
Simultaneous Disruption of Two DNA Polymerases, Polη and Polζ, in Avian DT40 Cells Unmasks the Role of Polη in Cellular Response to Various DNA Lesions
Replicative DNA polymerases are frequently stalled by DNA lesions. The resulting replication blockage is released by homologous recombination (HR) and translesion DNA synthesis (TLS). TLS employs specialized TLS polymerases to bypass DNA lesions. We provide striking in vivo evidence of the cooperation between DNA polymerase η, which is mutated in the variant form of the cancer predisposition disorder xeroderma pigmentosum (XP-V), and DNA polymerase ζ by generating POLη−/−/POLζ−/− cells from the chicken DT40 cell line. POLζ−/− cells are hypersensitive to a very wide range of DNA damaging agents, whereas XP-V cells exhibit moderate sensitivity to ultraviolet light (UV) only in the presence of caffeine treatment and exhibit no significant sensitivity to any other damaging agents. It is therefore widely believed that Polη plays a very specific role in cellular tolerance to UV-induced DNA damage. The evidence we present challenges this assumption. The phenotypic analysis of POLη−/−/POLζ−/− cells shows that, unexpectedly, the loss of Polη significantly rescued all mutant phenotypes of POLζ−/− cells and results in the restoration of the DNA damage tolerance by a backup pathway including HR. Taken together, Polη contributes to a much wide range of TLS events than had been predicted by the phenotype of XP-V cells.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001151
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001151
Summary
Replicative DNA polymerases are frequently stalled by DNA lesions. The resulting replication blockage is released by homologous recombination (HR) and translesion DNA synthesis (TLS). TLS employs specialized TLS polymerases to bypass DNA lesions. We provide striking in vivo evidence of the cooperation between DNA polymerase η, which is mutated in the variant form of the cancer predisposition disorder xeroderma pigmentosum (XP-V), and DNA polymerase ζ by generating POLη−/−/POLζ−/− cells from the chicken DT40 cell line. POLζ−/− cells are hypersensitive to a very wide range of DNA damaging agents, whereas XP-V cells exhibit moderate sensitivity to ultraviolet light (UV) only in the presence of caffeine treatment and exhibit no significant sensitivity to any other damaging agents. It is therefore widely believed that Polη plays a very specific role in cellular tolerance to UV-induced DNA damage. The evidence we present challenges this assumption. The phenotypic analysis of POLη−/−/POLζ−/− cells shows that, unexpectedly, the loss of Polη significantly rescued all mutant phenotypes of POLζ−/− cells and results in the restoration of the DNA damage tolerance by a backup pathway including HR. Taken together, Polη contributes to a much wide range of TLS events than had been predicted by the phenotype of XP-V cells.
Introduction
DNA replication involves a rapid but fragile enzymatic mechanism that is frequently stalled by damage in the DNA template. To complete DNA replication, DNA lesions are bypassed by specialized DNA polymerases, a process called translesion synthesis (TLS) (reviewed in [1], [2]). A number of TLS polymerases, including Polη and Polζ, that are conserved throughout eukaryotic evolution, have been identified in yeast and mammals. Polη deficiency is responsible for a variant form of xeroderma pigmentosum (XP-V) [3], [4] that is characterized by UV photosensitivity and a predisposition to skin cancer (reviewed in [5]). Deficiency in Rev3, the catalytic subunit of Polζ, results in a considerably more severe phenotype, compared with Polη. In fact, Rev3 disruption is lethal to mouse embryogenesis [6]. Chicken DT40 cells deficient in Rev3 exhibit significant chromosome instability and hypersensitivity to a wide variety of DNA-damaging agents [7]–[10]. In addition to their role in TLS, both Polη and Polζ can contribute to homologous DNA recombination (HR) in DT40 cells [9], [11], [12].
Exposure to UV induces cyclobutane pyrimidine dimers (CPDs) and 6-4 UV photoproducts in DNA. While Polη can efficiently and accurately bypass CPDs [4], [13]–[15], no single DNA polymerase has been shown to be capable of effectively bypassing 6-4 UV photoproducts in vitro. This suggests that the coordinate use of more than one polymerase is required to bypass such damage in vivo. In support of this notion, several biochemical studies have suggested that lesion bypass can be effected by two sequential nucleotide incorporation events [16], [17]. For example, to bypass the 6-4 UV photoproduct, Polη inserts a nucleotide opposite the damage as a first step, followed by extension from the inserted nucleotide as a second step. This extension process has been shown to be catalyzed by yeast Polζ and by human Polκ [1], [2], [18]. The contribution of mammalian Polζ to the extension step remains elusive, because functional Polζ has not been purified [19], [20]. Recently, replication of episomal plasmid DNA carrying various lesions was analyzed in mammalian cell lines to define the role of each DNA polymerase in TLS past individual DNA lesions [15], [21], [22]. Shachar et al. suggest the sequential usage of Polη and Polζ in TLS past the cisPt-GG lesion [21], while results of others do not support this two-step TLS model [15], [23]. Indeed, evidence for the two-step model in the replication of chromosomal DNA has so far been lacking. By contrast, both human and yeast Polη can bypass CPDs effectively in vitro without extension polymerases [4].
Combining genetic tractability with a number of sensitive phenotypic assays, the chicken DT40 B lymphocyte cell line provides a unique opportunity to precisely analyze the role of individual DNA polymerases in TLS as well as in HR. The immunoglobulin loci of DT40 cells undergo constitutive diversification in culture by a combination of gene conversion (which depends on HR) and point mutation (which depends on TLS [24]). This diversification is driven by activation-induced deaminase (AID) [25], [26], which catalyzes the deamination of cytosine to generate uracil in the immunoglobulin loci. The uracil is then eliminated by uracil glycosylase to form abasic sites, which are thought to be the lesions that trigger bypass, either by gene conversion or by mutagenic translesion synthesis (reviewed in [27]). To study TLS in a different context, an episomal plasmid-based system was recently developed to examine the replication of a plasmid carrying site-specific lesions, in this case 6-4 UV photoproducts, in DT40 cells [28].
To investigate the functional interaction between Polη and Polζ in the DT40 cell line, we created POLη−/−/POLζ−/− DT40 cells (hereafter called polη/polζ cells). Unexpectedly, depletion of Polη in the polζ cells suppressed virtually all mutant phenotypes associated with the loss of Polζ, including genome instability and hypersensitivity to DNA-damaging agents. Furthermore, the reconstitution of POLη−/−/POLζ−/− cells with intact human Polη, but not the polymerase-deficient mutant carrying D115A/E116A substitutions, increased their hypersensitivity to DNA-damaging agents to the level of the POLζ−/− cells, indicating that Polη-dependent DNA synthesis is toxic in the absence of Polζ. Remarkably, this alleviation of the polζ phenotype was associated with the restoration of effective translesion synthesis. These data provide in vivo support for the two-step model of lesion bypass, with Polζ playing a critical role in the extension step following nucleotide incorporation by Polη.
Results
Deletion of Polη reversed the hypersensitivity of the polζ mutant to UV, ionizing radiation, MMS, and cisplatin
We generated polη/polζ cells by inactivating both REV3 alleles of the polη DT40 cells using a previously published gene-targeting strategy (Figure 1A) [9], [12]. The growth properties of the mutant cells were examined by measuring their growth rate and cell-cycle profile. As reported previously, the polη cells had a normal growth rate, whereas the polζ cells proliferated more slowly, exhibiting an increase in the sub-G1 fraction, indicative of spontaneous cell death during the cell cycle (Figure 1C). The loss of Polζ caused a significant increase in the number of spontaneous arising γH2AX foci, which represent replication collapse (Figure S1). Interestingly, deletion of POLη in the polζ cells reversed their growth retardation and reduced the rate of cell death (Figure 1B and 1C). Similarly, the number of spontaneous chromosomal aberrations was significantly reduced in polη/polζ cells, compared with polζ cells (Table 1). Ectopic expression of human Polη in polη/polζ cells diminished their growth rate to the level of polζ cells. This observation does not reflect general toxicity of the overexpressed human Polη, since its ectopic expression caused pronounced growth retardation only in the polη/polζ double mutant but not in wild-type cells (Figure S2). These observations indicate that the growth defect of polζ cells is dependent on the presence of Polη.


The sensitivity of polη/polζ cells to genotoxic stresses was evaluated using a colony formation assay. polη cells showed a mild sensitivity to UV but not to the other genotoxic stresses, in agreement with the phenotype of mammalian XP-V cells [29], [30]. In contrast, polζ cells showed a marked sensitivity to UV, ionizing radiation, cis-diaminedichloroplatinum-II (cisplatin), and methylmethane sulfonate (MMS) (Figure 2), as previously described [9]. The polη/polζ mutant cells were less sensitive to UV than were the polζ cells. Furthermore, this double mutant showed significantly increased tolerance to ionizing radiation, cisplatin, and MMS, compared with the polζ cells. This increased tolerance of the polη/polζ cells was reversed by ectopic expression of human Polη. To investigate whether this reversion depended on the polymerase activity of human Polη, we expressed human POLη cDNA carrying D115A/E116A mutations in the polη/polζ cells. These mutations in the catalytic site abolish polymerase activity (data not shown), as previously reported in yeast [31]. The expression of the mutant Polη had no effect on the sensitivity of the polη/polζ cells to the DNA-damaging agents (Figure 2), indicating that Polη-dependent DNA synthesis sensitizes polζ cells to these DNA-damaging agents.

polζ cells have a more prominent defect in TLS past abasic site than polη/polζ cells
We wished to investigate if Polη and Polζ could collaborate in TLS past specific types of DNA damage. To this end, we performed two sets of experiments: analysis of immunoglobulin hypermutation, which in DT40 provides a readout of the bypass of abasic sites [24], and analysis of the replication of a T-T (6-4) photoproduct on an episomal plasmid [28].
To induce Ig hypermutation, we overproduced AID in DT40 cells using a retrovirus vector [32], [33]. This vector drives the monocistronic expression of AID and green fluorescent protein (GFP), allowing a comparative assessment of the level of ectopic AID expression. At 24 hours after infection with the AID retrovirus, virtually all cells from each line displayed a strong GFP signal, indicating that the deficiency of Polη and Polζ did not affect the expression of AID (Figure 3A). However, a substantial fraction of the polζ cells died at day 3, and with the surviving polζ cells at day 10 showed a decrease level of GFP signals (Figure 3A and 3B). Furthermore, polζ cells, but not polη or polη/polζ cells, displayed prominent chromosomal breaks at day 3 post-infection (Figure 3C). Since the break sites on the chromosome were randomly distributed, the overexpressed AID protein may be targeting a number of different loci in addition to the Ig locus, in DT40 cells. These observations suggest that TLS past abasic sites created by the combined action of AID and uracil glycosylase may be performed less effectively in polζ cells, compared with polη/polζ or polη cells, resulting in chromosome breakage and cell death.

To verify that Polη-dependent DNA synthesis was toxic to the AID-overproducing polζ cells, we reconstituted polη/polζ cells with either wild-type POLη or the catalytically inactive mutant polηD115A/E116A). Wild-type POLη expression sensitized the polη/polζ cells to the overexpression of AID, whereas the mutant POLη had no impact on cell survival (Figure 3A and 3B). We thus conclude that, in polζ cells, TLS past abasic sites may be less effective because neither Polη nor any other polymerase can extend DNA synthesis following the Polη-mediated insertion of nucleotides opposite the abasic site.
We have shown that AID overexpression results in increased TLS-mediated hypermutation at G/C base pairs in Ig V segments [32]. To define the role of Polη and Polζ in this TLS process, we determined the Ig Vλ nucleotide sequences of AID-overexpressing wild-type, polη, and polη/polζ cells. polζ cells were not analyzed because of the difficulty of ectopically expressing AID to the same extent as in the other lines. The number of non-templated point mutations (PM, Figure 4A) was somewhat lower in polη cells than in wild-type cells (This slight reduction is not significant (p = 0.15, t-test) [32]). Surprisingly, the level of Ig V mutations in polη/polζ cells was comparable to that of wild-type cells. This observation is in marked contrast with the fact that Rev1, an essential factor for the function of Polζ [20], plays a critical role in non-templated point mutations at the abasic site [34]. Moreover, Polζ played the critical role in cellular tolerance to AID-mediated abasic sites (Figure 3). These observations indicate that in the absence of both Polη and Polζ, other unidentified DNA polymerase(s) can participate in TLS past the abasic site. As the polη/polζ cells displayed a significant increase in the proportion of G/C to A/T transitions in the non-templated Ig V mutations (12/25; 48%), compared with wild-type cells (2/18; 11%)(Figure 4B), this unidentified DNA polymerase(s) may preferentially incorporate adenine opposite the abasic site in the absence of both Polη and Polζ.

Lack of TLS past the T-T (6-4) UV photoproduct in polζ cells is reversed by the inactivation of Polη
T-T (6-4) UV photoproducts represent the most formidable challenge to DNA replication, as they potently arrest replicative polymerases [35]. To analyze TLS past a T-T (6-4) photoproduct, we transfected two plasmids, pQTs and pQTo, [28] (Figure 5) carrying T-T (6-4) UV photoproducts into DT40 cells. At two days after transfection we recovered only replicated copies, and were thus able to analyze the replication of site-specific T-T (6-4) UV photoproducts in vivo. This photoproduct can be arranged in one of two ways. In the staggered conformation (pQTs) (Figure 5A), the lesions are separated by 28 intervening nucleotides and placed opposite a GpC mismatch. Replicated copies can thus result from TLS on the top or bottom strand. Error-free template switching should result in GpC at the site of the photoproduct, while TLS past this photoproduct may insert ApA (accurate TLS) or other nucleotides (inaccurate TLS) at this site. Note that our experiment was done in a nucleotide-excision repair-deficient (xpa-deficient) background, and thus excluded the recovery of replicated copies generated by error-free nucleotide-excision repair. In the second, unphysiological, replication template, the lesions are placed opposite to each other (pQTo) (Figure 5E). Using this conformation, replicated DNA copies can be recovered as a consequence of TLS, but not by template switching.
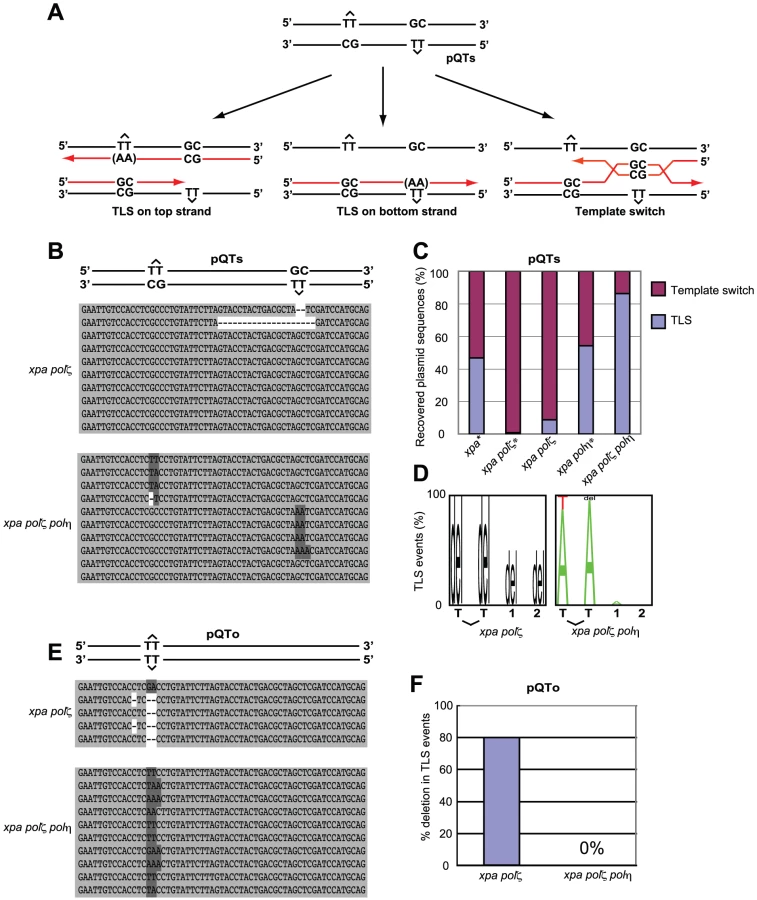
To assess the mode of bypass used when generating replicated copies of pQTs, we analyzed the nucleotide sequences of replicated copies of plasmids recovered from DT40 cells (Figure 5B) and determined the proportion of TLS relative to error-free template switching (Figure 5C). Overall replication efficiency was comparable among cells carrying the various genotypes used in the previous study and this study. Previous study found that 45% and 55% of the recovered plasmid copies resulted from TLS in xpa and xpa/polη cells, respectively [28]. In comparison, the efficiency of TLS in xpa/polζ cells was significantly reduced, with less than 10% of the recovered plasmid generated as a consequence of TLS. All TLS events observed in the xpa/polζ cells were associated with deletion at damaged sites (Figure 5D). Thus, as found previously [28] (Figure 5C), we conclude that Polζ is required for the successful bypass of T-T (6-4) UV photoproducts by TLS. Remarkably, the xpa/polη/polζ cells displayed a normal TLS efficiency, indicating that the failure of TLS in polζ cells is dependent on the presence of Polη.
We also classified the replication products obtained from the pQTo plasmid, where bypass can be effected only by TLS or deletion. As found in the previous study [28], the loss of Polζ was frequently associated with the deletion of two or more nucleotides covering the site of the T-T (6-4) UV photoproduct (Figure 5E and 5F). The loss of Polζ did not impair TLS in the absence of Polη, but severely compromised it in the presence of Polη. A possible explanation, discussed below, is that the Polη-dependent insertion of nucleotides opposite the T-T (6-4) UV photoproduct inhibits the completion of TLS in the absence of Polζ (presumably due to defective extension from inserted nucleotides), while other unidentified DNA polymerases can perform the complete TLS reaction in polη/polζ cells.
Polκ plays only a minor role in the damage tolerance of polη/polζ cells
The milder phenotype of the polη/polζ cells, compared with polζ single mutants, led us to investigate the contribution of other TLS polymerases to TLS in polη/polζ cells. We previously showed that polκ/polζ cells show a higher sensitivity to mono-alkylating agents, compared with polζ cells, though polκ cells show normal sensitivity [36], and thereby suggested that Polκ can partially substitute for the loss of Polζ. We therefore sought to determine whether Polκ contributes to damage tolerance in polη/polζ cells. To this end, we deleted the Polκ gene in polη/polζ cells and analyzed the phenotype of the resulting triple knockout polη/polκ/polζ clones (Figure 6A). Deletion of the Polκ gene tended to reduce growth kinetics. However, the polη/polκ/polζ clones exhibited only limited increase in DNA-damage sensitivity, compared with polη/polζ cells (Figure 6B). Likewise, overexpression of chicken Polκ did not increase cisplatin tolerance in polζ or polη/polζ cells (data not shown). These observations imply that Polκ does not play an important role in TLS in polη/polζ cells.

polη/polζ cells, but not polζ cells, displayed increased numbers of UV-induced sister chromatid exchange events
Replication arrest can be released by two major mechanisms: HR and TLS [37]. HR-dependent release is initiated by homologous pairing between the 3′ end of the arrested strand and the sister chromatid, followed by strand invasion and DNA synthesis to extend the invading 3′ strand (Figure 7D). To analyze the efficiency of HR-mediated release from replication blockage, we analyzed sister chromatid exchange (SCE) (Figure 6C and 6D) [38], [39]. The level of SCE is likely to be determined by two factors: the number of DNA lesions that cause a replication block and the efficiency of HR-dependent release from replication blockage. As previously reported [9], [39], the level of spontaneous SCE was slightly increased (1.5 to 2-fold) in all TLS mutants compared with wild-type cells, presumably because lesions are more frequently channeled to HR. The polη cells displayed a markedly greater increase in the level of UV-induced SCE (the number of spontaneous SCE subtracted from the number of SCE following UV irradiation shown in Figure 6D), which phenotype is attributable to defective TLS over UV-induced damage. In contrast, in the polζ cells, the UV-induced SCE level was similar to that of wild-type cells, suggesting that the defective TLS may not be adequately compensated by HR [9].

SCE was induced more efficiently in the polη/polζ cells than in the polζ cells, which is consistent with the increased UV tolerance of polη/polζ cells, compared with polζ cells. Reconstitution of the polη/polζ cells with wild-type POLη, but not with the catalytically inactive POLη, significantly reduced the number of UV-induced SCE events. Thus, the degree of UV tolerance correlated with the number of UV-induced SCE events at least in polζ and polη/polζ cells. This observation suggests that, in addition to TLS, HR-mediated release from replication blockages contributes to a significantly higher UV tolerance in polη/polζ cells than in polζ cells.
Discussion
Polη is required for TLS across a much wider range of DNA lesions than indicated by the sensitivity of Polη-deficient cells. Analysis of XP-V cells indicates that Polη plays a major role in TLS past cyclobutane dimers and certain bulky adducts, but few other lesions. We demonstrate here that, in DT40 cells, Polη is also involved in the interaction of the replication machinery with different types of damage, such as those induced by chemical cross-linking agents (Figure 2), abasic sites (Figure 3 and Figure 4), and T-T(6-4) UV photoproduct (Figure 5). Thus, the deletion of polη in the polζ cells reversed their hypersensitivity to UV, MMS, cisplatin, and the ectopic expression of AID. Our finding that polη/polζ cells were significantly more tolerant to all tested DNA-damaging agents compared with polζ cells, reveals that neither of these polymerases is absolutely required for the tolerance of these types of damage during DNA replication. However, when Polη is present, Polζ is also required for efficient recovery from the effects of these damaging agents.
XP-V cells show a modest phenotype, including moderate sensitivity to UV and cisplatin [40], only in the presence of caffeine, and do not show significant sensitivity to MMS. Consequently, it was once believed that Polη does not play a role in TLS past DNA lesions induced by alkylating agents. However, in vitro biochemical studies have shown that purified Polη can bypass a variety of lesions including 7, 8-dihydro-8-oxoguanine (8-oxoG), O6-methylguanine, abasic sites, benzo pyrene adducts, and cisplatin intrastrand crosslinks [2], [41]–[43]. The present study helps demonstrate that Polη can indeed be involved in TLS past a wide variety of DNA lesions, even without caffeine treatment in DT40 cells. This idea is relevant to mammalian cells, since Polη focus formation is observed in mammalian cells following treatment with cisplatin and UV in the absence of caffeine [40].
The collaborative action of Polη and Polζ in TLS
The improved damage tolerance of polη/polζ cells, compared with polζ cells, suggests following several possibilities. One possibility is that Polζ somehow inhibits Polη action and that Polη does not actually have a significant role when Polζ is present. However this possibility may be unlikely, since physical interaction between Polη and Rev1, and Polη dependent recruitment of Rev1 to the DNA damage site support the sequential actions of Polη followed by Polζ rather than inhibitory action of Polζ on Polη [44]–[47]. Thus, more likely possibility is that, Polη generates a replicative intermediate in an attempt to bypass the lesion, but cannot complete an effective bypass reaction without Polζ (Figure 7A). We suggest that the abortive intermediates generated by Polη in the absence of Polζ lead to a difficult-to-rescue replication collapse, thereby accounting for the hypersensitivity of the single polζ mutant (Figure 7B). The modest phenotype of XP-V cells indicates that Polζ may efficiently mediate TLS past a variety of DNA lesions, either in collaboration with other polymerases or possibly on its own.
This situation can be explored further in the light of current TLS models. In the canonical model for TLS replication, arrest by agents such as UV, MMS, and cisplatin, leads to PCNA becoming mono-ubiquitinated [40], [48]–[50]. This increases the affinity of PCNA for Polη and other Y-family TLS polymerases by virtue of their UBM and UBZ ubiquitin-binding motif [44], [48], [50]–[52] and likely contributes to the accumulation of Y-family polymerases at the sites of blocked replication forks. It has been suggested that these polymerases can compete with each other by mass action to attempt to carry out TLS. In the case of CPDs, if Polη wins this competition; bypass can occur without the need for a second polymerase. However, with other lesions such as a T-T (6-4) photoproduct, there is currently no evidence to show that any single polymerase can complete bypass. Polη may be able to start the bypass by incorporating opposite at least the 5′ base of the lesion, but it cannot extend from the resulting mismatch (Figure 7A). This would explain the abortive intermediate referred to above. To complete TLS, Polζ is required to extend from the inserted nucleotides (Figure 7A), an explanation that is consistent with the sequential action of Polη and Polζ demonstrated in in vitro studies [2]. A further implication of this model is that no other polymerase can effectively substitute for Polζ in this extension step (Figure 7B).
The successive action of Polη and Polζ might be mediated by the association of the two polymerases with Rev1 [45], [53]. The idea of a Rev1-mediated switch from Polη to Polζ is supported by the fact that Polη tightly interacts with Rev1 [1], [46], [47], [53], [54]. Moreover, DNA-damage-induced Rev1 focus formation appears to be dependent on Polη [46], [47]. Adding to these findings, the present work establishes a role for Polη in the bypass of a wider range of DNA damage than previously thought and demonstrates the in vivo importance of the two-step bypass of many lesions.
The effect on TLS of depleting both Polη and Polζ is distinctly different in DT40 cells than in human cells. Ziv et al. showed that depletion of Rev3 sensitized cells to UV more severely at two days after irradiation in Polη-deficient XPV fibroblasts than in Polη-proficient control cells [22], although its impact on Polη-proficient DT40 cells was considerably stronger than on Polη-deficient DT40 cells. There are several explanations for this apparent difference. First, we favor the idea that DT40 may be a more reliable cell line than others to evaluate TLS by measuring cellular survival due to following reasons. The cell cycle distribution is distinctly different between DT40 and other mammalian cell lines. In DT40 cells, ∼70% of the cells are in the S phase, and the G1/S checkpoint does not function at all [55]. In most of the mammalian cell lines, on the other hand, more than 50% of the cells are in the G1 phase, and G1/S checkpoint works at least partially. Therefore, environmental DNA damage interferes with DNA replication more significantly in DT40 cells than in mammalian cell lines. Accordingly, TLS contributes to the cellular survival of the colony formation assay to a considerably higher extent in DT40 cells than in mammalian cell lines. Ziv et al., on the other hand, evaluated TLS by measuring the number of living cells at 48 hours after UV irradiation [22]. Since a majority of UV irradiated cells may have stayed outside the S phase at 48 hours, it is unclear whether this survival reflects the efficiency of TLS. The same laboratory also analyzed TLS past a cisplatin G-G intrastrand crosslink located in a gapped episomal plasmid [21]. Depletion of Rev3 with or without codepletion of Polη in U2OS cells resulted in an 80% reduction in TLS past the lesion, irrespective of the presence or absence of Polη. The relevance of this finding to TLS during the replication of chromosomal DNA remains elusive. Yoon et al. investigated TLS in a double-stranded plasmid containing a single 6-4 photoproduct as well as replication origins derived from the SV40 virus [23]. Depletion of Rev3 or Rev7 in NER-deficient XP-A fibroblasts reduced the efficiency of TLS in the episomal plasmid by approximately 50%, with a similar reduction obtained in XP-V fibroblasts. This result using human fibroblasts is clearly different from the data we obtained using DT40 cells. Given the close sequence similarity between the two polymerases in human and chicken cells, we consider it unlikely that the mechanisms of lesion bypass are fundamentally different between the two organisms. The apparent difference between mammalian cells and DT40 may be caused by the incomplete si-RNA mediated inhibition in human cells versus the null mutation we have used in DT40. Another possible reason to explain this difference is the active HR system in DT40 cells, and the different usage order of TLS DNA polymerases because the DT40 B lymphocyte line undergoes Ig V hypermutation through TLS [32]. The usefulness of the three episomal plasmid systems to the analysis of TLS occurring during replication of chromosomal DNAs should be further investigated [15], [21], [22], [28]. Irrespective of the reason for this apparent difference, our data clearly indicate that, as discussed above, under some conditions Polη can hinder the efficient progress of the replication fork past lesions mediated by Polζ.
Rescue of failed translesion synthesis by homologous recombination
It is remarkable that deletion of the three major TLS polymerase genes, POLη, POLκ, and POLζ, results in only a mild reduction in the growth kinetics of DT40 cells (Figure 6A). This is in marked contrast with the immediate cell death associated with the massive chromosomal breaks generated upon deletion of Rad51 [56]. These observations imply that during DNA replication, if replication blocks are encountered, HR can at least partially compensate for defective TLS. The significant functional redundancy between TLS and HR is supported by our previous report, which concludes that the deletion of both RAD18 and RAD54, a gene involved in HR, as well as the deletion of both REV3 and RAD54, are synthetically lethal to cells [9], [39]. We show here that depletion of Polη irrespective of the status of Polζ markedly increases the level of UV-induced SCE (Figure 6C), suggesting that DNA damage that cannot be resolved by TLS because of the absence of Polη may be resolved by HR, leading to increased SCE levels (Figure 7D). However, SCE is not induced to the same level in polζ cells (Figure 6C). Thus, nucleotide incorporation by Polη appears to represent a point of commitment in the TLS reaction beyond which rescue by HR is problematic. This is likely to be explained by the creation of an intermediate, possibly the mismatched primer terminus, which can be efficiently extended by Polζ, but which cannot readily initiate HR.
In summary, the significant increase in the cellular tolerance of polη/polζ cells to DNA-damaging agents, compared with polζ cells, can be partially attributable to more efficient HR in polη/polζ cells than in polζ cells. However, the normal level of TLS-dependent Ig V mutation and restoration of TLS during 6-4 photoproduct bypass in polη/polζ cells (Figure 4B) suggests that one or more other unidentified TLS polymerases can act as a substitute and carry out TLS when both Polη and Polζ are missing (Figure 7C).
Materials and Methods
Cell lines and cell culture
Generation of polζ (rev3) - and polη-deficient DT40 cells was described previously [9], [12]. To generate polη/polζ cells, we sequentially introduced two rev3 gene-disruption constructs (rev3-hygro and rev3-His) [9] into polη (PuroR/BlsR) cells. A puromycin-resistant XPA disruption construct was used to disrupt the single XPA allele and recreate the xpa/rev3 cell line. After removal of the BlsR marker gene from the polη/polζ cells by the transient expression of CRE recombinase, a blasticidin-resistant XPA disruption construct was used to generate the xpa/polζ/polη cell line. A puromycin-resistant POLκ disruption construct was used to generate polη/polζ/polκ cells. To make a POLη expression plasmid, we inserted human POLη cDNA into the multi-cloning sites (MCS) of an expression vector, pCR3-loxP-MCS-IRES-GFP-loxP [57]. A mutant Polη that lacks polymerase activity (Mutant POLη) was generated by inserting D115A/E116A mutations into human Polη cDNA. The conditions for cell culture, selection, and DNA transfection were described previously[58]. The growth properties of cells were analyzed as described previously [58].
AID overexpression by retrovirus infection
For the retrovirus infection, the pMSCV-IRES-GFP recombinant plasmid was constructed by ligating the 5.2 kb BamHI-NotI fragment from pMSCVhyg (Clontech) with the 1.2 kb BamHI-Not1 fragment from pIRES2-EGFP (Clontech). Mouse AID cDNA [33] was inserted between the BglII and EcoRI sites of pMSCV-IRES-GFP. The preparation and infection of the retrovirus were carried out as previously described [33]. Expression of the GFP was confirmed by flow cytometry. The efficiency of infection was more than 90% as assayed by GFP expression.
Assay of TLS past a T-T (6–4) photoproduct on episomal plasmid
pQTs and pQTo plasmids containing a T-T (6-4) photoproduct were generated and transfected into DT40 cells as previously described [28].
Chromosome aberration analysis
Karyotype analysis was performed as described previously [56]. Cells were treated with colcemid for 3 hours to enrich mitotic cells.
Measurement of SCE level
Measurement of SCE level was performed as described previously [38]. For UV-induced SCE, cells were suspended in PBS and irradiated with 0.25 J/m2 UV followed by BrdU labeling.
Ig Vλ mutation analysis
Genomic DNA was extracted at 14 days after subcloning. The rearranged Vλ segments were PCR amplified using 5′-CAGGAGCTCGCGGGGCCGTCACTGATTGCCG-3′ as the forward primer in the leader-Vλ intron and 5′-GCGCAAGCTTCCCCAGCCTGCCGCCAAGTCCAAG-3′ as the reverse primer in the 3′ of the JCλ intron. To minimize PCR-introduced mutations, the high-fidelity polymerase, Phusion (Fynnzymes) was used for amplification (30 cycles at 94° C for 30 s; 60° C for 1 min; 72° C for 1 min). PCR products were cloned using a Zero Blunt TOPO PCR Cloning Kit (Invitrogen) and subjected to sequence analysis with the M13 forward (-20) or reverse primer. Sequence alignment using GENETYX-MAC (Software Development, Tokyo) allowed the identification of changes from the parental sequences in each clone.
As described previously [59], all sequence changes were assigned to one of three categories: point mutation, gene conversion, or ambiguous. This discrimination is based on the published sequences of Vλ pseudogenes that can act as donors for gene conversion. For each mutation, the database of Vλ pseudogenes was searched for a potential donor. If no pseudogene donor containing a string >9 bp could be found, the mutation was categorized as a non-templated point mutation. If such a string was identified and there were further mutations that could be explained by the same donor, then all these mutations were categorized as a single long-tract gene conversion event. If there were no further mutations, indicating that the isolated mutation could have arisen through a conversion mechanism or could have been non-templated, it was categorized as ambiguous.
Supporting Information
Zdroje
1. FriedbergEC
LehmannAR
FuchsRP
2005
Trading places: how do DNA polymerases switch during translesion DNA synthesis?
Mol Cell
18
499
505
2. PrakashS
JohnsonRE
PrakashL
2005
Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function.
Annu Rev Biochem
74
317
353
3. JohnsonRE
KondratickCM
PrakashS
PrakashL
1999
hRAD30 mutations in the variant form of xeroderma pigmentosum.
Science
285
263
265
4. MasutaniC
KusumotoR
YamadaA
DohmaeN
YokoiM
1999
The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta.
Nature
399
700
704
5. LehmannAR
2002
Replication of damaged DNA in mammalian cells: new solutions to an old problem.
Mutat Res
509
23
34
6. Van SlounPP
VarletI
SonneveldE
BoeiJJ
RomeijnRJ
2002
Involvement of mouse Rev3 in tolerance of endogenous and exogenous DNA damage.
Mol Cell Biol
22
2159
2169
7. MizutaniA
OkadaT
ShibutaniS
SonodaE
HocheggerH
2004
Extensive chromosomal breaks are induced by tamoxifen and estrogen in DNA repair-deficient cells.
Cancer Res
64
3144
3147
8. NojimaK
HocheggerH
SaberiA
FukushimaT
KikuchiK
2005
Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells.
Cancer Res
65
11704
11711
9. SonodaE
OkadaT
ZhaoGY
TateishiS
ArakiK
2003
Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates.
EMBO J
22
3188
3197
10. WuX
TakenakaK
SonodaE
HocheggerH
KawanishiS
2006
Critical roles for polymerase zeta in cellular tolerance to nitric oxide-induced DNA damage.
Cancer Res
66
748
754
11. LehmannAR
2006
New functions for Y family polymerases.
Mol Cell
24
493
495
12. KawamotoT
ArakiK
SonodaE
YamashitaYM
HaradaK
2005
Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis.
Mol Cell
20
793
799
13. JohnsonRE
WashingtonMT
PrakashS
PrakashL
2000
Fidelity of human DNA polymerase eta.
J Biol Chem
275
7447
7450
14. McCullochSD
KokoskaRJ
MasutaniC
IwaiS
HanaokaF
2004
Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity.
Nature
428
97
100
15. YoonJH
PrakashL
PrakashS
2009
Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells.
Proc Natl Acad Sci U S A
106
18219
18224
16. JohnsonRE
WashingtonMT
HaracskaL
PrakashS
PrakashL
2000
Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions.
Nature
406
1015
1019
17. WoodgateR
2001
Evolution of the two-step model for UV-mutagenesis.
Mutat Res
485
83
92
18. WolfleWT
JohnsonRE
MinkoIG
LloydRS
PrakashS
2006
Replication past a trans-4-hydroxynonenal minor-groove adduct by the sequential action of human DNA polymerases iota and kappa.
Mol Cell Biol
26
381
386
19. LawrenceCW
2007
Following the RAD6 pathway.
DNA Repair (Amst)
6
676
686
20. OkadaT
SonodaE
YoshimuraM
KawanoY
SayaH
2005
Multiple roles of vertebrate REV genes in DNA repair and recombination.
Mol Cell Biol
25
6103
6111
21. ShacharS
ZivO
AvkinS
AdarS
WittschiebenJ
2009
Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals.
EMBO J
28
383
393
22. ZivO
GeacintovN
NakajimaS
YasuiA
LivnehZ
2009
DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients.
Proc Natl Acad Sci U S A
106
11552
11557
23. YoonJH
PrakashL
PrakashS
2010
Error-free replicative bypass of (6-4) photoproducts by DNA polymerase zeta in mouse and human cells.
Genes Dev
24
123
128
24. SaleJE
2004
Immunoglobulin diversification in DT40: a model for vertebrate DNA damage tolerance.
DNA Repair (Amst)
3
693
702
25. ArakawaH
HauschildJ
BuersteddeJM
2002
Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion.
Science
295
1301
1306
26. HarrisRS
SaleJE
Petersen-MahrtSK
NeubergerMS
2002
AID is essential for immunoglobulin V gene conversion in a cultured B cell line.
Curr Biol
12
435
438
27. Di NoiaJM
NeubergerMS
2007
Molecular mechanisms of antibody somatic hypermutation.
Annu Rev Biochem
76
1
22
28. SzutsD
MarcusAP
HimotoM
IwaiS
SaleJE
2008
REV1 restrains DNA polymerase zeta to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo.
Nucleic Acids Res
36
6767
6780
29. WangYC
MaherVM
MitchellDL
McCormickJJ
1993
Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal, error-prone replication on a template containing photoproducts.
Mol Cell Biol
13
4276
4283
30. WatersHL
SeetharamS
SeidmanMM
KraemerKH
1993
Ultraviolet hypermutability of a shuttle vector propagated in xeroderma pigmentosum variant cells.
J Invest Dermatol
101
744
748
31. BebenekK
MatsudaT
MasutaniC
HanaokaF
KunkelTA
2001
Proofreading of DNA polymerase eta-dependent replication errors.
J Biol Chem
276
2317
2320
32. SaberiA
NakaharaM
SaleJE
KikuchiK
ArakawaH
2008
The 9-1-1 DNA clamp is required for immunoglobulin gene conversion.
Mol Cell Biol
28
6113
6122
33. ShinkuraR
ItoS
BegumNA
NagaokaH
MuramatsuM
2004
Separate domains of AID are required for somatic hypermutation and class-switch recombination.
Nat Immunol
5
707
712
34. ArakawaH
MoldovanGL
SaribasakH
SaribasakNN
JentschS
2006
A role for PCNA ubiquitination in immunoglobulin hypermutation.
PLoS Biol
4
e366
doi:10.1371/journal.pbio.0040366
35. FriedbergEC
WalkerGC
SiedeW
WoodRD
SchultzRA
2006
DNA repair and mutagenesis.
36. TakenakaK
OgiT
OkadaT
SonodaE
GuoC
2006
Involvement of vertebrate Polkappa in translesion DNA synthesis across DNA monoalkylation damage.
J Biol Chem
281
2000
2004
37. HocheggerH
SonodaE
TakedaS
2004
Post-replication repair in DT40 cells: translesion polymerases versus recombinases.
Bioessays
26
151
158
38. SonodaE
SasakiMS
MorrisonC
Yamaguchi-IwaiY
TakataM
1999
Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells.
Mol Cell Biol
19
5166
5169
39. YamashitaYM
OkadaT
MatsusakaT
SonodaE
ZhaoGY
2002
RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells.
EMBO J
21
5558
5566
40. AlbertellaMR
GreenCM
LehmannAR
O'ConnorMJ
2005
A role for polymerase eta in the cellular tolerance to cisplatin-induced damage.
Cancer Res
65
9799
9806
41. KusumotoR
MasutaniC
IwaiS
HanaokaF
2002
Translesion synthesis by human DNA polymerase eta across thymine glycol lesions.
Biochemistry
41
6090
6099
42. MasutaniC
KusumotoR
IwaiS
HanaokaF
2000
Mechanisms of accurate translesion synthesis by human DNA polymerase eta.
EMBO J
19
3100
3109
43. WashingtonMT
WolfleWT
SprattTE
PrakashL
PrakashS
2003
Yeast DNA polymerase eta makes functional contacts with the DNA minor groove only at the incoming nucleoside triphosphate.
Proc Natl Acad Sci U S A
100
5113
5118
44. GuoC
TangTS
BienkoM
ParkerJL
BielenAB
2006
Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage.
Mol Cell Biol
26
8892
8900
45. MurakumoY
OguraY
IshiiH
NumataS
IchiharaM
2001
Interactions in the error-prone postreplication repair proteins hREV1, hREV3, and hREV7.
J Biol Chem
276
35644
35651
46. AkagiJ
MasutaniC
KataokaY
KanT
OhashiE
2009
Interaction with DNA polymerase eta is required for nuclear accumulation of REV1 and suppression of spontaneous mutations in human cells.
DNA Repair (Amst)
8
585
599
47. TissierA
KannoucheP
ReckMP
LehmannAR
FuchsRP
2004
Co-localization in replication foci and interaction of human Y-family members, DNA polymerase pol eta and REVl protein.
DNA Repair (Amst)
3
1503
1514
48. KannouchePL
WingJ
LehmannAR
2004
Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage.
Mol Cell
14
491
500
49. NiimiA
BrownS
SabbionedaS
KannouchePL
ScottA
2008
Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells.
Proc Natl Acad Sci U S A
105
16125
16130
50. WatanabeK
TateishiS
KawasujiM
TsurimotoT
InoueH
2004
Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination.
EMBO J
23
3886
3896
51. BienkoM
GreenCM
CrosettoN
RudolfF
ZapartG
2005
Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis.
Science
310
1821
1824
52. PloskyBS
VidalAE
Fernandez de HenestrosaAR
McLeniganMP
McDonaldJP
2006
Controlling the subcellular localization of DNA polymerases iota and eta via interactions with ubiquitin.
EMBO J
25
2847
2855
53. GuoC
FischhaberPL
Luk-PaszycMJ
MasudaY
ZhouJ
2003
Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis.
EMBO J
22
6621
6630
54. YuasaMS
MasutaniC
HiranoA
CohnMA
YamaizumiM
2006
A human DNA polymerase eta complex containing Rad18, Rad6 and Rev1; proteomic analysis and targeting of the complex to the chromatin-bound fraction of cells undergoing replication fork arrest.
Genes Cells
11
731
744
55. YamazoeM
SonodaE
HocheggerH
TakedaS
2004
Reverse genetic studies of the DNA damage response in the chicken B lymphocyte line DT40.
DNA Repair (Amst)
3
1175
1185
56. SonodaE
SasakiMS
BuersteddeJM
BezzubovaO
ShinoharaA
1998
Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death.
EMBO J
17
598
608
57. FujimoriA
TachiiriS
SonodaE
ThompsonLH
DharPK
2001
Rad52 partially substitutes for the Rad51 paralog XRCC3 in maintaining chromosomal integrity in vertebrate cells.
EMBO J
20
5513
5520
58. TakataM
SasakiMS
SonodaE
MorrisonC
HashimotoM
1998
Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells.
EMBO J
17
5497
5508
59. SaleJE
CalandriniDM
TakataM
TakedaS
NeubergerMS
2001
Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation.
Nature
412
921
926
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance