much higher quantities of TNF-α ‒ does not promote premature PMN cell death (Figs 6A and 9). Finally, under the same experimental conditions Br-LPS did not induce the death of lymphocytes (Fig 6B) which are also susceptible to the pro-apoptotic effect of TNF-α [95].
It is worth noting that the amounts of IL-8 induced by B. abortus in purified PMNs were higher than other cytokines. It has been shown that this chemokine, rather that promoting cell death, delays spontaneous and TNF-α-induced apoptosis of human PMNs in a dose dependent manner [70]. The delay in apoptosis is mainly mediated through the interaction of IL-8 with its cognate RII receptor, while the RI receptor may provide an added effect. Still, PMNs died after Brucella infection or Br-LPS treatment, precluding the influence of IL-8 in a delimited population of PMNs. The different levels of cytokines detected in whole blood versus purified PMNs exposed to Brucella or Br-LPS may reflect the participation of Mo, DCs and serum components (e.g. complement) present in blood, which may have served as an additional stimuli and sources of cytokines, including IL-8.
For many years it has been recognized that a proportion of patients with chronic brucellosis display absolute neutropenia [27,28]. It has been also shown that the invasion of Brucella organisms induces significant hematological chages in the bone marrow, involving pancytopenia and phagocytosis of blood elements (including PMNs) by resident Mϕ [96–98]. In addition, during the accute phase of brucellosis there is a conspicuous absence of infected PMNs in the target organs, a phenomenon that is in clear contrast to the presence of Brucella inside Mϕ and DCs [29]. The fact that Brucella and its Br-LPS specifically induce the premature cell death of PMNs may explain, at least in part, these clinical signs.
Dying PMNs display “eat-me” signals. Therefore, they are readily removed by phagocytic cells. Then, it is likely that Brucella infected PMNs may serve as “Trojan horse” vehicles for dispersing the bacterium to other organs; hence, contributing to the long lasting infections observed in brucellosis [99]. The Brucella-induced cell death ‒without significant activation of PMNs and their non-phlogistic removal by Mϕ and DCs ‒ would help to hamper the promotion of proinflammatory signals (S7 Fig). This mechanism may represent a seminal component of the stealthy strategy used by Brucella organisms [16] to spread in its host while avoiding innate immunity.
Materials and Methods
Ethics
Human fresh blood was obtained in the blood bank of the Charité Hospital, Berlin, following a protocol approved by the Charité Hospital, Berlin Ethical Committee. Fresh blood was also obtained from normal healthy volunteer donors through the “Etablissement Français du Sang” following their approval and in agreement with the “French Ethics Committee on Human Experimentation F11”, within a convention EFS-08-21-2012 with Institut National de la Santé et de la Recherche Médicale, signed. All blood donors involved were informed about the study and provided written consents.
Bacterial strains, LPSs and lipid A preparations
Virulent B. abortus (2308), B. abortus-GFP (2308) [100], transgenic B. abortus-RFP (2308) with an integrated chromosomal gene coding for the red fluorescent protein from Discosoma coral (provided by Dr. Jean-Jacques Letesson; Unité de Recherche en Biologie Moléculaire, Facultés Universitaires Notre-Dame de la Paix, Namur, Belgium) and Salmonella enterica sv. Typhimurium (SL1344) were grown in tryptic Soy or Luria Bertani broths as previously described [16]. Bacterial cells were washed three times by centrifugation in Hanks or PBS solution before the assays. Purified LPSs were prepared from B. abortus (2308), B. abortus wadC (2308), E. coli (0127), Y. enterocolitica O:9 (MY79), O. anthropi (LMG 3331T) as reported before [43,77]. B. abortus lipid A was prepared by mild acid hydrolysis from Br-LPS and solubilized as described elsewhere [101]. All the Brucella Br-LPS and lipid A preparations were above 98% pure and devoid of contaminant proteins, free lipids and cyclic glucans.
Neutrophil purification
PMNs were purified by Histopaque and Percoll gradients from blood of healthy donors as previously described [16,50]. Cell preparations were composed from 95–98% of granulocytes. Cell viability was >90%. PMN preparations were maintained at 4°C in PBS or autologous plasma, and used within the first hour after extraction. Under our conditions, PMN spontaneous apoptosis was just evident after 5–7 hours after purification.
Bactericidal activity
Bactericidal activity was measured as previously described [16]. Briefly, B. abortus or S. enterica were mixed with 500 μL of purified human PMNs (1x106 PMNs/mL) at a MOI of 5 bacteria/PMN and incubated under mild agitation for 90 minutes. Control bacteria were incubated in the absence of PMNs to quantify bacterial replication during the experiment. Viable CFU were determined at 0, 45 and 90 minutes of incubation by lysing cells with 0.1% triton and plating samples in tripticase soy agar. The percentage of bacterial survival was calculated.
Phagocytosis assay
Human heparinized blood or purified PMNs were incubated with B. abortus-GFP or fluorescent latex beads for two hours at 37°C a multiplicity of infection (MOI) of 10–100 bacteria or beads/cell, under mild agitation. Smears were fixed with methanol and mounted with ProLong Gold Antifade Reagent with DAPI. One hundred PMNs were counted per sample and the number of particles determined to calculate the percentage of phagocytosis.
PMN cell death assays
PMN cell death was analyzed by treating human whole blood or isolated PMNs with different bacterial strains and LPSs. Human heparinized blood (100 or 500 μL) collected with lithium heparin was incubated for 2 hours at 37°C in agitation (200–300 rpm) with each treatment. Bacteria were tested at MOIs of 1, 10 or 100 bacteria/PMN. In the case of LPS or lipid A, blood samples were treated at concentrations from 3x10-3 to 3x101 pmol/mL. After incubation, blood samples were lysed for 5–10 min in 900 μL of red blood cell lysis buffer (NH4Cl 8.02 gm, NaHCO3 0.84gm and EDTA 0.37gm/L, pH 7.2). Cells were washed with ice cold PBS and re-suspended in 100 μL of Annexin V Binding Buffer (BD). 5 μL of Annexin V (BD) and 2 μL of AquaDead (Invitrogen) (diluted 1/20 in PBS) were added and incubated for 30 min on ice in the dark. Cells were washed once with ice cold PBS, re-suspended in 200 μL of paraformaldehyde 3% and incubated for 30 min at room temperature. Samples were then diluted 1 : 2 with PBS and acquired for analysis within 1 hour.
Intracellular detection of Br-LPS
For intracellular detection of Br-LPS a double labeling fluorescence approach was performed [36]. Human heparinized blood was incubated with B. abortus-RFP (red) for one hour (MOI 2) under mild agitation. Blood smears were fixed and permeabilized with methanol, stained with anti-Brucella LPS FITC (green) and mounted with ProLong Gold Antifade Reagent with DAPI (blue). Samples were observed by fluorescent microscopy (Olympus BH-2) under 1000 × magnification. Br-LPS shed by Brucella is shown in green staining around red bacteria.
Intracellular detection of Br-LPS was also performed in B. abortus infected PMNs by immunogold detection under the electron microscope. Briefly, purified human PMNs 5 ×106 were infected with B. abortus 2308 at MOI 20. After 1 hour incubation at 37°C under mild agitation, cells were washed and the pellet fixed with 200 μl of 2.5% glutaraldehyde in phosphate buffer 0.05M pH 7.4 (PB) at 4°C for 1 hour. Cells were pelleted at 3000 rpm for 10 min, washed in PB and suspended in 50 μL of PB. Then fixed cells were incubated at 40°C for 5 minutes, and 100 μL of 3% low melting agarose at 40°C added. The temperature was lowered, and 5 volumes of 2.5% glutaraldehyde in PB were added to the solid agarose block and incubated overnight at 4°C. Agarose blocks containing the fixed infected PMNs were processed for inclusion in Spurr resin for immunogold staining and for electron microscopy as described elsewhere [102]. For detection of Br-LPS, human IgG or mouse IgG with specificity against the O chain polysaccharide [103] were used in combination with protein-A/protein-G colloidal gold 15 nm (EY Laboratories, Inc.). Purified mouse and human IgGs from normal serum were used for controlling the specificity of the reaction. Finally, PMNs sections were stained following the led citrate procedure described by Reynolds [104] [101] and observed under a Hitachi H 7100 electron microscope.
Quantitation of Br-LPS interacting with PMNs
In order to determine the amount of Br-LPS interacting with human PMNs, 10 μg (0.3 pmol) of Br-LPS were incubated with 1x106 PMNs in 500 μL of HBSS at 37°C for 1 h under mild rotation in the presence or absence of human IgG anti-Br-LPS. PMNs were washed three times with HBSS to remove the excess of Br-LPS, and then the cell pellet lysed with deionized water containing 50 μg/mL of DNAase and 125 μg/mL of proteinase K (Fisher Scientific) at 37°C for 1 h under mild rotation. Cell lysate was incubated with SDS-PAGE sample buffer and subjected to Western blotting. Br-LPSs bands were revealed with a monoclonal antibody against the O chain polysaccharide of the Br-LPS conjugated with peroxidase [105]. Controls included the assay performed with Br-LPS in the absence or presence of human antibodies but in the absence of PMNs, and PMNs alone. Quantitation of Br-LPS bound to PMNs was estimated in relation to a standard curve of purified Br-LPS ranging from 0.1 ng to 12 ng. The read-out of the bands was performed by densitometry with the support of ImageJ software (http://imagej.net/).
ROS detection
Isolated PMNs (1x105) were re-suspended in 50 μL of Hanks Balanced Salt Solution (HBSS+1% FBS) per well of a 96-uncoated serum well plate. Cell suspension was supplemented with Reactive Oxygen Species (ROS) Detection Reagents (Invitrogen) and stimulated with phorbol myristate acetate (40 nM), Br-LPS (0.03–3 pmol/mL), Ec-LPS (0.09–7.5 pmol/mL) in 50 μl HBSS+1% FBS or left untreated. The kinetics of ROS production was monitored with a Victor Perkin Elmer luminometer at 37C for 90 min.
NET formation and cell cytotoxicity assay
Isolated PMNs (1x105) were re-suspended in 500 μL of RPMI medium (10 mM HEPES +1% FBS without glutamine) and let sit on 24-well plates for 30 min at 37°C. Cells were stimulated with phorbol myristate acetate (40 nM) (Sigma), Br-LPS (0.7–100 μg/ml), Ec-LPS (0.7–100 μg/ml) or left untreated in RPMI medium. After 6h 30 minutes, cell cytotoxicity was measured by Sytox (0.3 μM) (Invitrogen) staining with a fluorometer. Some cells were fixed in paraformaldehyde 8% and observed with a Leica inverted fluorescence microscope to evaluate the nuclear morphologies and NET spreading.
Cytokine quantitation
The levels of TNF-α, IL-8, IL-1β and IL-6 were measured by ELISA (eBioscience) in heparinized human blood (plasma) or supernatant of isolated PMNs treated with different stimuli according to manufacturer’s specifications.
Determination of caspase 8 and 9 activation
Heparinized human blood (500 μL) was incubated with B. abortus LPS (10 μg/mL) or PBS for 30 minutes under mild agitation and stained directly and incubated with anti-active caspase 8 or anti-active caspase 9 using Guava Caspase 8 FAM & Caspase 9 SR Kit (Millipore) according to manufacturer’s specifications and quantitated by flow cytometry. PMNs population was gated by forward light scatter and side light scatter parameters and analyzed by each caspase marker.
DNA fragmentation assays
PMNs were isolated as previously described [106] and incubated for one hour with B. abortus (MOI 100) or 10 μg/mL (0.3 pmol) of Br-LPS in the presence or absence of a pan-caspase inhibitor (Z-VAD-FMK). Cycloheximide (Sigma-Aldrich) was used as a positive control for DNA fragmentation. After incubation, PMN cell death was measured by using a DNA fragmentation ELISA (Roche) according to manufacturer’s specifications. For microscopic analysis, heparinized blood was incubated with B. abortus-RFP for 2 hours (MOI 100). Red blood cells were lysed and total leucocytes prepared, fixed and stained with APO-BrdU TUNEL Assay Kit (Invitrogen) according to manufacturer’s specifications. Cells were centrifuged on a microscope slide by using a Cytospin 2 (Shandon) and mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen).
TLR4 and CD14 neutralization
TLR4 and CD14 cell receptors were neutralized (before Br-LPS or Ec-LPS treatments) by incubating isolated PMNs or heparinized human blood with 1 μg of anti-hTLR4-IgG (clone W7C11) or 5 μg of anti-CD14-IgA (clone D3B8) antibodies (InvivoGen), for 20 minutes and one hour respectively. No inhibitory or stimulatory signals were observed with mouse monoclonal IgG1 (anti-bovine IgG, Sigma-Aldrich), or enriched mouse and human immunoglobulin preparations. Receptor blockage was verified in side controls by measuring TNF-α secretion after treating blood with 5 μg/mL Ec-LPS (Fig 9A).
PMN cell death inhibition assays
Heparinized human blood samples (350 μL) were pre-incubated with one of the following compounds for 1 hour: IM-54 (Enzo Life Sciences), Wortmanin, Genistein, Tyrphostin, PD098059, Necrostatin-5, Z-VAD-FMK, AZD7762, Catalase, Tiron, Acetovanillone (Sigma), BAPTA/AM, YVAD-CHO, NS3694, Thapsigargin (Calbiochem), Z-IETD-FMK, Z-WEHD-FMK, Z-LEVD-FMK, Z-LEHD-FMK (BioVision), Z-YVAD-FMK (Santa Cruz). Concentrations were utilized according to previous reports and standardized to our conditions for optimal inhibitory performance. After treatment with the inhibitory compounds, samples were incubated with Br-LPS (1.3 pmol/mL) for 2 hours. Samples were further processed and analyzed by cytometry for cell death with Annexin V as described above.
Flow cytometry and FACS analysis
PMN or lymphocyte populations were gated as indicated (S8 Fig) and analyzed for cell death of caspase activation by flow cytometry. FACS analysis was performed using a FACSCanto system (BD Biosciences) or Guava easyCyte (Millipore). FACS data were analyzed using FlowJo software (Tree Star, Inc.). For each experiment, control samples were included to define the proper gates.
Statistical analysis
Values were expressed as means ± standard error, and compared using Student’s t test for determining the statistical significance in the different assays. Values of p< 0.05 were considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010;116 : 625–7. doi: 10.1182/blood-2010-01-259028 20410504
2. Payne CM, Glasser L, Tischler ME, Wyckoff D, Cromey D, Fiederlein R, et al. Programmed cell death of the normal human neutrophil: an in vitro model of senescence. Microsc Res Tech. 1994;28 : 327–44. 7919520
3. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22 : 285–94. 15780986
4. Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83 : 865–75. 2921324
5. Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219 : 88–102. 17850484
6. Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1 : 1–13.
7. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330 : 362–6. doi: 10.1126/science.1195491 20947763
8. Anwar S, Whyte MKB. Neutrophil apoptosis in infectious disease. Exp Lung Res. 2007;33 : 519–28. 18075826
9. DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9 : 399–413. 15192322
10. Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189 : 1059–70. doi: 10.1083/jcb.201004096 20584912
11. François M, Le Cabec V, Dupont MA, Sansonetti PJ, Maridonneau-Parini I. Induction of necrosis in human neutrophils by Shigella flexneri requires type III secretion, IpaB and IpaC invasins, and actin polymerization. Infect Immun. 2000;68 : 1289–96. 10678940
12. Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, et al. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol. 2002;168 : 1861–8. 11823520
13. Dacheux D, Toussaint B, Richard M, Brochier G, Croize J, Attree I. Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect Immun. 2000;68 : 2916–24. 10768989
14. Scaife H, Woldehiwet Z, Hart CA, Edwards SW. Anaplasma phagocytophilum reduces neutrophil apoptosis in vivo. Infect Immun. 2003;71 : 1995–2001. 12654818
15. Van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, et al. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol. 2004;172 : 1768–1776. 14734760
16. Barquero-Calvo E, Chaves-Olarte E, Weiss DS, Guzmán-Verri C, Chacón-Díaz C, Rucavado A, et al. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One. 2007;2: e631. 17637846
17. Martirosyan A, Moreno E, Gorvel J-P. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol Rev. 2011;240 : 211–34. doi: 10.1111/j.1600-065X.2010.00982.x 21349096
18. Barquero-Calvo E, Conde-Alvarez R, Chacón-Díaz C, Quesada-Lobo L, Martirosyan A, Guzmán-Verri C, et al. The differential interaction of Brucella and Ochrobactrum with innate immunity reveals traits related to the evolution of stealthy pathogens. PLoS One. 2009;4: e5893. doi: 10.1371/journal.pone.0005893 19529776
19. Barquero-Calvo E, Martirosyan A, Ordoñez-Rueda D, Arce-Gorvel V, Alfaro-Alarcón A, Lepidi H, et al. Neutrophils exert a suppressive effect on Th1 responses to intracellular pathogen Brucella abortus. PLoS Pathog. 2013;9: e1003167. doi: 10.1371/journal.ppat.1003167 23458832
20. Gorvel JP, Moreno E. Brucella intracellular life: from invasion to intracellular replication. Vet Microbiol. 2002;90 : 281–97. 12414149
21. Roop RM, Gaines JM, Anderson ES, Caswell CC, Martin DW. Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Med Microbiol Immunol. 2009;198 : 221–38. doi: 10.1007/s00430-009-0123-8 19830453
22. Gross A, Terraza A, Ouahrani-Bettache S, Liautard JP, Dornand J. In vitro Brucella suis infection prevents the programmed cell death of human monocytic cells. Infect Immun. 2000;68 : 342–51. 10603407
23. Braude AI. Studies in the pathology and pathogenesis of experimental brucellosis. II. The formation of the hepatic granuloma and its evolution. J Infect Dis. 1951;89 : 87–94. 14861465
24. Ackermann MR, Cheville NF, Deyoe BL. Bovine ileal dome lymphoepithelial cells: endocytosis and transport of Brucella abortus strain 19. Vet Pathol. 1988;25 : 28–35. 3125659
25. Kreutzer DL, Dreyfus L a, Robertson DC. Interaction of polymorphonuclear leukocytes with smooth and rough strains of Brucella abortus. Infect Immun. 1979;23 : 737–42. 110680
26. Martínez de Tejada G, Pizarro-Cerdá J, Moreno E, Moriyón I. The outer membranes of Brucella spp. are resistant to bactericidal cationic peptides. Infect Immun. 1995;63 : 3054–61. 7622230
27. Crosby E, Llosa L, Miro Quesada M, Carrillo C, Gotuzzo E. Hematologic changes in brucellosis. J Infect Dis. 1984;150 : 419–24. 6481187
28. Ruiz-Castañeda M. Brucelosis. Tercera ed. Ediciones científcas, editor. Mexico, D.F.: La Prensa Médica Mexicana, S.A.; 1986.
29. Copin R, Vitry M-A, Hanot Mambres D, Machelart A, De Trez C, Vanderwinden J-M, et al. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 2012;8: e1002575. doi: 10.1371/journal.ppat.1002575 22479178
30. Prouty CC. Studies on the leucocyte content of milk drawn from Brucella abortus infected udders. J Bacteriol. 1934;27 : 293–301. 16559701
31. Riley LK, Robertson DC. Ingestion and intracellular survival of Brucella abortus in human and bovine polymorphonuclear leukocytes. Infect Immun. 1984;46 : 224–30. 6090315
32. Orduña A, Orduña C, Eiros JM, Bratos MA, Gutiérrez P, Alonso P, et al. Inhibition of the degranulation and myeloperoxidase activity of human polymorphonuclear neutrophils by Brucella melitensis. Microbiología. 1991;7 : 113–9. doi: 10.1128/IAI.01162-10 21300774
33. Forestier C, Moreno E, Pizarro-Cerda J, Gorvel J-P. Lysosomal accumulation and recycling of lipopolysaccharide to the cell surface of murine macrophages, an in vitro and in vivo study. J Immunol. 1999;162 : 6784–6791. 10352299
34. Moreno E, Gorvel J-P. Invasion, intracellular trafficking and replication of Brucella organisms in professional and non-professional phagocytes. In: López-Goñi I, Moriyón I, editors. Brucella: Molecular and Cellular Biology. United Kingdom: Horizon Scientific Press; 2004. pp. 287–312.
35. Forestier C, Deleuil F, Lapaque N, Moreno E, Gorvel JP. Brucella abortus lipopolysaccharide in murine peritoneal macrophages acts as a down-regulator of T cell activation. J Immunol. 2000;165 : 5202–10. 11046053
36. Chaves-Olarte E, Altamirano-Silva P, Guzmán-Verri C, Moreno E. Purification of intracellular bacteria: isolation of viable Brucella abortus from host cells. Host-Bacteria Interactions. Methods Protoc. 2014;1197 : 245–60 doi: 10.1007/978-1-4939-1261-2_14 25172285
37. Riley LK, Robertson DC. Brucellacidal activity of human and bovine polymorphonuclear leukocyte granule extracts against smooth and rough strains of Brucella abortus. Infect Immun. 1984;46 : 231–236. 6090316
38. Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 2008;4: e21. doi: 10.1371/journal.ppat.0040021 18266466
39. Caroff M, Bundle DR, Perry MB. Structure of the O-chain of the phenol-phase soluble cellular lipopolysaccharide of Yersinia enterocolitica serotype O:9. Eur J Biochem. 1984;139 : 195–200. 6199199
40. Caroff M, Bundle DR, Perry MB, Cherwonogrodzky JW, Duncan JR. Antigenic S-type lipopolysaccharide of Brucella abortus 1119–3. Infect Immun. 1984;46 : 384–8. 6437981
41. Pérez-Gutiérrez C, Llobet E, Llompart CM, Reinés M, Bengoechea JA. Role of lipid A acylation in Yersinia enterocolitica virulence. Infect Immun. 2010;78 : 2768–81. doi: 10.1128/IAI.01417-09 20385763
42. Iriarte M, González D, Delrue RM, Monreal D, Conde R, López-Goñi I, et al. Brucella lipopolysaccharide: structure, biosynthesis and genetics. Brucella: Molecular and Cellular Biology. Wymondham, UK: Horizon Bioscience; 2004. pp. 159–191.
43. Conde-Álvarez R, Arce-Gorvel V, Iriarte M, Manček-Keber M, Barquero-Calvo E, Palacios-Chaves L, et al. The lipopolysaccharide core of Brucella abortus acts as a shield against innate immunity recognition. PLoS Pathog. 2012;8: e1002675. doi: 10.1371/journal.ppat.1002675 22589715
44. Kubler-Kielb J, Vinogradov E. The study of the core part and non-repeating elements of the O-antigen of Brucella lipopolysaccharide. Carbohydr Res. 2013;366 : 33–7. doi: 10.1016/j.carres.2012.11.004 23261780
45. Velasco J, Bengoechea JA, Brandenburg K, Lindner B, Seydel U, González D, et al. Brucella abortus and its closest phylogenetic relative, Ochrobactrum spp., differ in outer membrane permeability and cationic peptide resistance. Infect Immun. 2000;68 : 3210–8. 10816465
46. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21 : 335–76. 12524386
47. Sabroe I, Dower SK, Whyte MKB. The role of Toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clin Infect Dis. Oxford University Press; 2005;41 Suppl 7: S421–6. 16237641
48. Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, et al. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460 : 264–8. doi: 10.1038/nature08118 19525933
49. Rasool O, Freer E, Moreno E, Jarstrand C. Effect of Brucella abortus lipopolysaccharide on oxidative metabolism and lysozyme release by human neutrophils. Infect Immun. 1992;60 : 1699–702. 1548094
50. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303 : 1532–5. 15001782
51. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176 : 231–41. 17210947
52. Geering B, Simon H-U. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. Nature; 2011;18 : 1457–69. doi: 10.1038/cdd.2011.75 21637292
53. Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11 : 95–102. 8018341
54. Bayraktutan U, Draper N, Lang D, Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res. 1998;38 : 256–62. 9683929
55. Roos D, Weening RS, Wyss SR, Aebi HE. Protection of human neutrophils by endogenous catalase: studies with cells from catalase-deficient individuals. J Clin Invest. 1980;65 : 1515–22. 7410555
56. Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci U S A. 2010;107 : 4230–5. doi: 10.1073/pnas.0913089107 20160104
57. Kamada S, Funahashi Y, Tsujimoto Y. Caspase-4 and caspase-5, members of the ICE/CED-3 family of cysteine proteases, are CrmA-inhibitable proteases. Cell Death Differ. 1997;4 : 473–8. 16465268
58. Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14 : 727–36. doi: 10.1038/nrm3683 24129419
59. Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77 : 58–63. 10101687
60. Zheng TS, Hunot S, Kuida K, Flavell RA. Caspase knockouts: matters of life and death. Cell Death Differ. 1999;6 : 1043–53. 10578172
61. Acorci MJ, Dias-Melicio LA, Golim MA, Bordon-Graciani AP, Peraçoli MTS, Soares AMVC. Inhibition of human neutrophil apoptosis by Paracoccidioides brasiliensis: role of interleukin-8. Scand J Immunol. 2009;69 : 73–9. doi: 10.1111/j.1365-3083.2008.02199.x 19144080
62. Afford SC, Pongracz J, Stockley R a, Crocker J, Burnett D. The induction by human interleukin-6 of apoptosis in the promonocytic cell line U937 and human neutrophils. J Biol Chem. 1992;267 : 21612–6. 1400472
63. Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80 : 2012–2020. 1382715
64. Ocaña MG, Asensi V, Montes AH, Meana A, Celada A, Valle-Garay E. Autoregulation mechanism of human neutrophil apoptosis during bacterial infection. Mol Immunol. 2008;45 : 2087–96. 18022234
65. Baran J, Guzik K, Hryniewicz W, Ernst M, Flad HD, Pryjma J. Apoptosis of monocytes and prolonged survival of granulocytes as a result of phagocytosis of bacteria. Infect Immun. 1996;64 : 4242–8. 8926095
66. Tumurkhuu G, Koide N, Takahashi K, Hassan F, Islam S, Ito H, et al. Characterization of biological activities of Brucella melitensis lipopolysaccharide. Microbiol Immunol. 2006;50 : 421–7. 16785713
67. Weiss DS, Takeda K, Akira S, Zychlinsky A, Moreno E. MyD88, but not toll-like receptors 4 and 2, is required for efficient clearance of Brucella abortus. Infect Immun. 2005;73 : 5137–5143. 16041030
68. Goldstein J, Hoffman T, Frasch C, Lizzio EF, Beining PR, Hochstein D, et al. Lipopolysaccharide (LPS) from Brucella abortus is less toxic than that from Escherichia coli, suggesting the possible use of B. abortus or LPS from B. abortus as a carrier in vaccines. Infect Immun. 1992;60 : 1385–9. 1548064
69. Dueñas AI, Orduña A, Crespo MS, García-Rodríguez C, Lps B, Ordun A, et al. Interaction of endotoxins with Toll-like receptor 4 correlates with their endotoxic potential and may explain the proinflammatory effect of Brucella spp. LPS. Int Immunol. 2004;16 : 1467–75. 15339879
70. Kettritz R, Gaido ML, Haller H, Luft FC, Jennette CJ, Falk RJ. Interleukin-8 delays spontaneous and tumor necrosis factor-alpha-mediated apoptosis of human neutrophils. Kidney Int. 1998;53 : 84–91. 9453003
71. Altstaedt J, Kirchner H, Rink L. Cytokine production of neutrophils is limited to interleukin-8. Immunology. 1996;89 : 563–8. 9014822
72. Sabroe I, Jones EC, Usher LR, Whyte MKB, Dower SK. Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol. 2002;168 : 4701–10. 11971020
73. Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, et al. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. 1999;274 : 4309–18. 9933632
74. Fadeel B, Ahlin A, Henter JI, Orrenius S, Hampton MB. Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood. 1998;92 : 4808–18. 9845548
75. Gamazo C, Moriyón I. Release of outer membrane fragments by exponentially growing Brucella melitensis cells. Infect Immun. 1987;55 : 609–15. 3818086
76. Zwerdling A, Delpino MV, Pasquevich K a, Barrionuevo P, Cassataro J, García Samartino C, et al. Brucella abortus activates human neutrophils. Microbes Infect. 2009;11 : 689–97. doi: 10.1016/j.micinf.2009.04.010 19376263
77. Lapaque N, Takeuchi O, Corrales F, Akira S, Moriyon I, Howard JC, et al. Differential inductions of TNF-alpha and IGTP, IIGP by structurally diverse classic and non-classic lipopolysaccharides. Cell Microbiol. 2006;8 : 401–13. 16469053
78. Lapaque N, Muller A, Alexopoulou L, Howard JC, Gorvel J-P. Brucella abortus induces Irgm3 and Irga6 expression via type-I IFN by a MyD88-dependent pathway, without the requirement of TLR2, TLR4, TLR5 and TLR9. Microb Pathog. 2009;47 : 299–304. doi: 10.1016/j.micpath.2009.09.005 19747534
79. Rodeberg DA, Morris RE, Babcock GF. Azurophilic granules of human neutrophils contain CD14. Infect Immun. 1997;65 : 4747–53. 9353060
80. Lapaque N, Forquet F, de Chastellier C, Mishal Z, Jolly G, Moreno E, et al. Characterization of Brucella abortus lipopolysaccharide macrodomains as mega rafts. Cell Microbiol. 2006;8 : 197–206. 16441431
81. Lei M, Du L, Jiao H, Cheng Y, Zhang D, Hao Y, et al. Inhibition of mCD14 inhibits TNFα secretion and NO production in RAW264.7 cells stimulated by Brucella melitensis infection. Vet Microbiol. 2012;160 : 362–8. doi: 10.1016/j.vetmic.2012.05.039 22770519
82. Lapaque N, Moriyon I, Moreno E, Gorvel J-P. Brucella lipopolysaccharide acts as a virulence factor. Curr Opin Microbiol. 2005;8 : 60–6. 15694858
83. Weersink JL, Antal-Szalmas P, Strijp JAG Van, Kessel KPM Van. Quantitation of surface neutrophils CD14 on human monocytes and. J Leukoc Biol. 1997;61 : 721–728. 9201263
84. Celli J, de Chastellier C, Franchini D-M, Pizarro-Cerda J, Moreno E, Gorvel J-P. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med. 2003;198 : 545–56. 12925673
85. Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5 : 415–8. 11256882
86. Almyroudis NG, Grimm MJ, Davidson BA, Röhm M, Urban CF, Segal BH. NETosis and NADPH oxidase: at the intersection of host defense, inflammation, and injury. Front Immunol. 2013;4 : 45. doi: 10.3389/fimmu.2013.00045 23459634
87. Meuth M. Chk1 suppressed cell death. Cell Div. 2010;5 : 21. doi: 10.1186/1747-1028-5-21 20813042
88. Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14 : 400–10. 17301840
89. Olofsson MH, Havelka AM, Brnjic S, Shoshan MC, Linder S. Charting calcium-regulated apoptosis pathways using chemical biology: role of calmodulin kinase II. BMC Chem Biol. 2008;8 : 2. doi: 10.1186/1472-6769-8-2 18673549
90. Miao EA, Rajan J V, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243 : 206–14. doi: 10.1111/j.1600-065X.2011.01044.x 21884178
91. Bakele M, Joos M, Burdi S, Allgaier N, Pöschel S, Fehrenbacher B, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. 2014;289 : 5320–9. doi: 10.1074/jbc.M113.505636 24398679
92. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11 : 1136–42. doi: 10.1038/ni.1960 21057511
93. Brodsky IE, Medzhitov R. Pyroptosis: macrophage suicide exposes hidden invaders. Curr Biol. 2011;21: R72–5. doi: 10.1016/j.cub.2010.12.008 21256438
94. Murray J, Barbara JAJ, Dunkley SA, Lopez AF, Van Ostade X, Condliffe AM, et al. Regulation of neutrophil apoptosis by tumor necrosis factor-alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. 1997;90 : 2772–2783. 9326245
95. Aggarwal S, Gollapudi S, Yel L, Gupta AS, Gupta S. TNF-alpha-induced apoptosis in neonatal lymphocytes: TNFRp55 expression and downstream pathways of apoptosis. Genes Immun. 2000;1 : 271–9. 11196704
96. Demir C, Karahocagil MK, Esen R, Atmaca M, Gönüllü H, Akdeniz H. Bone marrow biopsy findings in brucellosis patients with hematologic abnormalities. Chin Med J (Engl). 2012;125 : 1871–6. 22884045
97. El-Koumi MA, Afify M, Al-Zahrani SH. A prospective study of brucellosis in children: relative frequency of pancytopenia. Mediterr J Hematol Infect Dis. 2013;5: e2013011. doi: 10.4084/MJHID.2013.011 23505599
98. Kokkini G, Giotaki HG, Moutsopoulos HM. Transient hemophagocytosis in Brucella melitensis infection. Arch Pathol Lab Med. 1984;108 : 213–6. 6546508
99. Laskay T, van Zandbergen G, Solbach W. Neutrophil granulocytes—Trojan horses for Leishmania major and other intracellular microbes? Trends Microbiol. 2003;11 : 210–214. 12781523
100. Chacón-Díaz C, Muñoz-Rodríguez M, Barquero-Calvo E, Guzmán-Verri C, Chaves-Olarte E, Grilló MJ, et al. The use of green fluorescent protein as a marker for Brucella vaccines. Vaccine. 2011;29 : 577–82. doi: 10.1016/j.vaccine.2010.09.109 21056079
101. Moreno E, Berman DT, Boettcher L a. Biological activities of Brucella abortus lipopolysaccharides. Infect Immun. 1981;31 : 362–70. 6783538
102. Espinoza AM, Pereira R, Macaya-Lizano A V, Hernández M, Goulden M, Rivera C. Comparative light and electron microscopic analyses of tenuivirus major noncapsid protein (NCP) inclusion bodies in infected plants, and of the NCP in vitro. Virology. 1993;195 : 156–66. 8317091
103. Rojas N, Freer E, Weintraub A, Ramirez M, Lind S, Moreno E. Immunochemical identification of Brucella abortus lipopolysaccharide epitopes. Clin Diagn Lab Immunol. 1994;1 : 206–13. 7496947
104. Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17 : 208–12. 13986422
105. Bundle DR, Cherwonogrodzky JW, Gidney MA, Meikle PJ, Perry MB, Peters T. Definition of Brucella A and M epitopes by monoclonal typing reagents and synthetic oligosaccharides. Infect Immun. 1989;57 : 2829–36. 2474505
106. Chin AC, Lee WD, Murrin KA, Morck DW, Merrill JK, Dick P, et al. Tilmicosin induces apoptosis in bovine peripheral neutrophils in the presence or in the absence of Pasteurella haemolytica and promotes neutrophil phagocytosis by macrophages. Antimicrob Agents Chemother. 2000;44 : 2465–70. 10952596
107. Moreno E, Stackebrandt E, Dorsch M, Wolters J, Busch M, Mayer H. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J Bacteriol. 1990;172 : 3569–76. 2113907
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance