
Diced Triplets Expose Neurons to RISC
article has not abstract
Published in the journal:
. PLoS Genet 8(2): e32767. doi:10.1371/journal.pgen.1002545
Category:
Perspective
doi:
https://doi.org/10.1371/journal.pgen.1002545
Summary
article has not abstract
Expansions of short repeats—those with units of ≤12 bp—account for as many as 40 diseases [1]–[4]. About half of these disorders arise from expanded tracts of CAG/CTG triplets, many encoding polyglutamine. Since the discovery of the first polyglutamine-encoding CAG repeat disorder in 1991 [5], the predominant hypothesis has been that pathogenesis of the CAG category is a consequence of a toxic gain-of-function of excessively long strands of polyglutamine. Polyglutamine toxicity has been most systematically explored in Huntington's disease (HD), with evidence that it influences multiple processes, including transcriptional regulation, mitochondrial energy production, and calcium regulation. This proteocentric view is undergoing considerable revision, as mounting evidence suggests toxic roles for mutant transcripts in HD [6] (Figure 1). The initial clues regarding CNG transcript toxicity emerged from studies of myotonic dystrophy type 1 (DM1). DM1 is caused by an expanded CTG repeat located in the 3′ end of the DMPK gene. Transcripts with long CUG repeats dysregulate the splicing factors MBNL1 and CUGBP1, leading to aberrant splicing of numerous downstream transcripts; dysfunction of these proteins directly correlates with various features of the disease phenotype. Subsequently, multiple lines of evidence have emerged that RNA toxicity contributes to the pathogenesis of other CAG/CTG disorders. Structurally, RNA with sufficiently long stretches of CUG or CAG triplets can form hairpin structures likely to influence the affinity of RNA binding proteins [7]. At least partly as a consequence of these structures and changes in protein binding, transcripts with either type of repeat may aggregate into discrete foci that include MBNL1 [8], [9]. In HD, RNA foci and misregulation of splicing have been detected in peripheral HD tissue [9]. The potential toxicity of transcripts containing long CAG tracts has been demonstrated in fly, worm, and mouse systems [10]–[14]. HDL2, a disorder clinically and pathologically similar to HD, involves CUG transcript toxicity mediated by dysregulation of MBNL1 [15], [16]. A second potential mechanism of CAG/CTG toxicity emerged from evidence that bidirectional transcription through the HTT repeat region [17] is a source of Dicer-generated CAG/CUG repeat siRNAs capable of targeting cellular transcripts containing complementary repeats [18].
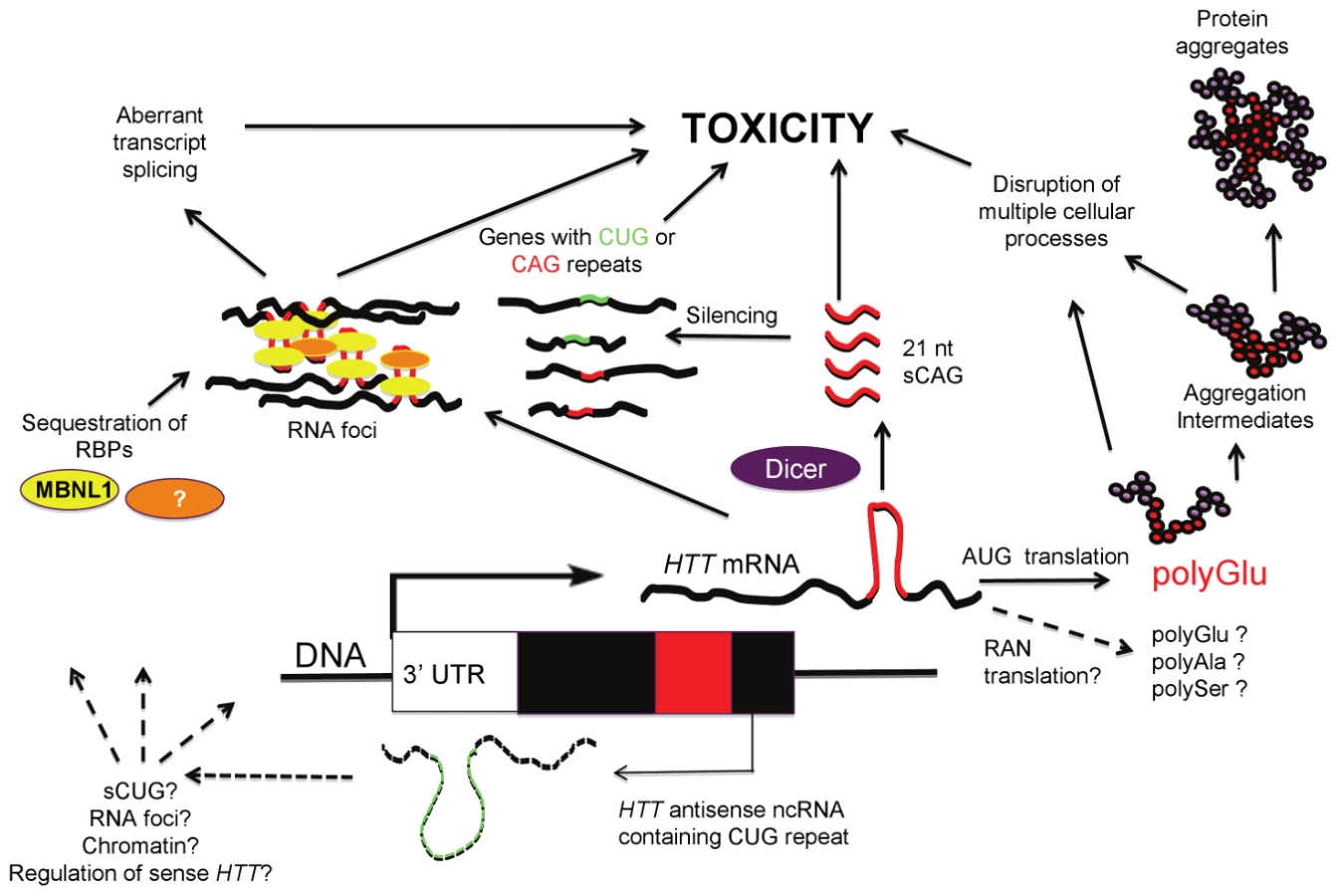
In this issue of PLoS Genetics, Bañez-Coronel and colleagues [19] provide further evidence for the involvement of HTT RNA and the RNAi pathway in HD pathogenesis. The authors demonstrate that overexpression of translatable and non-translatable HTT exon 1 constructs with expanded CAG repeats leads to Dicer-dependent production of short CAG repeat RNAs (sCAGs) with cytotoxic properties. Cytotoxic effects are triggered by expanded CAG repeats (which can form RNA hairpins), but not by expanded CAA repeats (which, like CAG, encode glutamine, but cannot form hairpins), consistent with recent findings in a fly model [11]. sCAG species were detected in Ago-2 complexes, supporting association with RNAi pathways. Antisense inhibitors of the sCAG species reverse cytotoxicity, and sCAGs were detected in R6/2 HD transgenic mice and in postmortem human HD brain tissue. sCAGs isolated from human HD tissue and then transfected into cells induced toxicity. The pathogenically relevant targets of the sCAGs remain to be determined, but initial experiments suggest several potential transcripts, including ADORA2A and MEIS2 (both reduced in HD brain tissue) and more variably DMPK, ASTN2, and ZFR, all containing either fully or partially complementary CUG and CAG repeats. Determining a more complete list of target sequences, and the extent to which downregulation is necessary or sufficient for toxicity, remain critical issues for further exploration. Curiously, the sCAG species isolated from HD models and human HD brain that induced toxicity were not a homogenous population of RNAs, but were identified in the <100-nt fraction. While cytotoxicity was sCAG-dependent (as toxicity was blocked with anti-sCAG), the relative contribution of sCAGs compared to other miRNAs in the isolated fraction is unknown. Whether Dicer is the only ribonuclease involved in sCAG production also remains to be determined.
It is noteworthy that a DM1 antisense transcript containing the repeat in the CAG orientation is also converted to 21-nt fragments that include CAG units [20], similar to the 21-nt sCAG fragments from the HD locus reported by Bañez-Coronel et al. [19]. While the function of the DM1 CAG fragments remains unknown, it was suggested that they may play a role in the abnormal chromatinization at the DM1 locus that occurs in the presence of the expansion mutation [20], raising the possibility that a similar phenomenon may also occur at other loci, such as HD, where sCNG fragments are generated. Both the DM1 and SCA7 antisense transcripts are thought to regulate their complementary sense transcripts [20], [21]. Conversely, the findings in HD by Bañez-Coronel et al. suggest that HD sCAG fragments might regulate non-HD CUG - and CAG-containing transcript levels (Figure 1), possibly through an RNA-RNA hybrid mechanism. This may occur through processes similar to the RNA-RNA hybrids formed between the expanded DMPK CUG repeats and the short CAG repeats in CUGBP1 mRNA, proposed to regulate the reduced CUGBP1 mRNA levels in DM1 patient muscles [22].
What regulates the expression of the HTT antisense and sCAG fragments is unknown but may involve epigenetic factors. The expression of the DM1 and SCA7 antisense transcripts are regulated by CTCF binding at sites proximal to the repeat and promoter regions coincident with localized chromatin modifications [20], [21]. Interestingly, CTCF binding also regulates SCA7 CAG instability [23]. The potential role of the HD CTCF site [17], [24] in regulating the expression of HTT or HTT antisense is yet to be determined.
Two recent observations complicate the interpretation of the Bañez-Coronel et al. findings [19]. First, homopolymeric polyalanine or polyserine proteins were found to be expressed via a mechanism termed Repeat Associated Non-ATG Translation (RAN) recently described by Zu et al. [25], reviewed in [26]; this raises the possibility that toxic RAN proteins could contribute to the pathogenesis induced by the AUG-free “untranslatable” HTT RNA fragment (Figure 1). Secondly, since the construct used by Bañez-Coronel et al. contains the entire HTT exon 1, it is also possible that the recently described antisense HTT transcript is coexpressed with the sense strand transcript [17]. Thus, the long CUG HTT antisense transcript might itself contribute to cytotoxicity, and/or lead to RAN-translated products, and/or influence the process by which sCAGs are generated (Figure 1).
While Bañez-Coronel et al. [19] primarily focus on the role of RNAi pathways and the toxicity of sCAGs, it is likely that toxicity induced by HTT sense and antisense RNAs, as in the case of HTT protein, involves multiple pathways, each warranting exploration. For example, what is the pathogenic effect of the aggregation of transcripts containing long CAG or CUG repeats? Might this lead to sequestration or dysregulation of splicing factors, as in DM1? Distinct proteins bind to mRNA containing CAG and CUG repeats—do properties of the transcripts, such as repeat length and the sequence of regions flanking the repeat, modulate this binding? Does the CAG repeat tract length affect transcript stability, or the efficiency of transcription or translation? What is the relationship between CAG expression level and CAG repeat length in inducing toxicity? Does RNA-mediated toxicity provide any clues to selective neuronal vulnerability in HD? Does RAN-translation arise in HD as it does in DM1 and SCA7 patient tissues? These questions demonstrate that every step of the mRNA life cycle in CAG/CTG disease warrants exploration.
Of utmost importance, the findings of Bañez-Coronel et al. [19] and others that implicate RNA in HD pathogenesis provide new leads in the search for therapeutic targets. Targeting only the mechanisms induced by expanded polyglutamine tracts may not be sufficient to stop disease pathogenesis. A comprehensive strategy to combat HD will require attention to RNA-mediated toxicity.
Zdroje
1. KobayashiHAbeKMatsuuraTIkedaYHitomiT 2011 Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 89 121 130
2. RentonAEMajounieEWaiteASimon-SanchezJRollinsonS 2011 A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 257 268
3. DeJesus-HernandezMMackenzieIRBoeveBFBoxerALBakerM 2011 Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 245 256
4. Lopez CastelAClearyJDPearsonCE 2010 Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11 165 170
5. La SpadaARWilsonEMLubahnDBHardingAEFischbeckKH 1991 Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352 77 79
6. WojciechowskaMKrzyzosiakWJ 2011 CAG repeat RNA as an auxiliary toxic agent in polyglutamine disorders. RNA Biol 8 565 571
7. MichlewskiGKrzyzosiakWJ 2004 Molecular architecture of CAG repeats in human disease related transcripts. J Mol Biol 340 665 679
8. HoTHSavkurRSPoulosMGManciniMASwansonMS 2004 Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci 18 2923 2933
9. MykowskaASobczakKWojciechowskaMKozlowskiPKrzyzosiakWJ 2011 CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39 8938 8951
10. LiLBYuZTengXBoniniNM 2008 RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453 1107 1111
11. LawlorKTO'KeefeLVSamaraweeraSEvan EykCLMcLeodCJ 2011 Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum Mol Genet 20 3757 3768
12. TsoiHLauCKLauKFChanHY 2011 Perturbation of U2AF65/NXF1-mediated RNA nuclear export enhances RNA toxicity in polyQ diseases. Hum Mol Genet 20 3787 3797
13. WangHLimPJYinCRieckherMVogelBE 2006 Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet 15 1025 1041
14. HsuRJHsiaoKMLinMJLiCYWangLC 2011 Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS ONE 6 e16417 doi:10.1371/journal.pone.0016417
15. HolmesSEO'HearnERosenblattACallahanCHwangHS 2001 A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29 377 378
16. RudnickiDDHolmesSELinMWThorntonCARossCA 2007 Huntington's disease–like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol 61 272 282
17. ChungDWRudnickiDDYuLMargolisRL 2011 A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet 20 3467 3477
18. de MezerMWojciechowskaMNapieralaMSobczakKKrzyzosiakWJ 2011 Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res 39 3852 3863
19. Bañez-CoronelMPortaSKagerbauerBMateuEPantanoL 2012 A pathogenic mechanism in Huntington's disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet 8 e10002481 doi:10.1371/journal.pgen.1002481
20. ChoDHThienesCPMahoneySEAnalauEFilippovaGN 2005 Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell 20 483 489
21. SopherBLLaddPDPinedaVVLibbyRTSunkinSM 2011 CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 70 1071 1084
22. WatanabeTTakagiASasagawaNIshiuraSNakaseH 2004 Altered expression of CUG binding protein 1 mRNA in myotonic dystrophy 1: possible RNA-RNA interaction. Neurosci Res 49 47 54
23. LibbyRTHagermanKAPinedaVVLauRChoDH 2008 CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet 4 e1000257 doi:10.1371/journal.pgen.1000257
24. FilippovaGNThienesCPPennBHChoDHHuYJ 2001 CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet 28 335 343
25. ZuTGibbensBDotyNSGomes-PereiraMHuguetA 2011 Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 108 260 265
26. PearsonCE 2011 Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet 7 e1002018 doi:10.1371/journal.pgen.1002018
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 2
Nejčtenější v tomto čísle
- Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA
- Contrasting Properties of Gene-Specific Regulatory, Coding, and Copy Number Mutations in : Frequency, Effects, and Dominance
- Homeobox Genes Critically Regulate Embryo Implantation by Controlling Paracrine Signaling between Uterine Stroma and Epithelium
- Nondisjunction of a Single Chromosome Leads to Breakage and Activation of DNA Damage Checkpoint in G2