
SOX9 Governs Differentiation Stage-Specific Gene Expression in Growth Plate Chondrocytes via Direct Concomitant Transactivation and Repression
Cartilage and endochondral bone development require SOX9 activity to regulate chondrogenesis, chondrocyte proliferation, and transition to a non-mitotic hypertrophic state. The restricted and reciprocal expression of the collagen X gene, Col10a1, in hypertrophic chondrocytes and Sox9 in immature chondrocytes epitomise the precise spatiotemporal control of gene expression as chondrocytes progress through phases of differentiation, but how this is achieved is not clear. Here, we have identified a regulatory element upstream of Col10a1 that enhances its expression in hypertrophic chondrocytes in vivo. In immature chondrocytes, where Col10a1 is not expressed, SOX9 interacts with a conserved sequence within this element that is analogous to that within the intronic enhancer of the collagen II gene Col2a1, the known transactivation target of SOX9. By analysing a series of Col10a1 reporter genes in transgenic mice, we show that the SOX9 binding consensus in this element is required to repress expression of the transgene in non-hypertrophic chondrocytes. Forced ectopic Sox9 expression in hypertrophic chondrocytes in vitro and in mice resulted in down-regulation of Col10a1. Mutation of a binding consensus motif for GLI transcription factors, which are the effectors of Indian hedgehog signaling, close to the SOX9 site in the Col10a1 regulatory element, also derepressed transgene expression in non-hypertrophic chondrocytes. GLI2 and GLI3 bound to the Col10a1 regulatory element but not to the enhancer of Col2a1. In addition to Col10a1, paired SOX9–GLI binding motifs are present in the conserved non-coding regions of several genes that are preferentially expressed in hypertrophic chondrocytes and the occurrence of pairing is unlikely to be by chance. We propose a regulatory paradigm whereby direct concomitant positive and negative transcriptional control by SOX9 ensures differentiation phase-specific gene expression in chondrocytes. Discrimination between these opposing modes of transcriptional control by SOX9 may be mediated by cooperation with different partners such as GLI factors.
Published in the journal:
. PLoS Genet 7(11): e32767. doi:10.1371/journal.pgen.1002356
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002356
Summary
Cartilage and endochondral bone development require SOX9 activity to regulate chondrogenesis, chondrocyte proliferation, and transition to a non-mitotic hypertrophic state. The restricted and reciprocal expression of the collagen X gene, Col10a1, in hypertrophic chondrocytes and Sox9 in immature chondrocytes epitomise the precise spatiotemporal control of gene expression as chondrocytes progress through phases of differentiation, but how this is achieved is not clear. Here, we have identified a regulatory element upstream of Col10a1 that enhances its expression in hypertrophic chondrocytes in vivo. In immature chondrocytes, where Col10a1 is not expressed, SOX9 interacts with a conserved sequence within this element that is analogous to that within the intronic enhancer of the collagen II gene Col2a1, the known transactivation target of SOX9. By analysing a series of Col10a1 reporter genes in transgenic mice, we show that the SOX9 binding consensus in this element is required to repress expression of the transgene in non-hypertrophic chondrocytes. Forced ectopic Sox9 expression in hypertrophic chondrocytes in vitro and in mice resulted in down-regulation of Col10a1. Mutation of a binding consensus motif for GLI transcription factors, which are the effectors of Indian hedgehog signaling, close to the SOX9 site in the Col10a1 regulatory element, also derepressed transgene expression in non-hypertrophic chondrocytes. GLI2 and GLI3 bound to the Col10a1 regulatory element but not to the enhancer of Col2a1. In addition to Col10a1, paired SOX9–GLI binding motifs are present in the conserved non-coding regions of several genes that are preferentially expressed in hypertrophic chondrocytes and the occurrence of pairing is unlikely to be by chance. We propose a regulatory paradigm whereby direct concomitant positive and negative transcriptional control by SOX9 ensures differentiation phase-specific gene expression in chondrocytes. Discrimination between these opposing modes of transcriptional control by SOX9 may be mediated by cooperation with different partners such as GLI factors.
Introduction
Chondrogenesis and the formation of bone by endochondral ossification depend on progressive steps of cell differentiation. Mesenchymal cells condense and differentiate into chondrocytes in a pattern that will define the eventual shape of the different skeletal elements. These chondrocytes proliferate, mature, exit the cell cycle and become prehypertrophic. The differentiation program culminates in the terminal differentiation and apoptosis of post-mitotic hypertrophic chondrocytes [1]. This differentiation program is controlled by members of the SOX and RUNX families of transcription factors and the integration of multiple signaling pathways mediated by Indian hedgehog (Ihh), parathyroid hormone-related protein (PTHrP), Wnts, BMPs, and Notch (reviewed in [2]). PTHrP and Ihh are two important players which interact to form a feedback loop that controls the pace of the differentiation program [3].
Sox9 is essential for chondrogenesis and chondrocyte differentiation [4]–[6]. It is essential for mesenchymal condensation prior to chondrogenesis, and in its absence chondrocyte differentiation fails. Inactivation of Sox9 in chondrocytes at different stages of differentiation suggests that its expression is essential for the survival of chondrocytes so that they can progress to hypertrophy [5]–[7]. Mutations in SOX9 are associated with the human skeletal malformation syndrome, campomelic dysplasia, in which skeletal abnormalities can be attributed to the disruption of the chondrogenic differentiation program due to failure to express SOX9 target genes. Upon hypertrophy, chondrocytes down-regulate Sox9 expression [8], [9], which is believed to mark the end of SOX9 control in the growth plate.
Despite the wealth of information about spatial and temporal gene expression patterns in the developing growth plate, it is not clear how transcriptional controls achieve appropriate and specific gene expression during chondrocyte differentiation. SOX9 activates many genes expressed in proliferating chondrocytes, including the extracellular matrix (ECM) genes Col2a1, Col9a1, Col11a2, Acan (aggrecan) and Cd-rap/Mia1 [10]–[15]. For the Col2a1 gene, which is expressed most strongly in proliferating chondrocytes, SOX9 directly transactivates the gene in vivo via a conserved enhancer sequence within the first intron [10], [11].
The collagen X gene, Col10a1, is a hypertrophic chondrocyte specific marker. The specificity and reciprocity of Sox9 and Col10a1 expression epitomise the strict control of temporal and differentiation phase-specific gene expression in the growth plate. Col10a1 is ideal for studying transcriptional regulation because as well as its highly specific expression pattern, over-expression or loss-of-function does not disrupt chondrocyte differentiation. These properties simplify interpretation of changes in gene expression resulting from perturbing transcriptional control [16]–[18]. Here, we examined the transcriptional controls that restrict Col10a1 expression to hypertrophic chondrocytes. We found that SOX9 coordinates gene expression during chondrocyte differentiation through both transcriptional activation and repression. Discrimination between these opposing actions is probably achieved by cooperation between SOX9 and different partners such as GLI factors.
Results
Proliferating and hypertrophic chondrocytes show overlapping and different protein binding domains in the Col10a1 enhancer
Previous cell transfection studies identified an enhancer element upstream of human COL10A1 [19]. This element is highly conserved in mammals and corresponds to a 640 bp region between −4.3 and −3.6 kb of the mouse Col10a1 gene (designated element A) (Figure 1A and 1B). We used DNase I footprinting assays to test the configurations in which the element A sequences could be directly bound by nuclear factors derived from chondrocytes at different differentiation states (Figure 2A). Extracts from hypertrophic chondrocytes MCTs, but not fibroblasts COS-1 or osteoblasts MC3T3-E1, protected six blocks of sequence (H1–H6). Noticeably, four different blocks (P1–P4) that partially overlap H1–H4 were protected by extracts from the proliferating chondrocyte/chondrosarcoma cell line CCL (Figure 1C and Figure 2A). Since proliferating chondrocyte/chondrosarcoma cells do not express Col10a1 (Figure S1A), these results suggest that in these cells, the proteins that bind to element A may contribute to the repression of Col10a1.


SOX9 binds to Col10a1 element A in proliferating chondrocytes
We and others previously showed that SOX9 regulates COL2A1/Col2a1 gene via a functional in vivo binding site in the intron 1 enhancer element [10], [11]. We identified the same SOX9-binding sequence within the Col10a1 element A (Figure 1C and Figure 2B). This site, COL2C1, lies on a region in block P3 that is not protected in hypertrophic chondrocytes, and is adjacent to a stretch of thymidine/guanine-rich (TG-rich) sequence. Electromobility shift assays revealed that SOX9 bound to this SOX9/TG-rich motif with a similar affinity as to the COL2A1 enhancer element (Figure S1C, S1E, S1F) [10] and the interaction involved dimeric binding (Figure 2C, cf. lane 3–4). SOX9 also interacted with the TG-rich motif but with a lower affinity than with the consensus SOX9 site. Mutation of the TG-rich motif reduced the overall SOX9 binding to the P3 element (Figure 2C, cf. lanes 6–10). The TG-rich motif resembles a RUNX binding consensus sequence, but we found that RUNX2 did not interact with this motif effectively compared with its binding to the RUNX site in the Bglap (osteocalcin) enhancer (Figure 2D). Chromatin immunoprecipitation (ChIP) assays using extracts from E13.5 mouse limb, a stage at which the cartilage anlagen is largely composed of immature chondrocytes, confirmed specific SOX9 binding to the Col10a1 element A and the Col2a1 enhancer in vivo (Figure 2E).
SOX9/TG-rich motif is required for appropriate Col10a1 expression
The paired SOX9 binding sequences in element A are separated by 4 bp, a distance similar to that between the paired SOX-like consensus sequences in Col2a1, Col9a1, and Acan that mediate transactivation of expression [15]. We tested the in vivo role of element A and the effects of SOX9/TG-rich motif mutations on the expression of Col10a1 mini-genes (Figure 3A) in transgenic mice. We have previously shown that a Flag-tagged Col10a1 vector Col10Flag (formerly known as FColX) is expressed in P10 hypertrophic chondrocytes [18]. Here, we show that in E15.5 humeri, the Col10Flag transgene was expressed in islands in prehypertrophic and hypertrophic chondrocytes in the upper hypertrophic zone (Figure 3B, e). In two independent mouse lines, a transgene comprising element A fused to the Col10Flag (Col10Flag-E) was expressed in a similar pattern as Col10Flag, but was significantly more strongly expressed than Col10Flag in all hypertrophic chondrocytes (Figure 3B, i), reflecting the enhancer activity of element A (see also Figure S2). However, mutation of the SOX9 site in element A (Col10Flag-EΔ1) resulted in marked expansion of the expression domain of the transgene, extending from the hypertrophic zone to the proliferating zone in the majority of transgenic fetuses (71.4%) (compare Figure 4, a with Figure 3B, i) and in almost all the Sox9-expressing chondrocytes in the rest (Figure 4, e). Expansion of transgene expression in proliferating chondrocytes was also noted but was less marked when the TG-rich motif in element A was mutated (Col10Flag-EΔ2) (compare Figure 4, m and i with a and e). Mutation of either SOX9 or the TG-rich motif did not abrogate transgene expression in hypertrophic chondrocytes. Together, these observations suggest that element A contains both positive and negative regulatory sequences, and that mutations in the SOX9/TG-rich motif in element A might disrupt SOX9-mediated repression in immature chondrocytes.


SOX9 is a negative regulator of Col10a1
To test whether SOX9 negatively regulates Col10a1, we established a cell line from hypertrophic chondrocytes MCTs which expressed the Col10Flag-E transgene at the non-permissive (growth-arrest) temperature (Figure 5A). Similar to previous findings for dedifferentiated chondrocytic cells MC615, over-expression of SOX9 in MCTs did not transactivate endogenous Col2a1 [20]. However, SOX9 over-expression significantly down-regulated expression of both endogenous Col10a1 and the exogenous Col10Flag-E reporter (Figure 5B) supporting the notion that SOX9 is a negative regulator of Col10a1. To study the regulation in vivo, SOX9 was ectopically expressed in hypertrophic chondrocytes in mice. Mice expressing Cre recombinase inserted into the endogenous Col10a1 gene (Col10a1-Cre) [21] were crossed with transgenic mice carrying a single copy of a Cre-inducible Sox9-IRES-EGFP expression construct (Z/Sox9) [22] (Figure 5C). In Col10a1-Cre;Z/Sox9 mice, Sox9 and the linked Egfp reporter gene were activated in the hypertrophic chondrocytes at E17.5 in the anterior ribs (Figure 5D, h, i). These mice displayed an expanded hypertrophic zone and reduced Col10a1 expression (Figure 5D, e, k). Interestingly, we also found that transcription of Cre from the Col10a1 locus was reduced in Col10a1-Cre;Z/Sox9 mice (Figure 5D, a, g). Col2a1 expression in Col10a1-Cre;Z/Sox9 mice hypertrophic chondrocytes was not up-regulated suggesting the reduction of Col10a1 expression in these cells was not because of a reversion to a more immature state (Figure 5D, d, j). In the ribs Runx2 was expressed predominantly in osteoblasts, the perichondrium flanking the hypertrophic chondrocytes, and in prehypertrophic chondrocytes (Figure 5D, f). Expression was low in hypertrophic chondrocytes. There was no significant change in Runx2 expression in prehypertrophic and hypertrophic chondrocytes in Col10a1-Cre;Z/Sox9 mice (Figure 5D, l). Collectively, these data suggest that SOX9 negatively regulates Col10a1 gene expression independent of Runx2.

GLI factors bind and regulate Col10a1 in proliferating chondrocytes
The specificity of SOX protein action is known to be achieved through interaction with cell-specific partners [23], [24]. We questioned whether concomitant transactivation of Col2a1 and repression of Col10a1 by SOX9 in proliferating chondrocytes could be mediated by different combinations of cofactors. ChIP assays in E13.5 mouse limb chondrocytes or CCL cells revealed similar interactions of TRAP230/MED12, a mediator of SOX9 activity [25], and of TRPS1, a GLI3-interacting repressor [26], [27], with both the Col10a1 element A and the Col2a1 enhancer (Figure 6A, upper panel). On the other hand, the transcriptional co-repressor, histone deacetylase HDAC4 [28] immunoprecipitated neither element. GLI1, GLI2 and GLI3 are effectors of Hh signaling which controls chondrocyte proliferation and maturation [29]. GLI1 is a transactivator expressed in proliferating chondrocytes and perichondrial tissue flanking the prehypertrophic and hypertrophic zones [30] whereas GLI2 and GLI3 can act as repressors and are predominantly expressed in non-hypertrophic chondrocytes and are down-regulated in hypertrophic chondrocytes [29], [31]. Since there is a conserved GLI-binding site near the SOX9/TG-rich motif in the same footprint block P3 (Figure 1C), we examined whether GLI1, GLI2 and GLI3 can interact with the element A. Strikingly, while SOX9 bound to both the Col10a1 element A and Col2a1 enhancer, GLI2 and GLI3 associated with only Col10a1 element A (Figure 6A, lower panel). GLI3 interacted the most with element A, while GLI1 interaction was much less. Quantitative ChIP assays confirmed the preferential interaction (Figure 6B). From these results we hypothesized that GLI proteins may repress Col10a1 expression. To test this in vivo, we examined the impact on transgene expression of mutating the GLI-binding site in element A (Col10Flag-EΔ3). Consistent with our hypothesis, the majority of fetuses (7 out of 10) expressing Col10Flag-EΔ3 showed distinct islands of transgene misexpression in non-hypertrophic chondrocytes (Figure 6C, e, f). Mutating all three sites (GLI, SOX9, TG-rich) in the transgene (Col10Flag-EΔ4) did not restrict the expansion of the expression domain to proliferating chondrocytes in all the expressing transgenic fetuses obtained (Figure 6C, i, j). Indeed in the majority of these expressing fetuses (3 out of 5), transgene expression extended throughout the entire cartilage zones. Thus mutation of the GLI site alone had a similar derepressing effect as mutating the SOX9/TG-rich motif and mutating all the motifs did not restrict expression but resulted in more extensive mis-expression. This is consistent with a model whereby SOX9 and GLI act cooperatively to repress Col10a1 transcription.

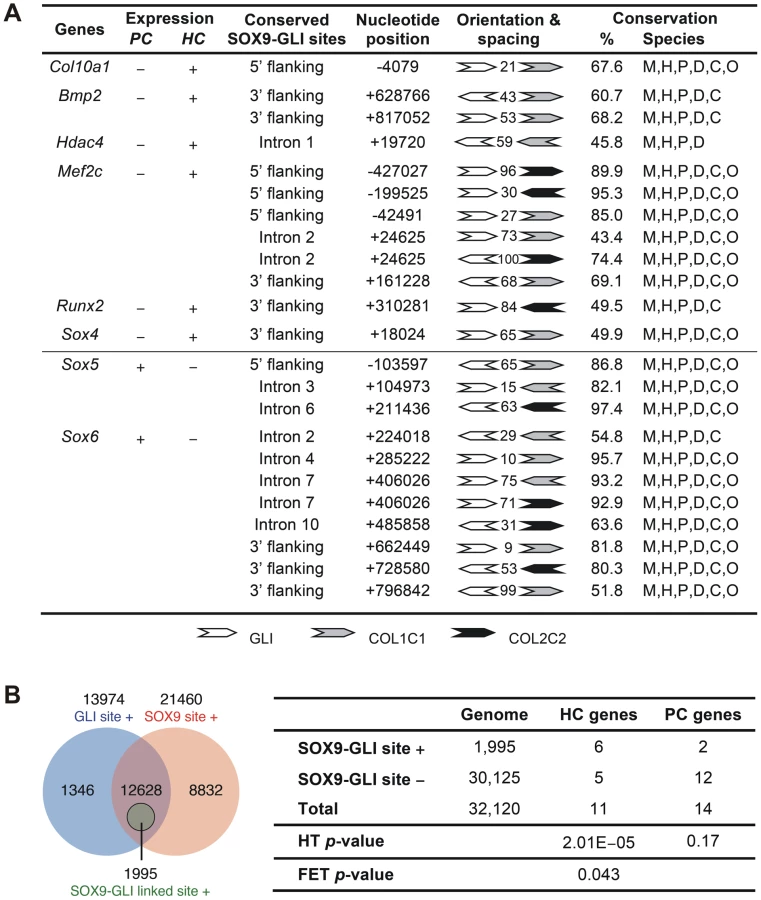
To assess whether the cooperation of SOX9 and GLI2/3 is a potential common mechanism for restricted or preferential gene expression in hypertrophic chondrocytes, we searched in silico for this configuration of binding sites in genes, other than Col10a1, that have strong and specific up-regulation in hypertrophic chondrocytes (HC genes) in the growth plate. Six of 11 HC genes analyzed, namely Col10a1, Bmp2, Hdac4, Mef2c, Runx2, and Sox4, possess the linked SOX9 and GLI sites (<100 nt spacing) in the inter - or intragenic conserved non-coding regions (Figure 7A and Figure S3). In contrast, these sites were absent from most of the genes tested (12 out of 14) that were expressed in proliferating but not (or down-regulated) in hypertrophic chondrocytes (PC genes). These include known SOX9 targets: Col2a1, Col9a1, Col11a2, Acan, and Mia1 (see Figure 7A legend for all negative genes). The exceptions were Sox5 and Sox6 (Figure 7A and Figure S3). To investigate whether the over-representation of linked SOX9-GLI sites in the HC genes but not the PC genes occurs by chance, we performed a hypergeometric test to calculate the probability of finding 6 or more SOX9-GLI site-containing genes out of 11 genes randomly sampled from the mouse genome. For the HC genes, the results showed that the occurrence of 6 or more genes with associated conserved SOX9-GLI sites is unlikely to occur by chance (p = 0.0000201) (Figure 7B). For the PC genes, the p-value was 0.17, which is comparable to random occurrence. Furthermore the frequency of the presence of SOX9-GLI sites for HC genes (6/11) was significantly higher than that for PC genes (2/14) (Fisher's test p = 0.043, one tailed). This suggests that the linked SOX9-GLI sites are preferentially associated with the HC genes.
Discussion
The positive and negative mechanisms mediating the stage-specific transcription of genes within the growth plate are not well defined, partly because of the difficulty in distinguishing direct effects on transcription from the consequences of abnormal differentiation. In this study we have exploited the specificity of Col10a1 expression in hypertrophic chondrocytes and the fact that manipulating its expression in vivo has no overt effect on differentiation, to dissect these transcriptional controls. We provide new insight into how differentiation stage-specific gene expression is achieved in the growth plate, presenting in vitro and in vivo evidence that SOX9, in addition to its known role as a transactivator of many genes preferentially expressed in non-hypertrophic chondrocytes, such as Col2a1, directly represses expression of Col10a1 at a stage prior to the onset of hypertrophy and subsequently in proliferating chondrocytes. This discovery extends our understanding of the mechanisms by which SOX9 controls chondrocyte differentiation phase-specific gene expression.
We have identified a conserved regulatory sequence, element A, that acts as an enhancer of Col10a1 expression in both cultured cells and in vivo. This element contains a SOX9 binding sequence that, when bound by SOX9, represses Col10a1 expression in immature and proliferating chondrocytes. Since Sox9 is expressed in non-hypertrophic chondrocytes but not in hypertrophic chondrocytes, this repressive action of SOX9 restricts Col10a1 expression to hypertrophic chondrocytes.
SOX9 has been proposed to direct chondrogenic fate in osteo-chondroprogenitor cells in part by interacting with RUNX2 [32], [33]. SOX9 may inhibit chondrocyte hypertrophy in part via activation of Bapx1 which represses Runx2 [34], [35]. Previous in vitro and in vivo studies suggest that Col10a1 expression is regulated positively by Mef2c, Runx2/Cbfa1, and AP-1 members, which are expressed in hypertrophic chondrocytes [19], [36]–[38]. RUNX2 has been shown to directly regulate the expression of Col10a1 [37]. The element A that we identified contains no conserved consensus RUNX site. The RUNX2 site revealed by Zheng et al. [37] is located within a poorly conserved region outside the element. Our data showed that the ectopic expression of Col10a1 transgene in non-hypertrophic chondrocytes does not require co-expression of Runx2. In addition, RUNX2 is not expressed in the costal hypertrophic chondrocytes and cultured hypertrophic chondrocytes MCTs (which is derived from costal cartilage), where Col10a1 expression is strong. Although real-time PCR showed levels of Col10a1 was markedly reduced in P1 Runx2-null mice [37], hypertrophic chondrocytes with strong Col10a1 expression do develop in many cartilages in Runx2 null fetuses [39], [40]. Collectively existing data suggest that RUNX2 together with other factors regulate Col10a1 in vivo via promoting chondrocyte hypertrophy or otherwise functions to initiate a cascade of regulatory pathways that sustain Col10a1 expression in hypertrophic chondrocytes.
Previous in vitro studies in chicken have suggested that a combined action of positive and negative DNA elements may contribute to the hypertrophic chondrocyte-specific expression of Col10a1 [41], [42]; however, these chick Col10a1 elements are not conserved in mammals. The enhancer element we identified is highly conserved in mammals, but not in chicken, which agrees with previous data [43]. This suggests that in both mammals and chicken, Col10a1 transcription is restricted to hypertrophic chondrocytes by repression, though by different cis-acting elements. In the chicken, this repression may extend to non-chondrogenic cell types [41]. We found no evidence to support such a mechanism in the mouse since when we abolished the interaction of SOX9 with the repressive element, we observed no ectopic Col10a1 expression in non-chondrogenic cells.
Consistent with a role for SOX9 in repressing Col10a1 in vivo, we have shown in Col10a1-Cre;Z-Sox9 mice, that activation of Sox9 expression in hypertrophic chondrocytes in costal cartilage caused down-regulation of Col10a1. It is also notable that in a recent report where Sox9 was over-expressed in hypertrophic chondrocytes in transgenic mice, expression of Col10a1 and the BAC-Col10-Sox9 transgene appeared reduced in the hypertrophic chondrocytes [44]. In the same report Sox9 knockdown in cultured chondrocytes did not affect Col10a1 expression [44]. By contrast Yamashita et al. showed that shRNA knockdown of Sox9 can up-regulate Col10a1 expression in primary costal chondrocyte culture, and that over-expression of Sox9 can down-regulate it [35]. These contradictory results may be related to the incomplete elimination of SOX9 protein and the known dosage dependent requirement for SOX9 action. A role for SOX9 as transcriptional repressor of Col10a1 in non-hypertrophic chondrocytes is consistent with the observation that COL10A1 expression was up-regulated in cartilage isolated from SOX9 haploinsufficient campomelic dysplasia patients [33].
How may SOX9 act both as a transactivator and a repressor in non-hypertrophic chondrocytes? It has been proposed that interactions between specific partner factors stabilize SOX protein binding to DNA and hence regulate target selection [24], thereby determining cell specification, as exemplified by SOX2 in embryonic stem cells and other systems [45]. Such selective cooperation of protein binding partners may mediate the concomitant positive and negative regulation of SOX9 target genes in the same cell that contributes to specification of the differentiation state (Figure 8). As illustrated in the schematic (Figure 8), we propose that SOX9 mediates repression of Col10a1 in proliferating chondrocytes by selective cooperation with GLI factors. Together with its role in activating Col2a1 and other matrix genes, SOX9 therefore plays an important role in maintaining chondrocytes in an immature non-hypertrophic state. RUNX2 by contrast promotes chondrocyte hypertrophy.

SOX9 cannot regulate chondrocyte differentiation appropriately without sonic hedgehog (Shh), which mediates the generation of chondrogenic precursor cells [46], and Indian hedgehog (Ihh), which regulates their proliferation and maturation [47]. GLI proteins are the effectors of Hh signaling. Double knockout mutants indicate that GLI2 has overlapping functions with GLI1 and GLI3 in skeletal and CNS development [48], [49]. Binding of Hh to its receptor, Patched, blocks the proteolytic processing of the GLI transcription factors from active (GLIA) to repressive (GLIR) forms, and the balance between these forms modulates hedgehog target gene expression [50]. In the growth plate, GLI2A can positively regulate chondrocyte hypertrophy and control vascularization of the hypertrophic cartilage in endochondral ossification [29], [51]. GLI3, which acts mainly as a repressor, has been suggested to inhibit chondrocyte hypertrophy [29], [52] and it is interesting that the highest interaction of element A was with GLI3. Mau et al. reported that Gli2/3 null mutations altered the expression domain of collagen X, but it was not possible to distinguish whether this was due to a direct effect on Col10a1 transcription or more general perturbation of hypertrophy [29]. How the GLI factors interact with other regulatory factors or genes in the chondrocyte differentiation program is not clear. Our results are consistent with cooperation between SOX9 and the Hh signaling pathway and suggest that SOX9 acts in synergy with GLI2 and GLI3, probably their repressive forms GLIR, to repress transcription in chondrocytes. Thus, reduced synergy between GLIR and SOX9 may explain the accelerated chondrocyte hypertrophy seen when Ptch1 is inactivated [53]. Over-representation of the SOX9-GLI paired consensus in a number of genes that are preferentially expressed in hypertrophic chondrocytes and not in proliferating chondrocytes, suggests that SOX9 may use this partnership to repress transcription of several genes in other chondrocyte types. However this partnership may not be the exclusive mechanism by which SOX9 acts to repress expression in chondrocytes.
Hattori et al. have recently shown that SOX9 directly represses Vegfa in cultured primary chondrocytes [44] by interacting with the 5′ untranslated region of the gene. This agrees with our findings of a repressive role of SOX9, however, we found no linked SOX9-GLI binding sites near the Vegfa gene and a recent SOX9 ChIP-on-chip study reported no in vivo interaction in Vegfa exon 1 [54].
A different mode by which SOX9 may repress gene expression in chondrocytes has been proposed by Huang et al [55]. In their model, SOX9 negatively regulates Ccn2 expression in non-hypertrophic chondrocytes via binding to overlapping binding sites for SOX and TCF/LEF, thereby interfering with binding of a TCF/LEF/β-catenin transactivation complex. Reduction of SOX9 upon hypertrophy allows this TCF/LEF/β-catenin complex to activate Ccn2 expression [55]. However, Ccn2 is also expressed in resting zone chondrocytes in the epiphyses of the growth plate and it is not clear why SOX9 does not repress the gene in these cells. We identified a conserved TCF consensus site in Col10a1 element A, but this is unlikely to interfere with SOX9 binding since it is located 59 bp downstream of the functional SOX9-GLI motif, unlike in Ccn2 where the SOX and TCF/LEF sites overlap (Figure 1C). This suggests that the model proposed by Huang et al. does not apply to Col10a1 element A-mediated repression of transcription.
It is also possible that SOX9 and GLI cooperate to activate or repress transcription depending on context. While our data implicate a cooperation of SOX9 with GLI factors in transcriptional repression, this association between SOX9 and GLI may not be restricted to negative regulation. Amano et al. have recently reported that GLI2 cooperates with SOX9 to transactivate the Pthlh gene (also known as PTHrP) in chondrocyte culture without direct binding to the gene [56]. However the expression patterns of Sox9 and Pthlh are mutually exclusive in the developing growth plate, Pthlh being expressed mainly in the perichondrium and only at extremely low levels in proliferating chondrocytes [57], [58]. This contradiction may reflect differences between in vitro assays and regulation in vivo, and it is also unclear whether the expressed GLI2 was processed to a repressor form or not in these cells. The observed stimulation of Pthlh promoter activity in the cultured chondrocytes could therefore be attributable to over-expression of GLI2 which persisted largely as the activated form GLI2A. However this report does raise the possibility of a context dependent SOX9-GLI partnership that mediates either transactivation or repression.
Sox9 has been suggested to act upstream of Sox5 and Sox6 in chondrogenesis [6], [46]. The presence of conserved SOX9–GLI sites in the Sox5 and Sox6 genes suggests their expression in proliferating chondrocytes may be positively controlled via cooperation of SOX9 with GLIA or GLI1, an activator that reinforces GLIA function. Hence, the roles played by SOX9 in transcriptional regulation may be determined by context—partnering with GLIA/GLI1 favours transactivation, with GLIR favours repression. Alternatively, as discussed above, the mode of regulation might depend on whether intermediate factors are present to interfere with the SOX9-GLI interaction. Interestingly, while there is a linked SOX9-GLI motif in Sox6, a conserved TCF site occurs between the SOX9 and GLI sites (Figure S3). Cooperation between SOX9 and TCF/LEF/β-catenin might therefore abrogate cooperative repression by SOX9 and GLI and transactivate Sox6 in proliferating chondrocytes.
Validation of these different modes of cooperative regulation by SOX9 and GLI factors in vivo would require the generation and analyses of compound null or conditional knockout mutants; however, the consequent dysregulation of chondrogenesis and impact on cell survival would make it impossible to distinguish changes in transcriptional control from effects on differentiation. For example, Sox9 is essential for chondrogenesis and Sox9 conditional null chondrocytes undergo apoptosis and as a consequence, hypertrophy with the characteristic activation of Col10a1 expression, fails to occur [4], [7], [59]. Because inactivation of Col10a1 does not disrupt the chondrogenic program, it provides an ideal system and tools to interrogate the transcriptional controls governing specificity of gene expression within the growth plate, independent of changes in chondrocyte differentiation. Important questions to be addressed in future are the identities and diversity of SOX9 partnerships and how the activity of SOX9 and its partners is modulated. In early myogenic differentiation, SOX9 and Smad3 have been reported to prevent premature expression of α-sarcoglycan gene expression by synergistic repression in a transforming growth factor β dependent manner [60] suggesting negative transcriptional regulation by SOX9 and its partners may be more common. The transactivation of Matn1 in chondrocytes by SOX9 can be modulated by combined action of L-SOX5, SOX6 and NFI factors [61]. Whether control of transcription by the SOX9-GLI partnership can be modulated by additional factors is an important question to be addressed in future.
In summary, our study implicates a complex regulatory function for SOX9 whereby it acts with different partners to orchestrate activation and repression of transcription in the chondrogenic differentiation pathway. Mutations in human SOX9 cause the skeletal malformation syndrome campomelic dysplasia which is attributed to the disruption of the chondrogenic differentiation program because of failure to express SOX9 target genes. This interpretation may need to be revised to include inappropriate expression of genes normally repressed by SOX9.
Materials and Methods
Bioinformatics analysis
COL10A1 sequences were aligned using LAGAN and PipMaker. The conserved SOX9 binding sites (COL2C1 and COL2C2) [10] were identified by rVISTA. The UCSC Mouse 30-Way Multiz Alignment data and a custom Perl script were used to identify human-mouse perfectly conserved SOX9, GLI, and TCF sites with a maximum spacing of 100 bp in the inter - and intra-genic non-coding regions. Conservation percentage of sequence spanning the SOX9 and GLI sites was calculated based on the number of perfectly matched nucleotides among all the aligned species (mouse, human, chimpanzee, canine, bovine, and opossum). From the 32,120 genes in the mouse genome, the number of genes containing conserved linked SOX9-GLI sites was found and used as the reference frequency of such genes in the genome. Whether the frequency of the presence of conserved SOX9-GLI sites in HC or PC genes exceeded this reference frequency was assessed by the hypergeometric distribution. The difference in the frequency of the presence of SOX9-GLI sites in the HC and PC genes was assessed by the Fisher's exact test.
Cell culture and transfection
Hypertrophic chondrocyte cell line MCTs (gift of Véronique Lefebvre [62]) was transfected with pCol10Flag-E, pSG-Sox9 (gift of Peter Koopman), or pSG5 expression vector using Fugene 6 (Roche). Expression of the exogenous collagen X from pCol10Flag-E in MCTs cells was examined by immunohistochemistry using anti-Flag M2 antibody (Sigma). CCL (gift of James Kimura [63]), MCT3T3-E1, and COS-1 cells were cultured in DMEM (Invitrogen) containing 10% FCS (Wisent) at 37°C at 5% CO2. MCTs cells were normally cultured at 32°C at 5% CO2 for expansion. Prior to assays, MCTs cells were cultured at 37°C for 1 day to induce growth arrest [62].
DNase I footprinting
A 300 bp DNA fragment within element A, corresponding to −4240 to −3935 bp of the mouse Col10a1, was used as probe. [γ-32P]ATP-labeled probes were incubated with nuclear extracts from CCL, MCTs, MCT3T3-E1, or COS-1 cells in the presence of poly(dA⋅dT) at room temperature, followed by DNase I digestion and denaturing PAGE.
Electromobility shift assays
COS-1 nuclear extracts over-expressing SOX9 and RUNX2 were pre-incubated with poly(dI⋅dC) or poly(dG⋅dC) at room temperature, followed by reaction with [γ-32P]ATP-labeled probes with or without the presence of nucleotide competitors, or antibodies for SOX9 (gift of Peter Koopman [64]) and OSF2/RUNX2 (gift of Gerard Karsenty [65]), then subjected to non-denaturing PAGE at room temperature. The sequences of oligonucleotide COL2C1 and OSE2 were as previously described [10], [65].
Col10Flag transgenic mice
The mouse Col10a1 element A, a 640 bp-fragment located between −4.2 and −3.6 kb, was cloned at the 5′ end of the pCol10Flag, previously known as FColX [18] consisting of −2070 to +7176 bp mouse Col10a1 genomic sequence, to generate pCol10Flag-E. The SOX9, TG-rich motif, and GLI binding sites in pCol10Flag-E were mutated to generate the single site mutants – respectively pCol10Flag-EΔ1, pCol10Flag-EΔ2, and pCol10Flag-EΔ3. All of these 3 motifs in pCol10Flag-E were mutated to generate pCol10Flag-EΔ4.
Col10a1-Cre;Z/Sox9 compound mutants
A 4.8 kb fragment of mouse genomic DNA (from 82 bp upstream of the start of transcription of Sox9 to 1119 bp downstream of the polyadenylation sequences), including the Sox9 coding region, its two introns and 1.1 kb of 3′ flanking DNA, together with an IRES2-EGFP (Clontech) sequence inserted between the Sox9 stop codon and the polyadenylation site (at +3237 bp), was cloned downstream of the loxP-flanked βgeo/3xpA of the pCall2 vector (gift of Andras Nagy [66]) to create the pZ/Sox9 expression vector. pZ/Sox9 was transfected into 129/SvEv-derived L4 embryonic stem (ES) cells by electroporation and ES clones containing a single copy of the transgene were injected into blastocysts followed by crossing of chimeras with C57BL/6N mice to generate the mouse line Z/Sox9. A mouse line carrying a single copy of pZ/Sox9 was generated which was then crossed with Col10a1-Cre mice [21] to obtain compound mutants.
Chromatin immunoprecipitation
CCL cell lysates or 13.5 dpc mouse limb tissue lysates were cross-linked followed by lysis and sonication to yield 200–500 bp DNA fragments then immunoprecipitation with antibodies against acetylated histone H3/H4 (Ac-H3, Ac-H4) (Upstate), HDAC4 (Abcam), GLI1, GLI2, GLI3 (all from Santa Cruz), SOX9 (gift of Robin Lovell-Badge [67]), TRAP230/MED12 (gift of Robert Roeder [25]), or TRPS1 (gift of Yasuteru Muragaki [26]). The target elements in Col10a1, Col2a1, or Crygb genes were amplified by real-time or standard PCR.
Gene expression analysis
In-situ hybridization was performed as previously described [9]. The probes used were pRK26 for Col10a1 [68], pWF21 for the Flag sequence [18], p88 for full-length Sox9 (gift of Peter Koopman), pBS-Cbfa1-Sh for full-length Runx2 (gift of Gerard Karsenty), pWF98 for full-length Egfp, pBSII-Cre-frag for Cre (gift of Andrew Groves), and pNJ61 for Col2a1 [9]. Gene expression in cell culture was analyzed by RT-PCR.
For additional details of all experiments, see the Text S1.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. AlmanBA 2008 Skeletal dysplasias and the growth plate. Clin Genet 73 24 30
2. KarsentyG 2008 Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet 9 183 196
3. KronenbergHM 2006 PTHrP and skeletal development. Ann N Y Acad Sci 1068 1 13
4. BiWDengJMZhangZBehringerRRDe CrombruggheB 1999 Sox9 is required for cartilage formation. Nat Genet 22 85 89
5. BiWHuangWWhitworthDJDengJMZhangZ 2001 Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A 98 6698 6703
6. AkiyamaHChaboissierMCMartinJFSchedlADe CrombruggheB 2002 The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16 2813 2828
7. IkegamiDAkiyamaHSuzukiANakamuraTNakanoT 2011 Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development 138 1507 1519
8. ZhaoQEberspaecherHLefebvreVDe-CrombruggheB 1997 Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn 209 377 386
9. NgLJWheatleySMuscatGEConwayCJBowlesJ 1997 SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol 183 108 121
10. BellDMLeungKKWheatleySCNgLJZhouS 1997 SOX9 directly regulates the type-II collagen gene. Nat Genet 16 174 178
11. LefebvreVHuangWHarleyVRGoodfellowPNDe-CrombruggheB 1997 SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 17 2336 2346
12. LiuYLiHTanakaKTsumakiNYamadaY 2000 Identification of an enhancer sequence within the first intron required for cartilage-specific transcription of the alpha2(XI) collagen gene. J Biol Chem 275 12712 12718
13. SekiyaITsujiKKoopmanPWatanabeHYamadaY 2000 SOX9 enhances aggrecan gene promoter/enhancer activity and is up-regulated by retinoic acid in a cartilage-derived cell line, TC6. J Biol Chem 275 10738 10744
14. XieWFZhangXSakanoSLefebvreVSandellLJ 1999 Trans-activation of the mouse cartilage-derived retinoic acid-sensitive protein gene by Sox9. J Bone Miner Res 14 757 763
15. HanYLefebvreV 2008 L-Sox5/Sox6 Drive Expression of the Aggrecan Gene in Cartilage by Securing Binding of Sox9 to a Far-upstream Enhancer. Mol Cell Biol 28 4999 5013
16. RosatiRHoranGSPineroGJGarofaloSKeeneDR 1994 Normal long bone growth and development in type X collagen-null mice. Nat Genet 8 129 135
17. KwanKMPangMKZhouSCowanSKKongRY 1997 Abnormal compartmentalization of cartilage matrix components in mice lacking collagen X: implications for function. J Cell Biol 136 459 471
18. HoMSTsangKYLoRLSusicMMakitieO 2007 COL10A1 nonsense and frame-shift mutations have a gain-of-function effect on the growth plate in human and mouse metaphyseal chondrodysplasia type Schmid. Hum Mol Genet 16 1201 1215
19. ChambersDYoungDAHowardCThomasJTBoamDS 2002 An enhancer complex confers both high-level and cell-specific expression of the human type X collagen gene. FEBS Lett 531 505 508
20. LefebvreVLiPDe CrombruggheB 1998 A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J 17 5718 5733
21. TsangKYChanDCheslettDChanWCSoCL 2007 Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol 5 e44
22. ScottCEWynnSLSesayACruzCCheungM 2010 SOX9 induces and maintains neural stem cells. Nature neuroscience 13 1181 1189
23. WilsonMKoopmanP 2002 Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12 441 446
24. KamachiYCheahKSEKondohH 1999 Mechanism of regulatory target selection by the SOX high-mobility-group domain proteins as revealed by comparison of SOX1/2/3 and SOX9 [In Process Citation]. Mol Cell Biol 19 107 120
25. ZhouRBonneaudNYuanCXde Santa BarbaraPBoizetB 2002 SOX9 interacts with a component of the human thyroid hormone receptor-associated protein complex. Nucleic Acids Res 30 3245 3252
26. NishiokaKItohSSuemotoHKannoSGaiZ 2008 Trps1 deficiency enlarges the proliferative zone of growth plate cartilage by upregulation of Pthrp. Bone 43 64 71
27. WuellingMKaiserFJBuelensLABraunholzDShivdasaniRA 2009 Trps1, a regulator of chondrocyte proliferation and differentiation, interacts with the activator form of Gli3. Dev Biol 328 40 53
28. VegaRBMatsudaKOhJBarbosaACYangX 2004 Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119 555 566
29. MauEWhetstoneHYuCHopyanSWunderJS 2007 PTHrP regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner utilizing hedgehog ligand dependent and independent mechanisms. Dev Biol 305 28 39
30. KoyamaEYoungBNagayamaMShibukawaYEnomoto-IwamotoM 2007 Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134 2159 2169
31. Ruiz i AltabaAMasCSteccaB 2007 The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends in cell biology 17 438 447
32. EamesBFSharpePTHelmsJA 2004 Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Dev Biol 274 188 200
33. ZhouGZhengQEnginFMunivezEChenY 2006 Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A 103 19004 19009
34. SaitoTIkedaTNakamuraKChungUIKawaguchiH 2007 S100A1 and S100B, transcriptional targets of SOX trio, inhibit terminal differentiation of chondrocytes. EMBO Rep 8 504 509
35. YamashitaSAndohMUeno-KudohHSatoTMiyakiS 2009 Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp Cell Res 315 2231 2240
36. RiemerSGebhardSBeierFPoschlEvon derM 2002 Role of c-fos in the regulation of type X collagen gene expression by PTH and PTHrP: Localization of a PTH/PTHrP-responsive region in the human COL10A1 enhancer. J Cell Biochem 86 688 699
37. ZhengQZhouGMorelloRChenYGarcia-RojasX 2003 Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol 162 833 842
38. ArnoldMAKimYCzubrytMPPhanDMcAnallyJ 2007 MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 12 377 389
39. GuoJChungUIYangDKarsentyGBringhurstFR 2006 PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol 292 116 128
40. InadaMYasuiTNomuraSMiyakeSDeguchiK 1999 Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn 214 279 290
41. LuVPIwamotoMFanningPPacificiMOlsenBR 1993 Multiple negative elements in a gene that codes for an extracellular matrix protein, collagen X, restrict expression to hypertrophic chondrocytes. J Cell Biol 121 1173 1179
42. MageeCNurminskayaMFavermanLGaleraPLinsenmayerTF 2005 SP3/SP1 transcription activity regulates specific expression of collagen type X in hypertrophic chondrocytes. J Biol Chem 280 25331 25338
43. GebhardSPoschlERiemerSBauerEHattoriT 2004 A highly conserved enhancer in mammalian type X collagen genes drives high levels of tissue-specific expression in hypertrophic cartilage in vitro and in vivo. Matrix Biol 23 309 322
44. HattoriTMullerCGebhardSBauerEPauschF 2010 SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137 901 911
45. KondohHKamachiY 2009 SOX-partner code for cell specification: Regulatory target selection and underlying molecular mechanisms. Int J Biochem Cell Biol 42 391 399
46. AkiyamaHStadlerHSMartinJFIshiiTMBeachyPA 2007 Misexpression of Sox9 in mouse limb bud mesenchyme induces polydactyly and rescues hypodactyly mice. Matrix Biol 26 224 233
47. St-JacquesBHammerschmidtMMcMahonAP 1999 Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13 2072 2086
48. MoRFreerAMZinykDLCrackowerMAMichaudJ 1997 Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124 113 123
49. ParkHLBaiCPlattKAMatiseMPBeeghlyA 2000 Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127 1593 1605
50. JiangJHuiCC 2008 Hedgehog signaling in development and cancer. Dev Cell 15 801 812
51. JoengKSLongF 2009 The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 136 4177 4185
52. KozielLWuellingMSchneiderSVortkampA 2005 Gli3 acts as a repressor downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development 132 5249 5260
53. MakKKKronenbergHMChuangPTMackemSYangY 2008 Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135 1947 1956
54. OhCDMaitySNLuJFZhangJLiangS Identification of SOX9 interaction sites in the genome of chondrocytes. PLoS One 5 e10113
55. HuangBLBruggerSMLyonsKM Stage-specific control of connective tissue growth factor (CTGF/CCN2) expression in chondrocytes by Sox9 and beta-catenin. J Biol Chem 285 27702 27712
56. AmanoKHataKSugitaATakigawaYOnoK 2009 Sox9 family members negatively regulate maturation and calcification of chondrocytes through up-regulation of parathyroid hormone-related protein. Mol Biol Cell 20 4541 4551
57. KobayashiTChungUISchipaniEStarbuckMKarsentyG 2002 PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 129 2977 2986
58. ChenXMacicaCMDreyerBEHammondVEHensJR 2006 Initial characterization of PTH-related protein gene-driven lacZ expression in the mouse. J Bone Miner Res 21 113 123
59. CheungMChaboissierMCMynettAHirstESchedlA 2005 The transcriptional control of trunk neural crest induction, survival, and delamination. Developmental cell 8 179 192
60. Hernandez-HernandezJMDelgado-OlguinPAguillon-HuertaVFurlan-MagarilMRecillas-TargaF 2009 Sox9 represses alpha-sarcoglycan gene expression in early myogenic differentiation. Journal of molecular biology 394 1 14
61. NagyAKenesiERentsendorjOMolnarASzenasiT 2011 Evolutionarily conserved, growth plate zone-specific regulation of the matrilin-1 promoter: L-Sox5/Sox6 and Nfi factors bound near TATA finely tune activation by Sox9. Molecular and cellular biology 31 686 699
62. LefebvreVGarofaloSDe CrombruggheB 1995 Type X collagen gene expression in mouse chondrocytes immortalized by a temperature-sensitive simian virus 40 large tumor antigen. J Cell Biol 128 239 245
63. MukhopadhyayKLefebvreVZhouGGarofaloSKimuraJH 1995 Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-alpha 1(II) collagen gene. J Biol Chem 270 27711 27719
64. WilhelmDMartinsonFBradfordSWilsonMJCombesAN 2005 Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev Biol 287 111 124
65. DucyPZhangRGeoffroyVRidallALKarsentyG 1997 Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89 747 754
66. NovakAGuoCYangWNagyALobeCG 2000 Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28 147 155
67. SekidoRLovell-BadgeR 2008 Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453 930 934
68. KongRYKwanKMLauETThomasJTBoot-HandfordRP 1993 Intron-exon structure, alternative use of promoter and expression of the mouse collagen X gene, Col10a-1. Eur J Biochem 213 99 111
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 11
Nejčtenější v tomto čísle
- Evidence-Based Annotation of Gene Function in MR-1 Using Genome-Wide Fitness Profiling across 121 Conditions
- De Novo Origins of Human Genes
- Capture of MicroRNA–Bound mRNAs Identifies the Tumor Suppressor miR-34a as a Regulator of Growth Factor Signaling
- TRY-5 Is a Sperm-Activating Protease in Seminal Fluid