
Conserved Genes Act as Modifiers of Invertebrate SMN Loss of Function Defects
Spinal Muscular Atrophy (SMA) is caused by diminished function of the Survival of Motor Neuron (SMN) protein, but the molecular pathways critical for SMA pathology remain elusive. We have used genetic approaches in invertebrate models to identify conserved SMN loss of function modifier genes. Drosophila melanogaster and Caenorhabditis elegans each have a single gene encoding a protein orthologous to human SMN; diminished function of these invertebrate genes causes lethality and neuromuscular defects. To find genes that modulate SMN function defects across species, two approaches were used. First, a genome-wide RNAi screen for C. elegans SMN modifier genes was undertaken, yielding four genes. Second, we tested the conservation of modifier gene function across species; genes identified in one invertebrate model were tested for function in the other invertebrate model. Drosophila orthologs of two genes, which were identified originally in C. elegans, modified Drosophila SMN loss of function defects. C. elegans orthologs of twelve genes, which were originally identified in a previous Drosophila screen, modified C. elegans SMN loss of function defects. Bioinformatic analysis of the conserved, cross-species, modifier genes suggests that conserved cellular pathways, specifically endocytosis and mRNA regulation, act as critical genetic modifiers of SMN loss of function defects across species.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001172
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001172
Summary
Spinal Muscular Atrophy (SMA) is caused by diminished function of the Survival of Motor Neuron (SMN) protein, but the molecular pathways critical for SMA pathology remain elusive. We have used genetic approaches in invertebrate models to identify conserved SMN loss of function modifier genes. Drosophila melanogaster and Caenorhabditis elegans each have a single gene encoding a protein orthologous to human SMN; diminished function of these invertebrate genes causes lethality and neuromuscular defects. To find genes that modulate SMN function defects across species, two approaches were used. First, a genome-wide RNAi screen for C. elegans SMN modifier genes was undertaken, yielding four genes. Second, we tested the conservation of modifier gene function across species; genes identified in one invertebrate model were tested for function in the other invertebrate model. Drosophila orthologs of two genes, which were identified originally in C. elegans, modified Drosophila SMN loss of function defects. C. elegans orthologs of twelve genes, which were originally identified in a previous Drosophila screen, modified C. elegans SMN loss of function defects. Bioinformatic analysis of the conserved, cross-species, modifier genes suggests that conserved cellular pathways, specifically endocytosis and mRNA regulation, act as critical genetic modifiers of SMN loss of function defects across species.
Introduction
Decreased Survival of Motor Neuron (SMN) protein function underlies most Spinal Muscular Atrophy (SMA) cases [1]. The SMN protein is ubiquitously expressed [2], [3], yet SMA pathology is remarkably specific. Patients lose spinal α-motorneurons and experience muscular dysfunction with atrophy. Mild cases result in slowly progressing muscular weakness, while severe cases dramatically perturb proximal neuromuscular function resulting in childhood death [4]. There is no effective treatment for SMA and at least 1 in 40 people in the US population are carriers of SMN loss of function disease alleles [5]–[7].
The SMN protein is a component of the well-characterized Gemin complex, which assembles splicing machinery in eukaryotes [8]–[10]. SMN also associates with β-actin mRNA during anterograde transport in neuronal processes suggesting a role for SMN in mRNA transport, sub-cellular localization and/or local translation [11]–[15]. In addition, SMN is found in post-synaptic densities and Z-discs of muscles along with other RNA processing proteins [11]–[19]. Roles for SMN in small nucleolar RNA (snoRNA) and microRNA (miRNA) pathways have also been suggested [20]–[22]. The relative contributions of SMN in these various compartments and the relative importance of SMN function in neurons and muscles for SMA pathology have been difficult to determine. Various tissue requirements for SMN function have been observed in different SMA model systems [23]–[27]. The diverse subcellular SMN localization and varied cellular requirements for SMN function suggest that this protein may act in multiple cellular compartments including the neuromuscular junction (NMJ) [28].
To determine in an unbiased fashion which cellular and molecular pathways are particularly relevant to SMA pathology, researchers have turned recently to genetic approaches in vertebrates and invertebrates. The identification of SMN loss of function modifier genes can reveal important biochemical pathways for SMA pathology. Studies in patients have already identified two genes that act as modifiers of SMA: SMN2 and Plastin 3 (PLS3).
Two genes encode human SMN protein: SMN1 and SMN2. The SMN1 gene encodes only full-length SMN protein while the SMN2 gene encodes two different transcripts; 10% of SMN2 transcripts encode a full-length SMN protein identical to the SMN1 gene product. However, due to a change in the splice consensus sequence, 90% of SMN2 transcripts contain a stop codon at the beginning of exon 7 and, therefore, encode a truncated protein (called SMNdeltaEx7 or Δ7SMN) of diminished function and stability [1], [29]–[31]. Humans have various numbers of SMN2 genes; patients with more copies of SMN2 generally have later onset/less severe symptoms than patients with fewer copies of SMN2. Decreased severity and delayed onset is usually attributed to increased full-length SMN levels from SMN2 in vivo [17], [32]–[36].
PLS3 may modulate the severity of SMA. In several families, daughters who lack SMN1 and over-express PLS3 were remarkably unaffected [37]. PLS3 encodes a conserved calcium-binding, actin-bundling/stabilizing protein that is broadly expressed in various tissues including blood, muscles and neurons [38]–[40]. Loss of the yeast PLS3 ortholog, Sac6p, results in defective endocytosis [41], [42]. Altering PLS3 levels modified SMN loss of function defects in zebrafish motorneurons consistent with results in human families and PLS3 co-precipitated with SMN from neuronal tissues [37]. However, increased PLS3 (due to profilin knockdown) did not decrease the defects in an SMA mouse model and it remains unclear how PLS3 might modify SMN neuromuscular defects [43].
Modifier genes identified in patient populations are clearly pertinent to SMA pathology. However, studies in humans are limited by kindred sizes and other considerations. As SMN orthologs are found in C. elegans and Drosophila melanogaster, it may be more efficient to identify SMA modifier genes in these powerful invertebrate models. SMN loss of function models have already been defined in C. elegans and Drosophila [18], [25], [44], [45]. Loss of Drosophila Smn (DmSmn) causes larval lethality and NMJ defects; DmSmn function is required in neurons and muscles in flies [26]. Loss of C. elegans SMN-1 (Cesmn-1) also causes neuromuscular function deficits followed by larval lethality [44]. Expression of Cesmn-1 in neurons dramatically restores neuromuscular function, whereas expression in muscles has little effect [44]. Given SMN conservation across species, genes that act as SMN loss of function modifiers in invertebrates could be important in SMA pathology in humans (e.g. PLS3) [37].
In a recent study, twenty-seven P-element transposon insertion lines were identified in Drosophila that modified SMN loss of function defects, and a role for the TGF-beta pathway in SMN loss of function pathology was delineated [26]. However, it remains unclear for several P-element lines which Drosophila gene near the transposon insertion site is responsible for modulating SMN phenotypic defects. The Drosophila P-element lines carried an inducible GAL4-UAS that could drive either over-expression or antisense RNAi expression of neighboring genes depending on transposon insertion site. Additionally, insertion of the P-element itself might perturb gene function. Eliminating ambiguity regarding modifier gene identity would increase the utility of the Drosophila study.
To explore the genetic circuitry affecting SMN activity in C. elegans, the Cesmn-1(lf) growth defect phenotype was used as a metric in a rapid large-scale genetic screen. Growth may be affected by a variety of changes, such as body length and longevity. Subsequently, modifier genes were tested using a C. elegans behavioral assay, the pharyngeal pumping, which is likely more pertinent to SMN loss of function neuromuscular defects. In addition, to identify conserved invertebrate SMN modifier genes, we utilized previously described Drosophila assays to assess genetic interaction of DmSmn with Drosophila orthologs of C. elegans modifier genes. In the study by Chang and co-workers, the DmSmn lethal phenotype correlated with NMJ defects for virtually all DmSmn modifier genes, suggesting that lethality and neuromuscular bouton number are effective measures of genetic interaction with the Drosophila SMN ortholog [26].
Here, we define conserved genetic modifiers of SMN loss of function using C. elegans and Drosophila. We find that PLS3 orthologs act as SMN modifier genes in both invertebrate species. A genome-wide RNAi screen in C. elegans identified four new SMN modifier genes, including ncbp-2 and flp-4, which also modify SMN loss of function defects in Drosophila. Candidate SMN modifier genes identified in a previous Drosophila screen were tested in C. elegans yielding twelve cross-species modifier genes. Examination of the literature for these genes suggested specific cellular pathways that are critical genetic modifiers of SMN function: endocytosis and RNA processing. These pathways may also be pertinent to SMN loss of function defects in patients with SMA.
Results
The previously described Cesmn-1(ok355) deletion allele causes a complete loss of Cesmn-1 function and is referred to herein as Cesmn-1(lf) [44]. Cesmn-1(lf) is recessive; heterozygous animals are overtly normal. To facilitate identification of heterozygous versus homozygous animals, we utilized the balanced strain hT2(bli-4(e937) let-?(q782) qIs48[myo-2p::GFP])/Cesmn-1(lf) (abbreviated +/Cesmn-1(lf) [44], [46]. Heterozygous +/Cesmn-1(lf) animals express pharyngeal GFP, homozygous Cesmn-1(lf) progeny do not express GFP, and progeny homozygous for the hT2 balancer die as GFP-expressing embryos.
Although complete loss of SMN function causes lethality, C. elegans that are homozygous mutant for Cesmn-1(lf) can survive for several days due to partial maternal rescue. It has been suggested that +/Cesmn-1(lf) hermaphrodites load sufficient Cesmn-1 maternal protein and/or perhaps mRNA into oocytes to support development through embryogenesis and early larval stages [44]. Accordingly, homozygous Cesmn-1(lf) larvae initially resemble wild type animals. Eventually maternally-loaded Cesmn-1 product is lost; Cesmn-1(lf) animals grow more slowly than +/Cesmn-1(lf) siblings, are shorter, sterile, and most Cesmn-1(lf) animals die before reaching adulthood (Figure 1A). Combined, these defects decrease the average size of the Cesmn-1(lf) population versus control animals; decreased average population size will be referred to herein as a growth defect. This growth defect was harnessed in an automated assay to identify Cesmn-1(lf) modifier genes in a genome-wide screen.

C. elegans growth assay
To validate growth as an assay for SMN modifier gene identification, we first demonstrated that RNAi knockdown of Cesmn-1 or the invertebrate ortholog of Plastin 3 (PLS3) altered Cesmn-1(lf) growth. The C. elegans gene plst-1 (PLaSTin (actin bundling protein) homolog-1) encodes a predicted protein similar to PLS3.
To knockdown gene function, C. elegans were reared on bacteria producing double stranded RNA corresponding to the gene of interest, a strategy known as ‘feeding RNAi’ [47]. Feeding RNAi decreases gene transcripts in most C. elegans tissues although knockdown in neurons is generally less effective than knockdown in muscles, germline, and other tissues [48]–[50]. Here, animals were reared for two generations on solid media and RNAi feeding bacteria corresponding to Cesmn-1 or plst-1, allowing knockdown of maternal and zygotic transcripts (Figure 1B). Bacteria containing the empty RNAi feeding vector were used as a negative control (empty(RNAi)).
An automated system was used to simultaneously measure growth and determine genotype for the progeny of +/Cesmn-1(lf) animals (Figure 1C). The COPAS BioSorter (Union Biometrica, Holliston, MA) measures C. elegans length as ‘time-of-flight’, which is the time required for the animal to pass through the fluorescence-detection chamber [51]. Cesmn-1(lf) homozygous animals do not express GFP while +/Cesmn-1(lf) heterozygous animals express GFP and are longer than Cesmn-1 homozygous animals of the same late larval or adult stage. Animals smaller than the L2 larval stage were excluded from this analysis to avoid bacterial debris. The percentage of large adult animals was determined for each genotype and RNAi treatment.
RNAi knockdown of Cesmn-1 decreased the percentage of large animals in both Cesmn-1(lf) homozygous and +/Cesmn-1(lf) heterozygous populations (Table 1, Rows 1 & 2). Initially, it seems counter-intuitive that the defects of Cesmn-1(lf) animals are exacerbated by Cesmn-1(RNAi). However, in this scenario, transcripts in both the somatic tissues and germline of +/Cesmn-1(lf) heterozygous animals are targeted and, consequently, maternally-loaded Cesmn-1 transcript and protein are depleted in homozygous Cesmn-1(lf) progeny, abrogating partially the observed maternal rescue. The ability of Cesmn-1(RNAi) to exacerbate Cesmn-1(lf) defects suggests that the effects of modifier genes can be assessed using RNAi feeding.

Knockdown of the C. elegans PLS3 ortholog, plst-1, increased the average length of the +/Cesmn-1(lf) population, but did not significantly alter the average length of Cesmn-1(lf) animals (Table 1, Rows 1 & 3). Genetic interaction with plst-1 was further confirmed by using the plst-1 (tm4255) mutant allele (Table 2). The average length of +/Cesmn-1(lf);plst-1(tm4255) adult animals was significantly increased in relation to +/Cesmn-1(lf) animals. In contrast, the average length of homozygous Cesmn-1(lf);plst-1(tm4255) was not altered, recapitulating the results of plst-1(RNAi). Increased average adult length is an overall growth metric thzat may encompass a variety of changes; decreased plst-1 function, by RNAi or mutant allele, could increase length, cause sterility, and/or increase longevity in +/Cesmn-1(lf) control animals. It appears that loss of Cesmn-1 function suppresses the effects of decreased plst-1 function (i.e. increased length was not observed in Cesmn-1(lf);plst-1(tm4255) homozygous mutant animals). The genetic and functional relationship between SMN and PLS3 bears further examination; as plst-1 and Cesmn-1 have opposing effects on the growth assay and since Cesmn-1(lf);plst-1(tm4255) animals resemble Cesmn-1(lf) single mutants, Cesmn-1 may act downstream of plst-1 in this growth assay [52].

Growth modifier genes identified in C. elegans genome-wide screen
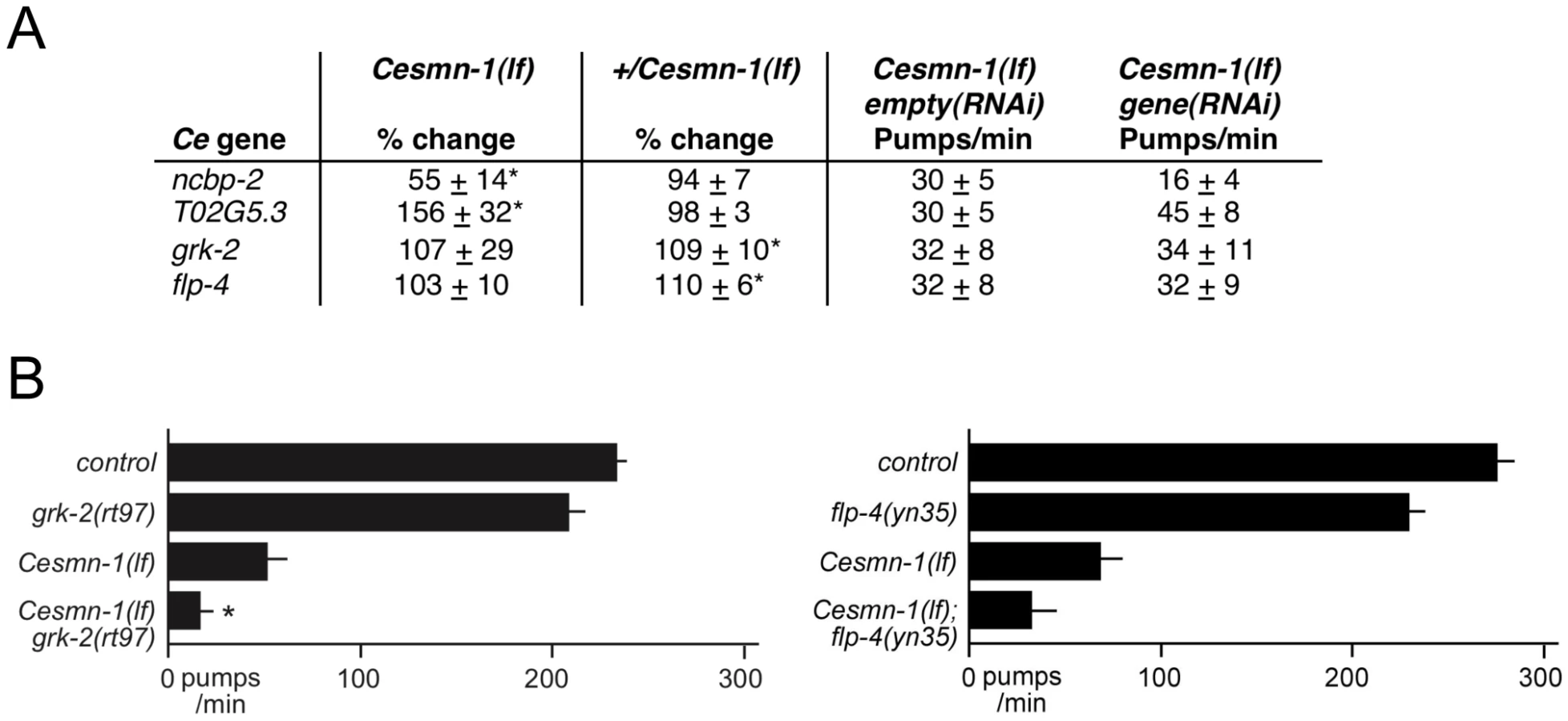
To identify additional genes that modify SMN loss of function defects, a large-scale genome-wide screen for enhancers and suppressors of the Cesmn-1(lf) growth defect was undertaken. The growth assay was adapted to a higher-throughput 96-well, liquid culture format and a previously described genome-wide C. elegans RNAi feeding library was used for gene knockdown (Figure 2A) [53]. Progeny of +/Cesmn-1(lf) animals were reared for two weeks (more than 2 generations) on RNAi feeding bacterial strains before assessment of growth using the COPAS Biosorter [51]. To identify RNAi clones that specifically altered the growth of Cesmn-1(lf) animals, a growth ratio of large to small animals was determined for each clone for Cesmn-1(lf) and for +/Cesmn-1(lf) genotypes. If the RNAi clone growth ratio was more than 2 standard deviations away from the mean for Cesmn-1(lf) animals and within 0.7 standard deviations of the mean for +/Cesmn-1(lf) animals in at least 40% of independent trials, then the corresponding gene was designated as an Cesmn-1(lf) modifier (Figure 2B). In the primary high-throughput screen, no suppressors were found, but four genes were identified as enhancers (Figure 2B). RNAi knockdown of these genes exacerbated homozygous Cesmn-1(lf) growth defects and did not significantly alter the growth of heterozygous +/Cesmn-1(lf) animals: ncbp-2, T02G5.3, grk-2, and flp-4. ncbp-2 encodes the C. elegans Cap Binding Protein 20 (CBP20 or Cbp20) ortholog [54]. T02G5.3 encodes a predicted protein of unknown function with no vertebrate orthologs based on BLAST analysis. grk-2 encodes one of two G-protein coupled receptor kinases. flp-4 encodes an FMRFamide family neuropeptide protein. The low number of modifiers identified in this screen versus the previous Drosophila screen may reflect the stringent criterion utilized here or the inefficiency of RNAi by feeding in neurons.

To determine if decreased adult body length accounts for the enhanced Cesmn-1(lf) growth defect upon knockdown of ncbp-2, T02G5.3, grk-2 and flp-4, the average body length of Cesmn-1(lf) young animals was determined (Text S1 and Table S4, top panel). Only ncbp-2(RNAi) significantly reduced the average body length of Cesmn-1(lf) animals suggesting that the enhanced growth defect caused by ncbp-2(RNAi) could be attributed to the Cesmn-1(lf) shorter body size. The other three enhancer genes may alter survival or growth as adult animals.
SMA is a neuromuscular disease and, therefore, our objective was the identification of modifier genes that impact SMN neuromuscular function. We then examined the impact of Cesmn-1 growth modifier genes on Cesmn-1 loss of function neuromuscular defects using RNAi and, when available, loss of function alleles of modifier genes.
RNAi knockdown of ncbp-2, grk-2, and T02G5.3 modified Cesmn-1(lf) neuromuscular defects
A recent study from the Sattelle laboratory demonstrated that loss of Cesmn-1 function causes progressive defects in C. elegans neuromuscular function in pharyngeal pumping [44]. C. elegans feeds on bacteria and other microorganisms using a small, discrete subset of neurons and muscles contained in the pharynx (Figure 3A) [55]. Pharyngeal cell specification, neuronal development, and myoblast fusion is completed within hours of hatching [56], [57]. The pharynx pumps continuously and symmetrically at over 250 beats per minute in wild type animals when food is present and larval pumping is interrupted only by molting under standard culture conditions. We confirmed a previous report [44] that in early larval stages, the pumping rates of Cesmn-1(lf) animals are indistinguishable from control animals, but at later larval stages Cesmn-1(lf) pumping rates drop (Figure 3B).Cesmn-1(lf) animals have progressive defects in pharyngeal pumping, which occur earlier than reported locomotion defects. At day 2, 62% of Cesmn-1(lf) animals are moving spontaneously, but pumping rates have dropped dramatically (Figure 3B). Restoration of Cesmn-1 function in neurons almost completely restores pumping rates suggesting that Cesmn-1 is required in neurons for this behavior [44].

The efficacy of RNAi by feeding in this neuromuscular assay was assessed for Cesmn-1 and plst-1 using Cesmn-1(lf) and +/Cesmn-1(lf) animals. Animals were allowed to hatch on RNAi feeding plates and pumping rates were determined after three days. Either plst-1(RNAi) or Cesmn-1(RNAi) decreased Cesmn-1(lf) pumping rates, but not +/Cesmn-1(lf) pumping rates (Figure 3C). In addition, plst-1(lf) significantly decreased the pumping rates of Cesmn-1(lf) animals, validating the genetic interaction of plst-1 with Cesmn-1 in the neuromuscular pharyngeal pumping assay (Figure 3D). This exacerbation of Cesmn-1 loss of function defects by plst-1 manipulation is consistent with results in other organisms [37]. The ability of Cesmn-1(RNAi) and plst-1(RNAi) to alter pumping of homozygous mutant Cesmn-1(lf) animals suggests that candidate modifier genes can be assessed using RNAi knockdown in this neuromuscular assay.
The four modifier genes from the C. elegans growth screen were tested for function as Cesmn-1 neuromuscular modifier genes using the pharyngeal pumping assay. Results are summarized in Figure 4A. ncbp-2(RNAi) and T02G5.3(RNAi) enhanced and suppressed the pharyngeal pumping defects of Cesmn-1(lf) animals, respectively; flp-4(RNAi) and grk-2(RNAi) had no significant effect compared to controls. We suggest that ncbp-2 and T02G5.3 are likely modifiers of Cesmn-1(lf) neuromuscular defects based on RNAi results.
Loss of grk-2 enhances Cesmn-1(lf) growth and neuromuscular defects
RNAi knockdown of C. elegans genes by feeding is robust in virtually all cell types but can often be inefficient and can result in only partial loss of gene function, especially in the nervous system [58], [59]. To address the specificity of the genetic modifiers, the RNAi results were confirmed by using mutant alleles when possible; alleles of ncbp-2 and T02G5.3 were not available.
A grk-2 loss of function allele has been previously described, grk-2(rt97) [60]. Loss of grk-2 significantly enhanced the growth defects of Cesmn-1(lf) animals (Table 2). Additionally, the pumping rates of grk-2(lf) animals derived from hT2 parents were not significantly lower than those of control animals, but the average pumping rates of Cesmn-1(lf) grk-2(lf) double mutant animals were significantly lower than the pumping rates of either single mutant (Figure 4B). This suggests that grk-2 loss enhances both Cesmn-1(lf) growth and pharyngeal pumping defects. A grk-2 gain of function allele is not available and transgenes are unstable in +/Cesmn-1(lf) animals (unpublished results and [44]).
To test the genetic interaction of flp-4 with Cesmn-1, we identified a flp-4 loss of function allele, flp-4(yn35), using PCR based screening techniques [61]–[65]. The flp-4(yn35) deletion removes all sequences encoding FLP-4 FMRFamide neuropeptides and likely causes a complete loss of flp-4 function ([66] and C. Li, in preparation). Although flp-4(yn35) reduced the percentage of Cesmn-1(lf) large animals in the growth assay, the difference was not statistical significant different (Table 2). Similar results were obtained using the pharyngeal pumping assay. The pumping rates of flp-4(lf) animals were slightly lower, but not significantly different than control animals. Loss of flp-4 function decreased pumping rates of Cesmn-1(lf) animals in five independent trials, but the difference was not statistically significant (p = 0.236, Figure 4B). Either flp-4 is not a bona fide modifier or flp-4(RNAi) may act off-target decreasing the function of more than one of the 32 other C. elegans FMRFamide genes [67].
Drosophila orthologs of plst-1, ncbp-2 and flp-4 modify SMN neuromuscular defects
SMN modifier genes that are conserved across species would be of considerable interest. Three of the candidate genes identified in the C. elegans screen encode conserved proteins with clear orthologs in other species: grk-2, flp-4, and ncbp-2. To determine if their orthologs modify SMN loss of function defects, we turned to the fruit fly Drosophila. Decreased function in the Drosophila SMN ortholog Smn (DmSmn) results in growth defects, early pupal arrest, and NMJ synaptic defects [26]. We utilized pre-existing Drosophila loss of function alleles and previously described Drosophila assays to assess genetic interaction of DmSmn with Drosophila orthologs of C. elegans modifier genes [18], [25], [26].
First, we determined if Fimbrin (Fim), the Drosophila ortholog of PLS3, modifies DmSmn loss of function defects in growth and NMJ assays. It has been shown that RNAi knockdown of DmSmn (DmSmn RNAi) results in 44% lethality in early pupal stages with 56% lethality at late pupal stages [26]. Loss of Fim alone does not cause larval or pupal lethality (data not shown). Three Fim loss of function alleles were crossed into the DmSmn(RNAi) background and each accelerated death compared to DmSmn(RNAi) control animals (Figure 5A).

In Drosophila, loss of SMN function results in a dose-dependant decrease in process arborization at the NMJ and diminished numbers of synaptic specializations, termed synaptic boutons [26]. Boutons are visualized as coincident pre-synaptic synaptotagmin and post-synaptic Discs large protein immunoreactivity. The number of synaptic boutons found between Drosophila neurons and muscles provides a simple and readily quantifiable assessment of phenotypic severity. We determined if the Drosophila PLS3 ortholog Fim might also modify the NMJ defects of DmSmn. RNAi knockdown of DmSmn using the ubiquitous tubulin promoter (TubGAL4;SmnRNAi) modestly decreased synaptic innervation in Drosophila larvae (reported as bouton numbers per muscle area, Figure 5B). Loss of Drosophila Fim function in Fimd02114 animals also modestly decreased bouton density. We found that effects of Fimd02114 and DmSmn knockdown were synergistic; bouton numbers were significantly decreased suggesting that loss of Fim function exacerbated DmSmn loss of function defects, being consistent with studies in vertebrate models of SMA [37]. These results suggest that PLS3 is a cross-species modifier of SMN function.
Next, Drosophila orthologs of candidate SMN modifier genes from C. elegans were examined. Cbp20 and Fmrf were selected as Drosophila orthologs of ncbp-2 and flp-4, respectively, based on similarity and Drosophila loss of function alleles were obtained. (There are 32 genes in C. elegans encoding 32 FMRFamide-related neuropeptides, in contrast, three FMRFamide genes exist in Drosophila. There may be less redundancy in FMRFamide gene function in Drosophila [68], [69], [70]).
Heterozygous loss of DmSmn function in +/Smn73Ao or +/Smnf01109 animals had no significant effect on bouton number as expected, Smn73Ao/Smnf01109 animals had dramatically decreased bouton numbers (Figure 6). Loss of one copy of Cbc20 or Fmrf modestly decreased synaptic bouton number compared to control animals. However, simultaneous loss of one copy of DmSmn and one copy of either modifier gene resulted in further synaptic bouton loss (Figure 6). The genetic interaction in trans-heterozygous animals is consistent with a strong genetic interaction between Smn and the two modifier genes. We were unable to obtain classical alleles of the grk-2 Drosophila ortholog. We conclude that Cbp20 and FMRFamide are conserved invertebrate enhancers of Smn loss of function defects and that this genetic interaction is conserved across species.

Testing Drosophila modifier genes in C. elegans assays
A previous Drosophila screen identified twenty-seven P-element insertion lines that altered Drosophila SMN (DmSmn) loss of function defects [26]. Cross-species validation of these genes might also help elucidate conserved pathways that are critical in SMN loss of function pathology. However, several genes flanked the P-element insertion site for many of these modifier lines and the precise DmSmn modifier gene could not be unambiguously identified. Therefore, 40 candidate modifier genes were reported [26]. We identified the likely C. elegans orthologs for 32 of these 40 genes using reciprocal BLAST similarity searching (Table S1). The ability of these genes to modify Cesmn-1(lf) growth defects was assessed by feeding Cesmn-1(lf) and +/Cesmn-1(lf) animals bacteria expressing the corresponding dsRNA; RNAi feeding clones were constructed for B0432.13, dhs-22 and ugt-49 [53]. Twelve genes crossed species and modified Cesmn-1(lf) defects in one or both C. elegans assays.
Orthologs of ten Drosophila genes modified Cesmn-1(lf) growth defects
Knockdown of seven C. elegans genes (uso-1, nhr-85, egl-15, atf-6, ape-1, kcnl-2 and nekl-3) orthologous to DmSmn modifier genes specifically enhanced Cesmn-1(lf) growth defects, but did not significantly alter the percentage of large heterozygous +/Cesmn-1(lf) animals. Knockdown of the C. elegans ortholog atn-1 significantly suppressed the growth defects of Cesmn-1(lf) animals without altering the percentage of large +/Cesmn-1(lf) siblings. Finally, C. elegans orthologs of two Drosophila genes were identified, whose genetic interaction with Cesmn-1 resembled the interaction of plst-1 with Cesmn-1: cash-1 and dlc-1. RNAi knockdown of these two genes increased the percentage of large animals in the +/Cesmn-1(lf) population without altering the Cesmn-1(lf) population. Growth assay results for these ten genes are found in Table 1 (Rows 4 through 13), results for all orthologs tested can be found in Table S1, and a discussion of modifier gene function is presented in Text S2.
For bona fide cross-species modifier genes, the impact of modifier genes on SMN loss of function defects should be conserved across species (i.e. enhancer genes should enhance in both species). For six cross-species genes, the impact of modifier gene loss on DmSmn and Cesmn-1 loss of function defects was conserved as expected. Specifically, the enhancement of Cesmn-1(lf) defects by RNAi knockdown of nhr-85, egl-15, and kcnl-2 was consistent with the effects of the corresponding Drosophila modifier genes on DmSmn [26]; the corresponding Drosophila insertion lines (d09801, f02864, and d03336) enhanced DmSmn defects and the transposon insertion in these lines are predicted to decrease function. The results for Drosophila orthologs of C. elegans uso-1 and nekl-3 were also consistent across species. The exacerbation of Cesmn-1(lf) growth defects observed after uso-1(RNAi) or nekl-3(RNAi) knockdown was consistent with the suppression of DmSmn defects observed after over-expression of the cognate Drosophila genes. There was also good concordance for the effect of actinin orthologs across invertebrate species. The d00712 Drosophila insertion line likely drives over-expression of the Drosophila gene Actinin (Actn) and enhances DmSmn defects [26], [71], [72], while suppression of Cesmn-1(lf) growth defects by RNAi knockdown of C. elegans atn-1 was observed here.
For four genes, it is unclear if the results for Drosophila orthologs are concordant across species: atf-6, ape-1, dlc-1 and cash-1. For atf-6 and ape-1, the corresponding Drosophila transposons (d05057 and d05779) are inserted into the 1st intron of one of the two transcripts predicted for the orthologous Drosophila genes; accordingly, these transposons may perturb gene function or may drive over-expression of the predicted 2nd shorter transcript. For the genes with complex genetic interactions with Cesmn-1 (i.e. dlc-1 and cash-1), the function of Drosophila orthologs ctp and CKA are likely decreased by Drosophila insertion lines f02345 and f04448, which suppressed and enhanced DmSmn defects, respectively [26]. Overall, six of ten genes that modified DmSmn growth defects are clearly concordant with the C. elegans growth data, suggesting conserved roles as SMN loss of function modifiers.
Orthologs of three Drosophila genes modified Cesmn-1(lf) neuromuscular defects
C. elegans orthologs of DmSmn modifier genes identified in the previous Drosophila screen [26] were also rescreened using the pharyngeal pumping assay. We found that RNAi knockdown daf-4 enhanced Cesmn-1(lf) pharyngeal pumping defects, while knockdown of kncl-2 or nhr-25 suppressed Cesmn-1(lf) pumping defects (Table 3, rows 3 through 5).

daf-4 encodes one of the C. elegans TGF-beta receptor subunits orthologous to Drosophila Wit (Witless). In C. elegans, daf-4 and TGF-beta/Dpp pathway function is required for cell specification at numerous stages and for transit through the stress-resistant, long-lived dauer stage [73]. RNAi knockdown of daf-4 exacerbated Cesmn-1(lf) pumping defects, consistent with the effect of TGF-beta pathway manipulation in Drosophila [26].
RNAi knockdown of two C. elegans genes diminished Cesmn-1(lf) pumping defects: kcnl-2 and nhr-25. kcnl-2 encodes a likely C. elegans SK channel subunit and nhr-25 is one of the two C. elegans proteins most similar to Drosophila Usp (Ultraspiracle). No clear ortholog of Usp is found in the C. elegans genome. The corresponding Drosophila d00712 transposon insertion line likely drives over-expression of Usp resulting in enhancement of DmSmn defects [26]. This is consistent with C. elegans results. By contrast, the impact of SK/kcnl-2 loss in growth versus pumping assays is discordant. The d03336 transposon insertion is located in the SK gene, likely perturbs SK function, and enhances DmSmn growth and Drosophila NMJ defects [26]. This is consistent with kcnl-2(RNAi) enhancement of C. elegans growth defects described above. The suppression of Cesmn-1(lf) pumping defects observed here after kcnl-2 knockdown may reflect differences in the requirement for kcnl-2 function in neuromuscular tissue and/or the relative inefficiency of RNAi knockdown in neurons.
Modifier gene interactions implicate specific pathways critical in neurodegenerative disease
To address the specificity of the invertebrate SMN modifier genes, the impact of their RNAi knockdown was examined on an unrelated pharyngeal pumping defective strain. Loss of egl-30 (ad805), which perturbs Gqα function in C. elegans, decreases their pharyngeal pumping rates [74]. RNAi knockdown did not significantly alter egl-30 pharyngeal pumping rates for any modifier gene (Table S3), suggesting that these genes are likely specific modifiers of SMN loss of function defects.
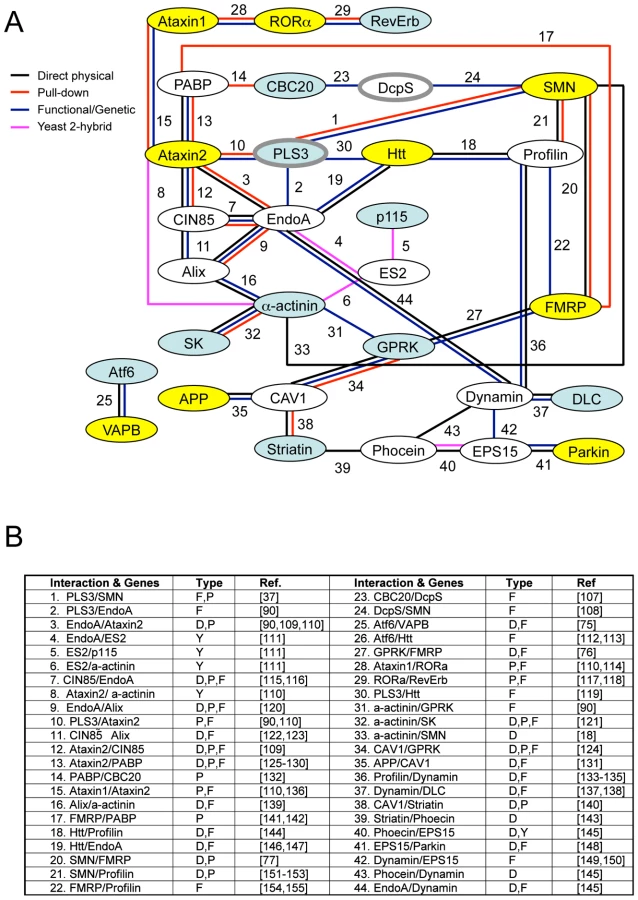
Combined the results described here define eleven conserved genes that modify invertebrate SMN ortholog function in at least one assay in both C. elegans and Drosophila (summarized in Table 4). A subset of these cross-species modifier genes interact, directly or indirectly, with previously described neurological or neuromuscular disease proteins suggesting common neurodegenerative pathways may be at work (i.e. ATF6 with VAPB/ALS8 or GPRK2 and SMN1 with FMRP) [75]–[77]. To determine if specific cellular mechanisms could be implicated in SMN loss of function pathology, the published literature and public databases were examined for physical and/or functional interactions between cross-species SMN modifier genes, SMN and neuromuscular disease genes. A protein/genetic interaction map was assembled and is presented in Figure 7 with references. We note that genes implicated in endocytosis and mRNA translational regulation unexpectedly predominate in this interaction map. These two cellular pathways may be pertinent to SMN loss of function pathology.

Discussion
Enormous effort over the last few decades has resulted in the successful identification of numerous neurodegenerative disease genes and the proteins they encode. However, in many cases there remains considerable controversy as to why perturbation of these genes results in neurodegeneration [78]–[81]. SMN plays a well-described and ubiquitous role in the Gemin complex and snRNP assembly [8]–[10], yet SMA specifically affects neuromuscular function, motorneuron survival, and leads to muscle atrophy. Given this neuromuscular specificity, it seems likely that loss of SMN function impacts cellular pathways outside of the Gemin complex. In addition, given the complexity of cellular signaling pathways, genetic pathways that are not directly involved in SMN activity may impact SMN loss of function pathology. To identify SMN modifier pathways, we have used a genetic approach. Unbiased genetic screens are powerful tools as they utilize functional criteria for the identification of genes critical for cellular function. In the case of SMN loss of function, genetic screens can reveal conserved genes and pathways that are important for neuromuscular dysfunction and pathology independent of initial assumptions about the roles of SMN in neurons and muscles. The identification of hitherto unsuspected molecular pathways that modulate SMN neuropathology, directly or indirectly, is expected to widen the range of targets for SMA therapy development.
Conserved genes that modify SMN loss of function defects in disparate species likely represent pathways that are important for SMN loss of function defects or pathology. C. elegans and Drosophila models have been used here to identify SMN loss of function modifier genes that ‘cross species’. It is difficult to estimate how many modifiers of SMN loss of function were missed in the genome-wide C. elegans RNAi screen. ‘Growth’ encompasses a variety of factors; slow progression through the larvae stages, reduced growth in the adult stage, longevity, body size, different culture format (liquid versus plates), or a combination of these. Additionally, identification of genetic modifiers for a null allele can be more challenging as compared to identification of genetic modifiers for partial loss of function alleles [82]. No genetic screen can identify all modifier genes pertinent to a pathway and important players can be missed (e.g. miRNAs). Despite this, there is excellent concordance of modifier gene action in C. elegans and Drosophila. In most cases, genes that enhanced SMN loss of function defects in Drosophila also enhanced SMN loss of function defects in C. elegans and vice versa. This concordance suggests that the genetic relationships between SMN and these modifier genes are conserved across species. Orthologous genes are likely also important in SMN loss of function pathology in vertebrate species, as suggested by other invertebrate modifier screens that have identified conserved human disease-related genes and/or functional pathways [83]–[88].
Thus far, there are only two published human SMA modifier genes: Plastin 3 (PLS3) and SMN2. The role of SMN2 is clear as it provides a modicum of functional SMN protein. However, the role of PLS3 in SMA is controversial and it is unclear how PLS3 levels might modulate severity in SMA patients [37], [43]. We find that invertebrate PLS3 orthologs act as modifiers in C. elegans and Drosophila models. This cross-species interaction of PLS3 and SMN both increases confidence in the invertebrate models and suggests that plastin-associated pathways are important for SMN function at a fundamental level in multiple contexts.
In the bioinformatic analysis presented in Figure 7, we independently identified two cellular pathways that connect multiple modifier genes with SMN: endocytosis and RNA processing/translational control pathways. Regarding the former, it is of note that the yeast ortholog of PLS3, Sac6p, is a key player in endocytosis and Sac6p levels are critical when expanded polyglutamine neurodegenerative disease proteins are expressed in this system [42], [89]–[91]. We suggest that 1) these two cellular mechanisms may be of particular importance in SMA pathology and 2) that unexpected and intimate connections exist between these two pathways. A pair of recently published studies found that the microRNA regulatory RISC complex and endocytosis are physically and functionally coupled in non-neuronal cells [92], [93]. Interestingly, the RISC complex also contains Gemin complex proteins; the function of the Gemin and RISC complexes may be related, directly or indirectly [20]. We speculate that in normal animals, physically coupling the seemingly disparate pathways of endocytosis and local translational regulation may help coordinate synaptic activity and receptor signaling with protein translation during both synaptic development and neuron maintenance [94]. Defects in endocytosis have been suggested previously to play a pivotal role in neurodegenerative diseases in numerous scenarios. In such diseases, including SMA, perturbation of endocytosis may result in RNA translational control defects, or vice versa [92], [93]. A recent study has demonstrated impaired synaptic vesicle release at the NMJs in severe SMA mice consistent with defects in synaptic vesicle endocytosis/recycling and/or defects in active zone organization [94]. Further studies are warranted to ascertain the interdependence of endocytosis with translational control pathways and to explore the relevance of these pathways in neurodegenerative disease.
Materials and Methods
Genetic analysis
Cesmn-1(lf) homozygous strains cannot be maintained due to infertility; hT2(lethal)[myo-2p::GFP]/Cesmn-1(lf) (hT2[bli-4(e937) let-?(q782)qIs48] (I;III)) animals are fertile and were maintained using standard techniques [95]. The lack of homologous pairing for the rearranged chromosomes LGI and LGIII in hT2 animals likely results in increased maternal/zygotic expression of Cesmn-1 and other balanced genes [96]. As expected, we found that the progeny of hT2 animals were relatively resistant to Cesmn-1(RNAi) compared to wild type control strains in our assays (data not shown). Consequently, to keep the genetic background invariant, all animals were tested herein were the progeny of hT2(lethal)[myo-2p::GFP] parents. The use of RNAi sensitive C. elegans mutant strains was avoided as their behavior is not normal in many assays (Hart, unpublished observations) and because SMN complex/Sm proteins have been implicated in miRNA pathways [20], [97], [98]. We note that RNAi knockdown is not always effective. To control for genetic background effects, animals tested in these studies were either heterozygous for hT2 balancer chromosome or progeny of hT2 parents unless otherwise noted.
plst-1(tm4255) animals were obtained from the Japanese National Bioresource Project and were backcrossed four times before further study. The tm4255 allele is a 368 base pair deletion that removes one of the calponin-like, actin-binding homology (CH) domains; plst-1(tm4255) is likely a partial loss of function allele. To test the genetic interaction of plst-1 with Cesmn-1, the backcrossed plst-1(tm4255) allele was used to create a double mutant with Cesmn-1(lf). The flp-4(yn35) deletion allele was isolated by PCR-based screening of EMS-mutagenized animals. The yn35 allele is a 928 base pair deletion that removes exon 3 of flp-4 gene along with 5′ sequences (flanking sequences, ttctgaaaaacttttaataa and agctcgccgagccgagtctt) [66]. The grk-2(rt97) loss of function allele was previously characterized [60].
Drosophila stocks were maintained on standard cornmeal/yeast/molasses/agar medium at 25°C. The mutations of Smn73Ao and Smnf01109 have been described previously [25]. Cbp20e02787 is a Piggy-Bac insertion mutation from the Exelixis collection. The insertion location is 5′ upstream and adjacent to the start codon of the Cbp20 transcript. FmrfKG1300 and Fim alleles are loss of function alleles (Flybase). The line d03334 may have an unlinked lethal mutation on another chromosome. Fimd02114 and SmnRNAi; Fimd02114 have Tubulin:Gal4 in the background; this Gal4 transgene does not alter Smn defects (data not shown).
Bioinformatics
C. elegans orthologs of Drosophila and human genes were identified by BLAST searching at NCBI. When a clear ortholog was not identified by reciprocal BLAST analysis, the most similar C. elegans genes were generally tested. plst-1 corresponds to exons of predicted adjacent genes Y104H12BR.1 and Y104H12BL.1 based on similarity searching. T02G5.3 corresponds to exons of T02G5.3, T02G5.2, and T02G5.1 based on high-throughput cDNA sequencing and gene prediction programs [99]. New gene predictions have been reported to Wormbase. To assemble the interaction map in Figure 7, literature pertaining to each modifier gene was examined at NCBI, AceView, C. elegans and yeast on-line databases (Wormbase and SGD) to identify functional or direct interactions between modifier genes and neurodegenerative disease genes.
Construction of RNAi feeding clones for B0432.13, dhs-22 and ugt-49
The L4440 vector [47] was used to clone PCR products corresponding to B0432.13, dhs-22 and ugt-49 genes. Plasmids were transformed into the bacterial strain HT115(DE3) [47], [49]. Primers used for cloning were: B0432.13 forward 5′-acaagctctcgacatcgctg-3′, reverse 5′ - ttaatcgccgcatcctcttg -3′; dhs-22 forward 5′-tatgctgtgcagaagcgaag-3′, reverse 5′-ctgcttgattcctggtgtattc-3′; ugt-49 forward 5′-acgtggatgtagctgaatgg-3′, reverse 5′ - acgtgaagaacagcaacgaac-3′.
C. elegans growth studies
For analysis of modifier genes, animals were reared for two generations/5 days on plates spread with bacterial RNAi strains from the Ahringer or Vidal RNAi libraries [53]. RNAi clones corresponding to modifier genes in Table 4 were sequenced to confirm accuracy. The hlh-4(RNAi) clone in the feeding library was incorrect. A Cesmn-1(ok355);hlh-4(tm604) double mutant strain was generated. hlh-4(tm604) did not affect the pharyngeal pumping rates of Cesmn-(lf) (data not shown) and hlh-4 was excluded from further analysis. Length and GFP fluorescence was determined using the COPAS Biosorter (Union Biometrica, Holliston, MA) and the percentage of large animals was determined for each genotype [51]. Three to six independent determinations were undertaken for each genotype/RNAi culture. Significant changes from empty(RNAi) were calculated for each RNAi/genotype using the two-tailed Mann-Whitney U test.
C. elegans pharyngeal pumping
The average pharyngeal pumping rates of animals were determined after 3 days (at 25, 25 and 20°C) post-hatching on empty vector (empty(RNAi)) or candidate gene RNAi bacterial feeding strains. Animals were videotaped while feeding for 10 seconds with an AxioCam ICc1 camera on a Zeiss Stemi SV11 at 20 to 66× magnification. Movies were slowed before counting pumping rates. Pharyngeal grinder movements in any axis were scored as a pumping event. Average pumping rates (± standard error of the mean, S.E.M) for each genotype/treatment were calculated independently in two to four separate experiments. The percent change in pumping rate on empty vector versus candidate gene RNAi was determined for each trial for both Cesmn-1(lf) homozygous and +/Cesmn-1(lf) heterozygous animals and used to calculate the mean, S.E.M, and significance.
C. elegans genome-wide RNAi screen
hT2(bli-4(e937) let-?(q782) qIs48[myo-2p::GFP]) (I;III) animals were reared in liquid cultures in a 96-well plate format on RNAi feeding strains [53]. At least two independent cultures corresponding to each C. elegans RNAi feeding clone were established. Concentrated dsRNA expressing bacteria was added to cultures as necessary to prevent starvation. Cultures were maintained for 8 days at 25°C to generate sufficient animals for analysis. Length and fluorescence were determined using the COPAS BioSorter (Union Biometrica, Holliston, MA). Data was exported to Excel (Microsoft Corp.) for analysis. Thirty-one clones were identified that modified the average length of Cesmn-1(lf) animals relative to +/Cesmn-1(lf) siblings in both trials. Four of these genes altered Cesmn-1(lf) size relative to +/Cesmn-1(lf) siblings in at least 40% of subsequent trials and these were selected as candidate modifier genes for neuromuscular analysis as described in the text.
Drosophila pupal lethality
Three males and three virgin females were placed on fresh food at 25°C on day 1. Eggs were collected for next 2 days (Set 1), and the parents transferred to fresh food. Eggs were collected for another 2 days (Set 2), and the parents discarded. The F1 animals were scored after 15 days, - on the 16th day for the first set, and the 19th day for the second set, from day 1. White pupae were scored as early stage death and black pupae were scored as late stage death. Control crosses of tubGAL4:FL26B(Smn RNAi) out-crossed to the wild type strain were used as a control for every experimental set. Significance was determined by Chi-square analysis.
Drosophila NMJ analysis
Primary antibodies were used at the following dilutions: monoclonal anti-DLG (1∶500) (Developmental Studies Hybridoma Bank), polyclonal anti-Synaptotagmin (1∶1000) (a gift from Hugo Bellen). FITC - (1∶40) and Cy5 - (1∶40) conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Jackson Immunoresearch Laboratories. Anti-Disc large used at 1∶100 (Hybridoma) and anti-HRP used at 1∶1000 (Cappell). 3rd instar larvae were dissected and fixed for 5 minutes in Bouin's fixative. Stained specimens were mounted in FluoroGuard Antifade Reagent (Bio-Rad), and images were obtained with a Zeiss LSM510 confocal microscope. Bouton numbers were counted based on the Discs large and Synaptotagmin staining in the A2 segment between muscles 6 and 7 or muscle 4 as indicated. The ratio of muscle area for the various genotypes was normalized to wild type. At least 10–12 animals of each genotype were dissected for the bouton analysis. The ANOVA multiple comparison test was used for statistical analysis.
Supporting Information
Zdroje
1. LefebvreS
BurglenL
ReboulletS
ClermontO
BurletP
1995 Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80 155 165
2. BodaB
MasC
GiudicelliC
NepoteV
GuimiotF
2004 Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur J Hum Genet 12 729 737
3. MonaniUR
McPhersonJD
BurghesAH
1999 Promoter analysis of the human centromeric and telomeric survival motor neuron genes (SMNC and SMNT). Biochim Biophys Acta 1445 330 336
4. PearnJ
1980 Classification of spinal muscular atrophies. Lancet 1 919 922
5. CusinV
ClermontO
GerardB
ChantereauD
ElionJ
2003 Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 40 e39
6. McAndrewPE
ParsonsDW
SimardLR
RochetteC
RayPN
1997 Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 60 1411 1422
7. OginoS
LeonardDG
RennertH
EwensWJ
WilsonRB
2002 Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet 110 301 307
8. GubitzAK
FengW
DreyfussG
2004 The SMN complex. Exp Cell Res 296 51 56
9. OhnT
KedershaN
HickmanT
TisdaleS
AndersonP
2008 A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat Cell Biol 10 1224 1231
10. HuaY
ZhouJ
2004 Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett 572 69 74
11. FanL
SimardLR
2002 Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet 11 1605 1614
12. RossollW
JablonkaS
AndreassiC
KroningAK
KarleK
2003 Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163 801 812
13. FrancisJW
SandrockAW
BhidePG
VonsattelJP
BrownRHJr
1998 Heterogeneity of subcellular localization and electrophoretic mobility of survival motor neuron (SMN) protein in mammalian neural cells and tissues. Proc Natl Acad Sci U S A 95 6492 6497
14. GiavazziA
SetolaV
SimonatiA
BattagliaG
2006 Neuronal-specific roles of the survival motor neuron protein: evidence from survival motor neuron expression patterns in the developing human central nervous system. J Neuropathol Exp Neurol 65 267 277
15. PagliardiniS
GiavazziA
SetolaV
LizierC
Di LucaM
2000 Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet 9 47 56
16. GennarelliM
LucarelliM
CaponF
PizzutiA
MerliniL
1995 Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun 213 342 348
17. CoovertDD
LeTT
McAndrewPE
StrasswimmerJ
CrawfordTO
1997 The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6 1205 1214
18. RajendraTK
GonsalvezGB
WalkerMP
ShpargelKB
SalzHK
2007 A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol 176 831 841
19. WalkerMP
RajendraTK
SaievaL
FuentesJL
PellizzoniL
2008 The SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum Mol Genet
20. MourelatosZ
DostieJ
PaushkinS
SharmaA
CharrouxB
2002 miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16 720 728
21. PellizzoniL
BacconJ
CharrouxB
DreyfussG
2001 The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol 11 1079 1088
22. WhiteheadSE
JonesKW
ZhangX
ChengX
TernsRM
2002 Determinants of the interaction of the spinal muscular atrophy disease protein SMN with the dimethylarginine-modified box H/ACA small nucleolar ribonucleoprotein GAR1. J Biol Chem 277 48087 48093
23. Cifuentes-DiazC
FrugierT
TizianoFD
LaceneE
RoblotN
2001 Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol 152 1107 1114
24. ShafeyD
CotePD
KotharyR
2005 Hypomorphic Smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp Cell Res 311 49 61
25. ChanYB
Miguel-AliagaI
FranksC
ThomasN
TrulzschB
2003 Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 12 1367 1376
26. ChangHC
DimlichDN
YokokuraT
MukherjeeA
KankelMW
2008 Modeling spinal muscular atrophy in Drosophila. PLoS ONE 3 e3209 doi:10.1371/journal.pone.0003209
27. McWhorterML
MonaniUR
BurghesAH
BeattieCE
2003 Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162 919 931
28. KariyaS
ParkGH
Maeno-HikichiY
LeykekhmanO
LutzC
2008 Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17 2552 2569
29. LorsonCL
AndrophyEJ
2000 An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet 9 259 265
30. MonaniUR
CoovertDD
BurghesAH
2000 Animal models of spinal muscular atrophy. Hum Mol Genet 9 2451 2457
31. MonaniUR
LorsonCL
ParsonsDW
PriorTW
AndrophyEJ
1999 A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 8 1177 1183
32. LefebvreS
BurletP
LiuQ
BertrandyS
ClermontO
1997 Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16 265 269
33. FeldkotterM
SchwarzerV
WirthR
WienkerTF
WirthB
2002 Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70 358 368
34. WirthB
HerzM
WetterA
MoskauS
HahnenE
1999 Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 64 1340 1356
35. LefebvreS
BurglenL
FrezalJ
MunnichA
MelkiJ
1998 The role of the SMN gene in proximal spinal muscular atrophy. Hum Mol Genet 7 1531 1536
36. BurghesAH
1997 When is a deletion not a deletion? When it is converted. Am J Hum Genet 61 9 15
37. OpreaGE
KroberS
McWhorterML
RossollW
MullerS
2008 Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320 524 527
38. BretscherA
1981 Fimbrin is a cytoskeletal protein that crosslinks F-actin in vitro. Proc Natl Acad Sci U S A 78 6849 6853
39. BretscherA
WeberK
1980 Fimbrin, a new microfilament-associated protein present in microvilli and other cell surface structures. J Cell Biol 86 335 340
40. GlenneyJRJr
KaulfusP
MatsudairaP
WeberK
1981 F-actin binding and bundling properties of fimbrin, a major cytoskeletal protein of microvillus core filaments. J Biol Chem 256 9283 9288
41. AdamsAE
BotsteinD
DrubinDG
1989 A yeast actin-binding protein is encoded by SAC6, a gene found by suppression of an actin mutation. Science 243 231 233
42. KublerE
RiezmanH
1993 Actin and fimbrin are required for the internalization step of endocytosis in yeast. EMBO J 12 2855 2862
43. BowermanM
AndersonCL
BeauvaisA
BoylPP
WitkeW
2009 SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci 42 66 74
44. BrieseM
EsmaeiliB
FrabouletS
BurtEC
ChristodoulouS
2009 Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet 18 97 104
45. Miguel-AliagaI
CulettoE
WalkerDS
BaylisHA
SattelleDB
1999 The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum Mol Genet 8 2133 2143
46. McKimKS
PetersK
RoseAM
1993 Two types of sites required for meiotic chromosome pairing in Caenorhabditis elegans. Genetics 134 749 768
47. TimmonsL
FireA
1998 Specific interference by ingested dsRNA. Nature 395 854
48. KamathRS
Martinez-CamposM
ZipperlenP
FraserAG
AhringerJ
2001 Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2 RESEARCH0002
49. TimmonsL
CourtDL
FireA
2001 Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263 103 112
50. RualJF
CeronJ
KorethJ
HaoT
NicotAS
2004 Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14 2162 2168
51. PulakR
2006 Techniques for analysis, sorting, and dispensing of C. elegans on the COPAS flow-sorting system. Methods Mol Biol 351 275 286
52. HuangLS
SternbergPW
2006 Genetic dissection of developmental pathways. WormBook 1 19
53. KamathRS
AhringerJ
2003 Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30 313 321
54. LallS
PianoF
DavisRE
2005 Caenorhabditis elegans decapping proteins: localization and functional analysis of Dcp1, Dcp2, and DcpS during embryogenesis. Mol Biol Cell 16 5880 5890
55. AveryL
1993 The genetics of feeding in Caenorhabditis elegans. Genetics 133 897 917
56. SulstonJE
HorvitzHR
1977 Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56 110 156
57. SulstonJE
SchierenbergE
WhiteJG
ThomsonJN
1983 The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100 64 119
58. SimmerF
MoormanC
van der LindenAM
KuijkE
van den BerghePV
2003 Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol 1 e12 doi:10.1371/journal.pbio.0000012
59. KennedyS
WangD
RuvkunG
2004 A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 427 645 649
60. FukutoHS
FerkeyDM
ApicellaAJ
LansH
SharmeenT
2004 G protein-coupled receptor kinase function is essential for chemosensation in C. elegans. Neuron 42 581 593
61. BarrettPL
FlemingJT
GobelV
2004 Targeted gene alteration in Caenorhabditis elegans by gene conversion. Nat Genet 36 1231 1237
62. Gengyo-AndoK
MitaniS
2000 Characterization of mutations induced by ethyl methanesulfonate, UV, and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem Biophys Res Commun 269 64 69
63. GilchristEJ
O'NeilNJ
RoseAM
ZetkaMC
HaughnGW
2006 TILLING is an effective reverse genetics technique for Caenorhabditis elegans. BMC Genomics 7 262
64. JansenG
HazendonkE
ThijssenKL
PlasterkRH
1997 Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat Genet 17 119 121
65. LesaGM
2006 Isolation of Caenorhabditis elegans gene knockouts by PCR screening of chemically mutagenized libraries. Nat Protoc 1 2231 2240
66. LiuT
KimK
LiC
BarrMM
2007 FMRFamide-like neuropeptides and mechanosensory touch receptor neurons regulate male sexual turning behavior in Caenorhabditis elegans. J Neurosci 27 7174 7182
67. LiC
KimK
2008 Neuropeptides. WormBook 1 36
68. MountSM
SalzHK
2000 Pre-messenger RNA processing factors in the Drosophila genome. J Cell Biol 150 F37 44
69. LaskoP
2000 The Drosophila melanogaster genome: translation factors and RNA binding proteins. J Cell Biol 150 F51 56
70. NambuJR
Murphy-ErdoshC
AndrewsPC
FeistnerGJ
SchellerRH
1988 Isolation and characterization of a Drosophila neuropeptide gene. Neuron 1 55 61
71. RoulierEM
FyrbergC
FyrbergE
1992 Perturbations of Drosophila alpha-actinin cause muscle paralysis, weakness, and atrophy but do not confer obvious nonmuscle phenotypes. J Cell Biol 116 911 922
72. FyrbergE
KellyM
BallE
FyrbergC
ReedyMC
1990 Molecular genetics of Drosophila alpha-actinin: mutant alleles disrupt Z disc integrity and muscle insertions. J Cell Biol 110 1999 2011
73. InoueT
ThomasJH
2000 Targets of TGF-beta signaling in Caenorhabditis elegans dauer formation. Dev Biol 217 192 204
74. BrundageL
AveryL
KatzA
KimUJ
MendelJE
1996 Mutations in a C. elegans Gqalpha gene disrupt movement, egg laying, and viability. Neuron 16 999 1009
75. GkogkasC
MiddletonS
KremerAM
WardropeC
HannahM
2008 VAPB interacts with and modulates the activity of ATF6. Hum Mol Genet 17 1517 1526
76. WangH
WuLJ
KimSS
LeeFJ
GongB
2008 FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59 634 647
77. PiazzonN
RageF
SchlotterF
MoineH
BranlantC
2008 In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem 283 5598 5610
78. BauerPO
NukinaN
2009 The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110 1737 1765
79. CrawfordTO
PardoCA
1996 The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3 97 110
80. GoedertM
SpillantiniMG
2006 A century of Alzheimer's disease. Science 314 777 781
81. LangAE
LozanoAM
1998 Parkinson's disease. First of two parts. N Engl J Med 339 1044 1053
82. JorgensenEM
MangoSE
2002 The art and design of genetic screens: Caenorhabditis elegans. Nat Rev Genet 3 356 369
83. DimitriadiM
HartAC
2010 Neurodegenerative disorders: Insights from the nematode Caenorhabditis elegans. Neurobiol Dis
84. FiuzaUM
AriasAM
2007 Cell and molecular biology of Notch. J Endocrinol 194 459 474
85. KimSK
2007 Common aging pathways in worms, flies, mice and humans. J Exp Biol 210 1607 1612
86. LeichtDT
BalanV
KaplunA
Singh-GuptaV
KaplunL
2007 Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta 1773 1196 1212
87. SchlegelA
StainierDY
2007 Lessons from “lower” organisms: what worms, flies, and zebrafish can teach us about human energy metabolism. PLoS Genet 3 e199 doi:10.1371/journal.pgen.0030199
88. SilvermanGA
LukeCJ
BhatiaSR
LongOS
VeticaAC
2009 Modeling molecular and cellular aspects of human disease using the nematode Caenorhabditis elegans. Pediatr Res 65 10 18
89. KaksonenM
ToretCP
DrubinDG
2005 A modular design for the clathrin - and actin-mediated endocytosis machinery. Cell 123 305 320
90. RalserM
NonhoffU
AlbrechtM
LengauerT
WankerEE
2005 Ataxin-2 and huntingtin interact with endophilin-A complexes to function in plastin-associated pathways. Hum Mol Genet 14 2893 2909
91. Singer-KrugerB
NemotoY
DaniellL
Ferro-NovickS
De CamilliP
1998 Synaptojanin family members are implicated in endocytic membrane traffic in yeast. J Cell Sci 111 ( Pt 22) 3347 3356
92. GibbingsDJ
CiaudoC
ErhardtM
VoinnetO
2009 Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 11 1143 1149
93. LeeYS
PressmanS
AndressAP
KimK
WhiteJL
2009 Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol 11 1150 1156
94. KongL
WangX
ChoeDW
PolleyM
BurnettBG
2009 Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29 842 851
95. BrennerS
1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
96. BeanCJ
SchanerCE
KellyWG
2004 Meiotic pairing and imprinted X chromatin assembly in Caenorhabditis elegans. Nat Genet 36 100 105
97. BilinskiSM
JaglarzMK
SzymanskaB
EtkinLD
KlocM
2004 Sm proteins, the constituents of the spliceosome, are components of nuage and mitochondrial cement in Xenopus oocytes. Exp Cell Res 299 171 178
98. DostieJ
MourelatosZ
YangM
SharmaA
DreyfussG
2003 Numerous microRNPs in neuronal cells containing novel microRNAs. RNA 9 180 186
99. HillierLW
ReinkeV
GreenP
HirstM
MarraMA
2009 Massively parallel sequencing of the polyadenylated transcriptome of C. elegans. Genome Res 19 657 666
100. TownendJ
2002 Practical statistics for environmental and biological scientists Chichester, UK John Wiley and Sons Ltd
101. GunawardenaS
GoldsteinLS
2001 Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32 389 401
102. CaoYJ
DreixlerJC
CoueyJJ
HouamedKM
2002 Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 449 47 54
103. BensimonG
LacomblezL
MeiningerV
1994 A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330 585 591
104. CouratierP
SindouP
EsclaireF
LouvelE
HugonJ
1994 Neuroprotective effects of riluzole in ALS CSF toxicity. Neuroreport 5 1012 1014
105. HaddadH
Cifuentes-DiazC
MiroglioA
RoblotN
JoshiV
2003 Riluzole attenuates spinal muscular atrophy disease progression in a mouse model. Muscle Nerve 28 432 437
106. NishimuraAL
Mitne-NetoM
SilvaHC
Richieri-CostaA
MiddletonS
2004 A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75 822 831
107. ShenV
LiuH
LiuSW
JiaoX
KiledjianM
2008 DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. RNA 14 1132 1142
108. SinghJ
SalciusM
LiuSW
StakerBL
MishraR
2008 DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem Biol 3 711 722
109. NonisD
SchmidtMH
van de LooS
EichF
DikicI
2008 Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal 20 1725 1739
110. LimJ
HaoT
ShawC
PatelAJ
SzaboG
2006 A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125 801 814
111. LiS
ArmstrongCM
BertinN
GeH
MilsteinS
2004 A map of the interactome network of the metazoan C. elegans. Science 303 540 543
112. JeongH
ThenF
MeliaTJJr
MazzulliJR
CuiL
2009 Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137 60 72
113. JiaK
HartAC
LevineB
2007 Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3 21 25
114. SerraHG
DuvickL
ZuT
CarlsonK
StevensS
2006 RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127 697 708
115. PetrelliA
GilestroGF
LanzardoS
ComoglioPM
MigoneN
2002 The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature 416 187 190
116. KanekoT
MaedaA
TakefujiM
AoyamaH
NakayamaM
2005 Rho mediates endocytosis of epidermal growth factor receptor through phosphorylation of endophilin A1 by Rho-kinase. Genes Cells 10 973 987
117. GuillaumondF
DardenteH
GiguereV
CermakianN
2005 Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythms 20 391 403
118. FormanBM
ChenJ
BlumbergB
KliewerSA
HenshawR
1994 Cross-talk among ROR alpha 1 and the Rev-erb family of orphan nuclear receptors. Mol Endocrinol 8 1253 1261
119. FreemanJL
PitcherJA
LiX
BennettV
LefkowitzRJ
2000 alpha-Actinin is a potent regulator of G protein-coupled receptor kinase activity and substrate specificity in vitro. FEBS Lett 473 280 284
120. Chatellard-CausseC
BlotB
CristinaN
TorchS
MissottenM
2002 Alix (ALG-2-interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J Biol Chem 277 29108 29115
121. LuL
ZhangQ
TimofeyevV
ZhangZ
YoungJN
2007 Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res 100 112 120
122. ChenB
BorinsteinSC
GillisJ
SykesVW
BoglerO
2000 The glioma-associated protein SETA interacts with AIP1/Alix and ALG-2 and modulates apoptosis in astrocytes. J Biol Chem 275 19275 19281
123. SchmidtMH
HoellerD
YuJ
FurnariFB
CaveneeWK
2004 Alix/AIP1 antagonizes epidermal growth factor receptor downregulation by the Cbl-SETA/CIN85 complex. Mol Cell Biol 24 8981 8993
124. CarmanCV
LisantiMP
BenovicJL
1999 Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem 274 8858 8864
125. RalserM
AlbrechtM
NonhoffU
LengauerT
LehrachH
2005 An integrative approach to gain insights into the cellular function of human ataxin-2. J Mol Biol 346 203 214
126. LessingD
BoniniNM
2008 Polyglutamine genes interact to modulate the severity and progression of neurodegeneration in Drosophila. PLoS Biol 6 e29 doi:10.1371/journal.pbio.0060029
127. CioskR
DePalmaM
PriessJR
2004 ATX-2, the C. elegans ortholog of ataxin 2, functions in translational regulation in the germline. Development 131 4831 4841
128. SatterfieldTF
PallanckLJ
2006 Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet 15 2523 2532
129. NonhoffU
RalserM
WelzelF
PicciniI
BalzereitD
2007 Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18 1385 1396
130. KozlovG
TrempeJF
KhaleghpourK
KahvejianA
EkielI
2001 Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc Natl Acad Sci U S A 98 4409 4413
131. IkezuT
TrappBD
SongKS
SchlegelA
LisantiMP
1998 Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein. J Biol Chem 273 10485 10495
132. MassenetS
PellizzoniL
PaushkinS
MattajIW
DreyfussG
2002 The SMN complex is associated with snRNPs throughout their cytoplasmic assembly pathway. Mol Cell Biol 22 6533 6541
133. GareusR
Di NardoA
RybinV
WitkeW
2006 Mouse profilin 2 regulates endocytosis and competes with SH3 ligand binding to dynamin 1. J Biol Chem 281 2803 2811
134. SchaferDA
WeedSA
BinnsD
KarginovAV
ParsonsJT
2002 Dynamin2 and cortactin regulate actin assembly and filament organization. Curr Biol 12 1852 1857
135. WitkeW
PodtelejnikovAV
Di NardoA
SutherlandJD
GurniakCB
1998 In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J 17 967 976
136. Al-RamahiI
PerezAM
LimJ
ZhangM
SorensenR
2007 dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet 3 e234 doi:10.1371/journal.pgen.0030234
137. Ghosh-RoyA
DesaiBS
RayK
2005 Dynein light chain 1 regulates dynamin-mediated F-actin assembly during sperm individualization in Drosophila. Mol Biol Cell 16 3107 3116
138. Navarro-LeridaI
Martinez MorenoM
RoncalF
GavilanesF
AlbarJP
2004 Proteomic identification of brain proteins that interact with dynein light chain LC8. Proteomics 4 339 346
139. PanS
WangR
ZhouX
HeG
KoomenJ
2006 Involvement of the conserved adaptor protein Alix in actin cytoskeleton assembly. J Biol Chem 281 34640 34650
140. GaillardS
BartoliM
CastetsF
MonneronA
2001 Striatin, a calmodulin-dependent scaffolding protein, directly binds caveolin-1. FEBS Lett 508 49 52
141. KhandjianEW
HuotME
TremblayS
DavidovicL
MazrouiR
2004 Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci U S A 101 13357 13362
142. GagneJP
BonicalziME
GagneP
OuelletME
HendzelMJ
2005 Poly(ADP-ribose) glycohydrolase is a component of the FMRP-associated messenger ribonucleoparticles. Biochem J 392 499 509
143. CastetsF
BartoliM
BarnierJV
BaillatG
SalinP
1996 A novel calmodulin-binding protein, belonging to the WD-repeat family, is localized in dendrites of a subset of CNS neurons. J Cell Biol 134 1051 1062
144. ShaoJ
WelchWJ
DiprosperoNA
DiamondMI
2008 Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol Cell Biol 28 5196 5208
145. BaillatG
GaillardS
CastetsF
MonneronA
2002 Interactions of phocein with nucleoside-diphosphate kinase, Eps15, and Dynamin I. J Biol Chem 277 18961 18966
146. SittlerA
WalterS
WedemeyerN
HasenbankR
ScherzingerE
1998 SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol Cell 2 427 436
147. QinZH
WangY
SappE
CuiffoB
WankerE
2004 Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci 24 269 281
148. FallonL
BelangerCM
CoreraAT
KontogianneaM
Regan-KlapiszE
2006 A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol 8 834 842
149. SalciniAE
HilliardMA
CroceA
ArbucciS
LuzziP
2001 The Eps15 C. elegans homologue EHS-1 is implicated in synaptic vesicle recycling. Nat Cell Biol 3 755 760
150. KrishnanKS
RikhyR
RaoS
ShivalkarM
MoskoM
2001 Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron 30 197 210
151. SharmaA
LambrechtsA
Hao leT
LeTT
SewryCA
2005 A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res 309 185 197
152. GiesemannT
Rathke-HartliebS
RothkegelM
BartschJW
BuchmeierS
1999 A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J Biol Chem 274 37908 37914
153. BowermanM
ShafeyD
KotharyR
2007 Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J Mol Neurosci 32 120 131
154. ReeveSP
BassettoL
GenovaGK
KleynerY
LeyssenM
2005 The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol 15 1156 1163
155. TessierCR
BroadieK
2008 Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135 1547 1557
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance