
Regulating Repression: Roles for the Sir4 N-Terminus in Linker DNA Protection and Stabilization of Epigenetic States
Silent information regulator proteins Sir2, Sir3, and Sir4 form a heterotrimeric complex that represses transcription at subtelomeric regions and homothallic mating type (HM) loci in budding yeast. We have performed a detailed biochemical and genetic analysis of the largest Sir protein, Sir4. The N-terminal half of Sir4 is dispensable for SIR–mediated repression of HM loci in vivo, except in strains that lack Yku70 or have weak silencer elements. For HM silencing in these cells, the C-terminal domain (Sir4C, residues 747–1,358) must be complemented with an N-terminal domain (Sir4N; residues 1–270), expressed either independently or as a fusion with Sir4C. Nonetheless, recombinant Sir4C can form a complex with Sir2 and Sir3 in vitro, is catalytically active, and has sedimentation properties similar to a full-length Sir4-containing SIR complex. Sir4C-containing SIR complexes bind nucleosomal arrays and protect linker DNA from nucleolytic digestion, but less effectively than wild-type SIR complexes. Consistently, full-length Sir4 is required for the complete repression of subtelomeric genes. Supporting the notion that the Sir4 N-terminus is a regulatory domain, we find it extensively phosphorylated on cyclin-dependent kinase consensus sites, some being hyperphosphorylated during mitosis. Mutation of two major phosphoacceptor sites (S63 and S84) derepresses natural subtelomeric genes when combined with a serendipitous mutation (P2A), which alone can enhance the stability of either the repressed or active state. The triple mutation confers resistance to rapamycin-induced stress and a loss of subtelomeric repression. We conclude that the Sir4 N-terminus plays two roles in SIR–mediated silencing: it contributes to epigenetic repression by stabilizing the SIR–mediated protection of linker DNA; and, as a target of phosphorylation, it can destabilize silencing in a regulated manner.
Published in the journal:
. PLoS Genet 8(5): e32767. doi:10.1371/journal.pgen.1002727
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002727
Summary
Silent information regulator proteins Sir2, Sir3, and Sir4 form a heterotrimeric complex that represses transcription at subtelomeric regions and homothallic mating type (HM) loci in budding yeast. We have performed a detailed biochemical and genetic analysis of the largest Sir protein, Sir4. The N-terminal half of Sir4 is dispensable for SIR–mediated repression of HM loci in vivo, except in strains that lack Yku70 or have weak silencer elements. For HM silencing in these cells, the C-terminal domain (Sir4C, residues 747–1,358) must be complemented with an N-terminal domain (Sir4N; residues 1–270), expressed either independently or as a fusion with Sir4C. Nonetheless, recombinant Sir4C can form a complex with Sir2 and Sir3 in vitro, is catalytically active, and has sedimentation properties similar to a full-length Sir4-containing SIR complex. Sir4C-containing SIR complexes bind nucleosomal arrays and protect linker DNA from nucleolytic digestion, but less effectively than wild-type SIR complexes. Consistently, full-length Sir4 is required for the complete repression of subtelomeric genes. Supporting the notion that the Sir4 N-terminus is a regulatory domain, we find it extensively phosphorylated on cyclin-dependent kinase consensus sites, some being hyperphosphorylated during mitosis. Mutation of two major phosphoacceptor sites (S63 and S84) derepresses natural subtelomeric genes when combined with a serendipitous mutation (P2A), which alone can enhance the stability of either the repressed or active state. The triple mutation confers resistance to rapamycin-induced stress and a loss of subtelomeric repression. We conclude that the Sir4 N-terminus plays two roles in SIR–mediated silencing: it contributes to epigenetic repression by stabilizing the SIR–mediated protection of linker DNA; and, as a target of phosphorylation, it can destabilize silencing in a regulated manner.
Introduction
The eukaryotic genome is organized into euchromatic and heterochromatic domains that generally reflect their potential for gene expression. Chromatin repressed by the Silent information regulator (SIR) complex in the budding yeast Saccharomyces cerevisiae shares many key features with heterochromatin in higher eukaryotes. Notably, it has hypoacetylated nucleosomes [1], [2], is less accessible to DNA-binding enzymes than is euchromatin [3]–[5], it replicates late in S phase [6] and is spatially sequestered at the nuclear envelope or near the nucleolus [7]. The genes found within heterochromatin are generally silent, and in complex organisms this gene repression is crucial for the proper development of differentiated tissues and organs [8].
Unlike the situation in other eukaryotes, where histone H3 lysine 9 methylation and its specific ligands mediate repression, heritable transcriptional silencing in S. cerevisiae relies on the association of a trimeric SIR complex with unmodified histones (reviewed in [9]–[12]). This heterotrimeric complex contains equimolar amounts of Sir2, Sir3 and Sir4 [13], each of which is essential for the repression of promoters at the homothallic mating type loci, HMR and HML [14] and in subtelomeric domains [15]. In analogy to centromeric position effect variegation in flies, repression at telomeres has been called telomere position effect, or TPE.
The SIR complex is targeted to the genes it represses by interacting with sequence-specific DNA-binding proteins that bind silencers or telomeric TG repeats. This binding initiates or “nucleates” the formation of silent chromatin on adjacent genes. Repressor activator protein 1 (Rap1; [16]) is a key factor for SIR-mediated repression, because it has high affinity sites both at telomeres and in silencer elements [16], [17]. Furthermore, Rap1 interacts with both Sir3 and Sir4 [18]. HM silencer elements contain sites for two further sequence-specific factors, namely Abf1 (ARS-binding factor 1) and ORC (Origin recognition complex) [19], [20]. Abf1 recruits the SIR complex by binding to Sir3 [10], and the largest subunit of ORC, Orc1, enhances SIR recruitment by binding Sir1, an intermediary protein that in turn binds Sir4 [21].
The initial recruitment of Sir4 or Sir3 to telomeric TG-repeats or to silencers, brings in Sir2, a histone deacetylase [22]–[24], which generates high-affinity binding sites for Sir3 by removing acetylation from the histone N-termini of nearby nucleosomes [25]–[27]. Sir3 binds nucleosomes in a manner that is highly sensitive to histone H4 K16 acetylation [28]. The sequential activation of this NAD-dependent histone deacetylase, its generation of high affinity binding sites for Sir3, and their occupancy by the trimeric SIR complex, allow a repressive chromatin structure to propagate along the chromatin fiber [29], [30]. Whereas Sir4 can be recruited to silencer elements independently of Sir2 and Sir3, the spreading of the SIR complex and formation of a silent domain require all three proteins [30], [31]. Mutations that disrupt the interaction between Sir3 and Sir4 compromise repression of the HM loci and of genes at telomeres [32], [33].
At 152 kDa, Sir4 is the largest and the least well conserved of the Sir proteins [34]. Its non-globular structure has rendered it refractory to biochemical analysis, except when expressed together with Sir2 [13]. Sir2 and Sir4 form a stable heterodimer, which is mediated by residues 737–839 of Sir4 and a large pocket situated between Sir2's non-conserved N-terminus and its C-terminal catalytic domain (R. Sternglanz and R-M. Xu, personal communication). This tight interaction enhances the de-acetylation activity of Sir2 in vitro [13], [35]. Sir4 also interacts with an array of additional factors that are required for efficient repression, leading to its designation as a scaffold for silent chromatin assembly [10], [11]. Importantly, the C-terminal coiled-coil of Sir4 (residues 1257–1358) dimerizes to generate Sir3-binding sites on its outer surface [36], [37], and this interphase is essential for SIR-mediated repression [32]. This coiled-coil domain also binds Yku70 and Rap1 [38]–[41]. Yku70's interaction partner, Yku80, binds two sites within Sir4, one at the Sir4 N-terminus and one in the C-terminal 627 residues [42], [43]. The Ku heterodimer (Yku70/Yku80) not only facilitates SIR recruitment at telomeres, but helps anchor telomeres and silent chromatin at the nuclear envelope, which can enhance the efficiency of SIR-mediated repression [40], [44], [45]. A second, more central domain of Sir4 called PAD (residues 950–1262; partitioning and anchoring domain) also mediates anchorage to the nuclear envelope [42], [46], [47]. The PAD domain of Sir4 binds a nuclear envelope-associated protein called Esc1 (Establishes silent chromatin 1) [47], [48]. Disruption of ESC1 and YKU70 or YKU80 releases telomeres from the nuclear envelope, and selectively de-represses TPE, while repression at HM loci remains intact [42], [49], [50].
It is not surprising that the C-terminal half of Sir4 is crucial for silencing, given that it mediates protein-protein interactions with Rap1, Sir2, Sir3, Sir4, Yku70/Yku80 and Esc1. Although we know much less about the functions of the N-terminal part of Sir4, Marshall et al. [51] reported that the N-terminus of Sir4 was required for silencing at the HM loci. They showed that expression in trans of an N-terminal fragment restored mating in the presence of a silencing-deficient C-terminal fragment of Sir4 (the final 45%, starting from about residue 744) [51]. Since then, the first 270 residues of Sir4 (Sir4N) were shown to bind DNA in vitro [52] and to interact with three proteins: Sir1 [21], Yku80 [43] and Sif2 [53], a component of the SET3C deacetylase complex [53], [54]. Although Sir4 binding to Sir1 or Yku80 facilitates SIR complex recruitment to HM loci and telomeres, neither interaction is essential for SIR-mediated silencing [49], [55], [56]. Thus, it remained mysterious what function the Sir4 N-terminus might have.
Here we have explored the function of the N - and C-terminal domains of Sir4 in silencing at both the HM loci and yeast telomeres by means of biochemical and genetic assays. We re-examined the ability of the N - and C-termini to work together in trans and found, surprisingly, that a slightly shorter C-terminal fragment (Sir4C; residues 747–1358) than that used by Marshall et al. [51], is sufficient to silence HMR and HML in a sir4Δ background. Neither this C-terminal domain nor a fusion protein of Sir4C to the N-terminal 270 residues, however, was sufficient to complement fully a sir4 deletion for TPE. From this we conclude that the Sir4 N-terminus is dispensable for formation of a repressed chromatin structure, yet it is needed at telomeres or in situations in which SIR complex recruitment is compromised.
We confirmed by biochemical reconstitution assays that recombinant Sir4C is sufficient to form a complex with Sir2 and Sir3 that binds nucleosomal arrays in vitro and deacetylates histone H4 K16ac. However, Sir4C-containing complexes bind with a four-fold lower affinity and confer less protection of linker DNA from micrococcal nuclease attack. Thus, the DNA binding affinity of Sir4N contributes substantially to the tight association of the SIR complex with chromatin, which becomes important when recruitment is compromised. To see if silencing is regulated through Sir4, we mapped phosphorylation sites within Sir4N in vivo and in vitro, and found that this domain is a major target for phosphorylation in living cells. Two key phosphoacceptor sites for the cyclin-dependent kinase, serine 63 and serine 84, influence the stability of repression at most telomeres showing TPE. We propose that Sir4N phosphorylation regulates the stability of subtelomeric repression during the cell cycle and possibly in response to environmental stress.
Results
Sir4C is sufficient for silencing at intact HML and HMR loci
To examine the function of the N-terminus of Sir4, we first repeated the assay of Marshall et al. [51] in which N - and C-terminal fragments of Sir4 were expressed in trans and scored for the restoration of silencing at HML in a sir4Δ background. We created strains with either a full deletion of SIR4 (sir4Δ) or with a partial deletion of the endogenous SIR4 locus (sir4N), such that only its N-terminal 270 amino acids were expressed. We then expressed full-length Sir4 or various C-terminal fragments of the protein from CEN-ARS plasmids (pRS) carrying the full SIR4 promoter and terminator (Figure 1A, 1B). Because overexpression of either full-length protein or fragments of Sir4 derepress gene silencing [53], we chose conditions that reproduced as closely as possible the endogenous Sir4 protein levels (Figure S1B and data not shown). Quantitative mating assays can be used to determine the degree of repression at HML, because mating is compromised by coincident expression of a and α mating type information. In contrast to the findings of Marshall et al. [51], expression of a C-terminal fragment of Sir4 (Sir4C, residues 747–1358) alone was sufficient to repress HML, as indicated by the restoration of mating in a MATa sir4Δ strain (Figure 1C, Table 1). Consistently, expression of Sir4C also repressed a TRP1 reporter inserted at HMR in both the sir4Δ and sir4N backgrounds (Figure 1D, Table 1).

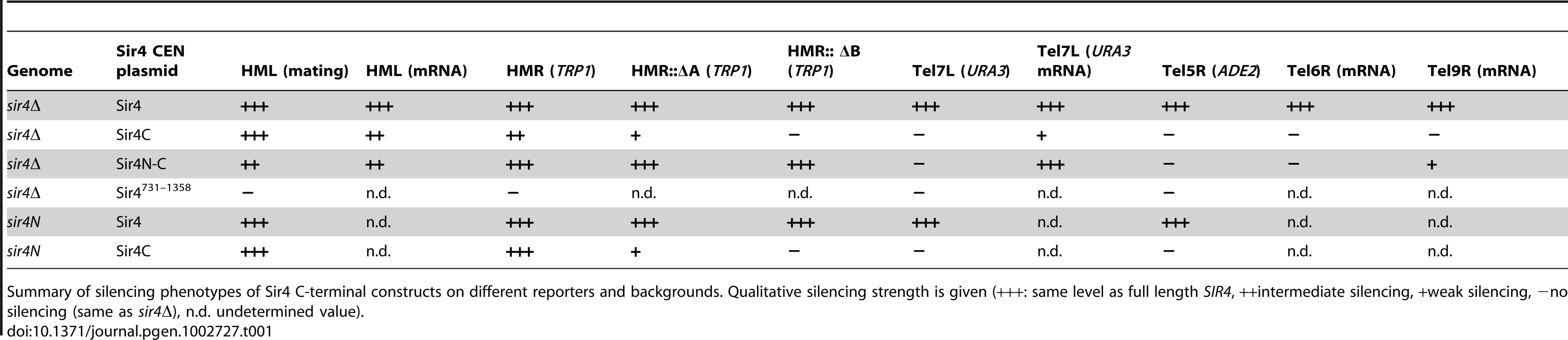
In trying to explain the discrepancy between our findings and those of Marshall and colleagues, we noticed that they had used a galactose-inducible Sir4 C-terminal fragment that was a few amino acids longer than ours, and co-expressed as well a slightly longer N-terminal fragment than we used [51]. Intriguingly, our analysis of a longer C-terminal fragment (residues 731–1358; Sir4731–1358), showed that it failed to repress an HMR::TRP1 reporter, either alone (in a sir4Δ background) or when expressed with Sir4N (in a sir4N background; Figure S1A). Immunoblotting showed that steady-state levels of the Sir4731–1358 fragment were much lower than of those of the shorter Sir4C (Figure S1B). The instability of the Sir4731–1358 fragment would explain its inability to repress HMR; indeed, it is likely that the fragment used by Marshall and colleagues was also unstable, and therefore did not silence on its own. We tried also expressing a longer (330 residue) N-terminal fragment with both long and short Sir4C fragments, but observed no differences in the mating assay compared to the shorter Sir4N fragment (data not shown). Our results suggest that a stable 611-residue C-terminal fragment of Sir4 is sufficient to repress both HM loci.
The N-terminus of Sir4 contributes to repression at HM loci with incomplete silencers
To date, the N-terminus of Sir4 was implicated in recruiting the SIR complex to silencers or to telomeres through its affinity for Sir1 or Yku80, respectively [21], [40], [41], [56], [57]. The interactions that recruit the SIR complexes to silencers are, however, redundant [20]. Therefore, we next tested the impact of Sir4N on silencing under conditions of compromised recruitment, that is, in strains lacking either Sir1 or Yku70 which eliminates Yku80 function as well (Figure 2). The expression of Sir4C in a sir1Δ strain could still restore silencing of HML, either in the absence (sir4Δ) or the presence of Sir4N (sir4N, Figure 2A). Consistent with it being a direct binding partner of Sir1, Sir4N expression did not enhance silencing in the sir1Δ background (Figure 2A). On the other hand, in the sir4Δ yku70Δ background, Sir4C only supported mating at 30% of wild-type levels. In this case, mating efficiency was indeed enhanced by co-expression of Sir4N (compare SIR4C in sir4Δ and sir4N, Figure 2A). This confirms that the N - and C-termini of Sir4 can complement in trans at the HML locus, as reported by Marshall et al. (1987), although in our hands, this is true only in yku70Δ cells.
To assess more directly whether Sir4N compensates for the absence of other recruitment sites at silencers, we used a HMR::TRP1 reporter strain that lacks either the A sequence (ORC–Sir1-binding site) or the B sequence (Abf1-binding site) within the E silencer (Figure 2B) [19], [20]. When we deleted the ORC–Sir1-binding element (HMR-EΔA), Sir4C was no longer sufficient to repress the reporter gene at HMR (Figure 2B, Table 1). Similarly, in the absence of the Abf1-binding site (HMR-EΔB, Figure 2C, Table 1), Sir4C did not restore silencing, either with or without the Sir4N fragment. Thus, Sir4C is not sufficient for silencing at an HMR locus in which the silencers are weakened by deletion of a binding site for one of the recruitment factors.
The co-expression of Sir4C and Sir4N in trans did not enhance silencing at compromised silencers, as they did in the yku70 mutant (Figure 2A–2C). This may be explained if Sir4N interacts only weakly with the SIR complex. To test this possibility, we tethered the Sir4N and Sir4C domains with a short linker peptide, to form a stable fusion protein (Sir4N–C; Figure 1A). Importantly, when expressed in a sir4Δ background, Sir4N-C repressed the reporter gene as effectively as full-length Sir4 at the silencer-compromised HMR loci (HMR-EΔA and HMR-EΔB; Figure 2D, Table 1). The strains expressing Sir4N-C were also competent for mating (Figure S2A), albeit with lower efficiency than cells expressing Sir4C alone, possibly due to an altered growth rate (see legend, Figure S2). These data confirm a role for the N-terminus of Sir4 in silencing the HM loci when the binding of recruitment factors is compromised. Indeed, at HMR with weakened silencers, the expression of a Sir4 N-terminal fragment along with Sir4C allows repression, whereas Sir4C alone does not.
Linking Sir4N to Sir4C increases but does not fully restore telomeric silencing
To test whether silencing at telomeres requires the N-terminus of Sir4, we monitored expression of a URA3 reporter gene at telomere 7L (Tel7L::URA3) [4] by assaying growth in the absence of uracil in a strain that lacks Ppr1, the transcription factor responsible for inducing URA3 in auxotrophic conditions [58]. In contrast to repression at the HM loci, telomeric silencing could not be established by expressing Sir4C (Figure 3A, Table 1) nor by co-expressing Sir4C with Sir4N in trans (Figure S2B, Table 1). This was true not only for URA3 expression at Tel7L, but also for the ADE2 reporter gene expression at Tel5R (Figure S2C, Table 1). We also monitored Tel7L::URA3 repression by counter-selecting with the drug 5-FOA, with similar results (Figure 3A).

Given that repression at HMR with weakened silencers was enhanced by expression of a Sir4N-C fusion, we tested the effect of this hybrid on TPE. Surprisingly, expression of the Sir4N-C fusion in a sir4Δ strain failed to repress either Tel7L::URA3 or Tel5R::ADE2 reporters in the standard drop assay (Figure 3A, Figure S2C, Table 1). We also assayed silencing by measuring mRNA levels of subtelomeric genes from telomeres 6R and 9R by quantitative PCR (QPCR; Figure 3C). Both genes were derepressed in cells expressing only Sir4C or Sir4N-C. For Tel9R we observed partial repression by Sir4N–C compared to sir4Δ (Figure 3B, Table 1). Intriguingly, the levels of the HMLα1 gene were only partial reduced when Sir4C was expressed, whereas expression of Sir4N–C conferred repression to near-background levels (Figure 3B). A similar effect was observed at Tel7L::URA3 when URA3 was transcribed at basal levels (i.e. growth in the presence of uracil and in the absence of Ppr1; Figure 3B). In this case, Sir4N–C repressed transcription to background levels, unlike in the drop assay in the absence of uracil (Figure 3A), which strongly induces transcription from the URA3 promoter.
Resistance to rapamycin is a sensitive means to monitor native telomeric silencing, as growth on the drug requires expression of multiple stress genes located near telomeres, which are normally silenced by the SIR complex [59]. We therefore monitored the level of stress gene expression by scoring for resistance to rapamycin in Sir4-, Sir4C - and Sir4N–C-expressing cells. Whereas SIR4+ cells fail to grow on rapamycin, intriguingly, both Sir4C - and Sir4N–C-expressing cells behaved like sir4Δ when grown in the presence of rapamycin (Figure 3A). This argues that neither Sir4C nor Sir4N–C can prevent the induction of natural subtelomeric genes by stressful conditions (Figure 3A), suggesting that full-length Sir4 is needed for native subtelomeric repression, although not at HM loci.
Transcriptional repression generally correlates with the binding of Sir proteins throughout the silent domain, and we therefore tested the binding of Sir4 to HML and telomeres by chromatin immunoprecipitation (ChIP). We detected a clear enrichment of Sir4, Sir4C and Sir4N-C at HML-E and HML-α1 (Figure S2F). Consistent with the silencing assays, on the other hand, only full length Sir4 was strongly enriched at telomeres (Figure S2F). This confirms that full-length and truncated Sir4 proteins are bound at the sites that are silenced robustly, and shows again that Sir4C is not sufficient for binding in subtelomeric domains.
To see if Sir4C would be sufficient for silencing at telomeres if we enhanced SIR recruitment by Rap1, we monitored TPE in the absence of the Rap1-interacting factor 1 (Rif1), which competes for Sir3 and Sir4 recruitment by the Rap1 C-terminus [60]. Whereas deletion of RIF1 increased telomere length and SIR recruitment, leading to enhanced silencing [18], [41], it did not increase Sir4C - or Sir4N–C-mediated repression at Tel7L or Tel5R::ADE2 (Figure S2D, S2E; compare to Figure 3A). Taken together, these data indicate that Sir4C is insufficient for TPE, and that Sir4N can contribute weakly to improve repression at native telomeric genes and reporters, yet only full-length Sir4 supports robust TPE.
Sir4C can form a stable and active SIR complex
Because Sir4C can silence HM loci, we asked whether Sir4C forms a stable complex with Sir2 and Sir3. To test this, we co-expressed Sir4C with Sir2 and Sir3 in baculovirus-infected insect cells. Using conditions identical to those used to purify the full-length Sir2–Sir3–Sir4 complex, we were able to purify a SIR complex containing Sir4C (Figure 4A, 4C; [13], [52]). Upon glycerol density gradient sedimentation the complex migrated in two distinct complexes: one containing Sir2, Sir3 and Sir4C and the other containing only Sir2 and Sir4C, exactly like the complex with full-length Sir4 (Figure 4C, 4D; [13]). We conclude that Sir4C is sufficient to form a complex similar to the wild-type SIR complex, when expressed in insect cells.

To confirm that the Sir4 N-terminus is dispensable for the deacetylation activity of the SIR complex, we incubated recombinant protein complexes of either full-length Sir2–Sir3–Sir4 or truncated Sir2–Sir3–Sir4C with histone octamers that were fully acetylated on histone H4K16. We assayed H4K16ac deacetylation over time by Western blotting [28], and found that the two complexes had similar deacetylation activities (Figure 4B). We conclude that Sir4C forms a stable and active SIR complex, consistent with its ability to confer HM repression.
Sir4N promotes high-affinity binding to chromatin and linker DNA protection
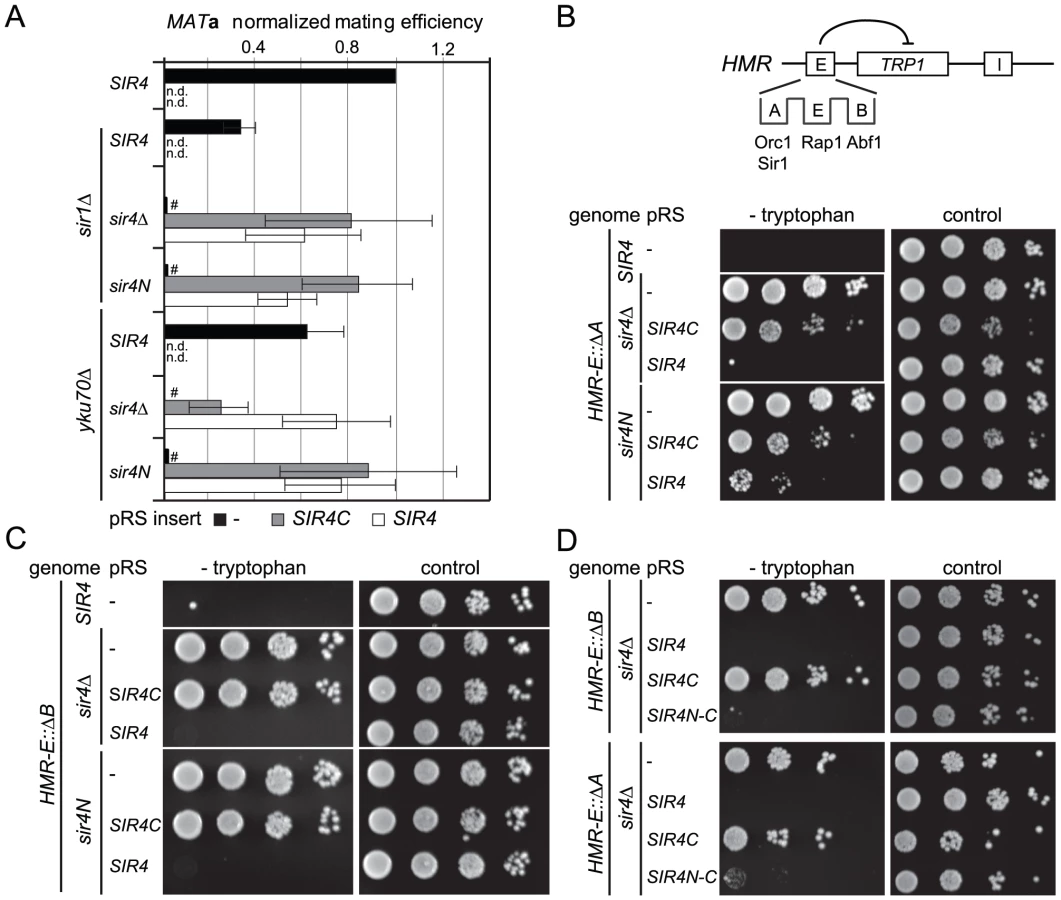
To explain the contributions of the Sir4 N-terminus for repression in biochemical terms, we examined the contribution of Sir4N to SIR complex loading onto nucleosomal arrays in vitro. In a previous study, we showed that recombinant Sir4N has considerable non-specific affinity for DNA [52]. To test whether this contributes to the affinity of the SIR complex for chromatin, we first compared the DNA-binding properties of the Sir2–Sir4 and Sir2–Sir4C complexes. Increasing amounts of each complex were titrated into a constant amount of a high-affinity histone octamer-binding sequence (Widom 601; [61]). By using the binary Sir2–Sir4 complex rather than ternary complexes with Sir3, we could avoid contributions of Sir3 to DNA binding [26]. SIR complex association with DNA leads to the appearance of higher molecular weight species after native gel electrophoresis, and the disappearance of unbound DNA. We quantified the disappearance of the unbound DNA as a function of Sir2–Sir4 complex concentration. This showed that the truncated Sir2–Sir4C complex has about four–fold lower affinity than the full-length Sir2–Sir4 complex for naked DNA (Figure 5A).
We next examined the contribution of the Sir4 N-terminus to nucleosome binding, by titrating the complexes onto hexameric nucleosomal arrays assembled in vitro, as previously described [52]. Again, by quantifying the disappearance of unbound nucleosomes, we found that the Sir2–Sir4C complex has roughly two-fold lower affinity for chromatin than the full-length Sir2–Sir4 complex (Figure 5B). This effect was even more pronounced when we compared the binding of holo-SIR complex with that of the Sir2–Sir3–Sir4C complex. The complex carrying the truncated Sir4C had a much lower affinity for nucleosomal arrays than that containing full-length Sir4 (Figure 5C), possibly because Sir3 sterically masks part of Sir4C's chromatin-binding surface [27], [33].
Since the SIR complex is known to protect nucleosomal linker DNA from micrococcal nuclease (MNase) attack [28], [52], we examined the contribution of Sir4N to linker DNA protection. Importantly, we used two - to four-fold more of the truncated Sir2–Sir4C complex than of wild-type Sir2–Sir4 complex, to ensure that equal amounts of chromatin-SIR complex were formed (Figure 5B). The Sir2–Sir4C complex showed less linker DNA protection than the full-length Sir2–Sir4 complex (Figure 5D), despite the fact that equal fractions of nucleosomes were bound in each reaction. These data suggest that the affinity of Sir4N for DNA promotes a tighter binding of SIR complexes to chromatin, thereby enhancing linker DNA protection. This attributes a function to the Sir4 N-terminus beyond recruitment by Sir1 or Yku80.
Truncated Sir4 mediates formation of Sir3 foci independently of silencing
Silencing at telomeres is sensitive to the anchorage and clustering of the telomeres at the nuclear envelope [44], [45], [50]. Since Sir4C can restore silencing at HM loci but not at telomeres, we wondered whether Sir3 focus formation, as an indication of telomere clustering, might depend on Sir4N. To test this hypothesis, we expressed a Sir3–EGFP fusion protein in yeast cells in which the endogenous SIR4 gene was either deleted (sir4Δ) or truncated (sir4N), and either SIR4 or SIR4C was expressed from a CEN-ARS plasmid. Similar to a strain without tagged Sir3 (Figure 3A, S2B), both Sir4C and full-length Sir4 restored mating-type repression in the SIR3–EGFP background, but only full-length Sir4 was able to restore TPE fully (Figure S3). Although Sir4C-expressing cells are competent to mate, they grow more slowly (Figure 2C) and occasionally had larger and obviously distorted nuclei compared to wild-type cells. We imaged Sir3-EGFP in living cells and found that Sir3–EGFP foci formed when either Sir4 or the truncated Sir4C protein was expressed, as determined by counting the number of cells containing at least three Sir3–EGFP foci (Figure 6A, 6C). In about 20% of the Sir4C-expressing cells, we observed one to three very intense Sir3 foci in addition to the small telomere clusters, whether or not the endogenous SIR4 gene was present (Figure 6B). We conclude that the N-terminus of Sir4 is not necessary for the formation of Sir protein clusters, and thus that large Sir3–EGFP foci can form in the absence of TPE. The dissociation of Sir3 foci from TPE confirms a recent report showing that non-perinuclear Sir3 clusters can form in cells unable to support SIR-mediated repression [62].

The Sir4 N-terminal domain is its major site of phosphorylation in G2/M-phase cells
SIR complex binding at telomeres appears to be modulated in response to the physiological state of the cells. For instance, Sir proteins are released from telomeres both in mitotic cells [63], [64], and in response to genotoxic stress [65], [66]. Indeed, activation of the DNA damage checkpoint affects TPE, but not HM repression, much like the deletion of Sir4N. Moreover, subtelomeric domains contain a number of genes that are regulated in response to nutrient stress [67], [68] by a kinase cascade that targets, among other things, Sir3 [59]. Finally Sir4N harbors many potential phosphoacceptor sites, and whole phosphoproteome studies suggested that Sir4 is modified in a manner that fluctuates with the activity of the cell-cycle regulated cyclin-dependent kinase (CDK; [69], [70]). Thus it was proposed that the N-terminal half of Sir4 might act as a phosphorylation-dependent regulatory domain [34].
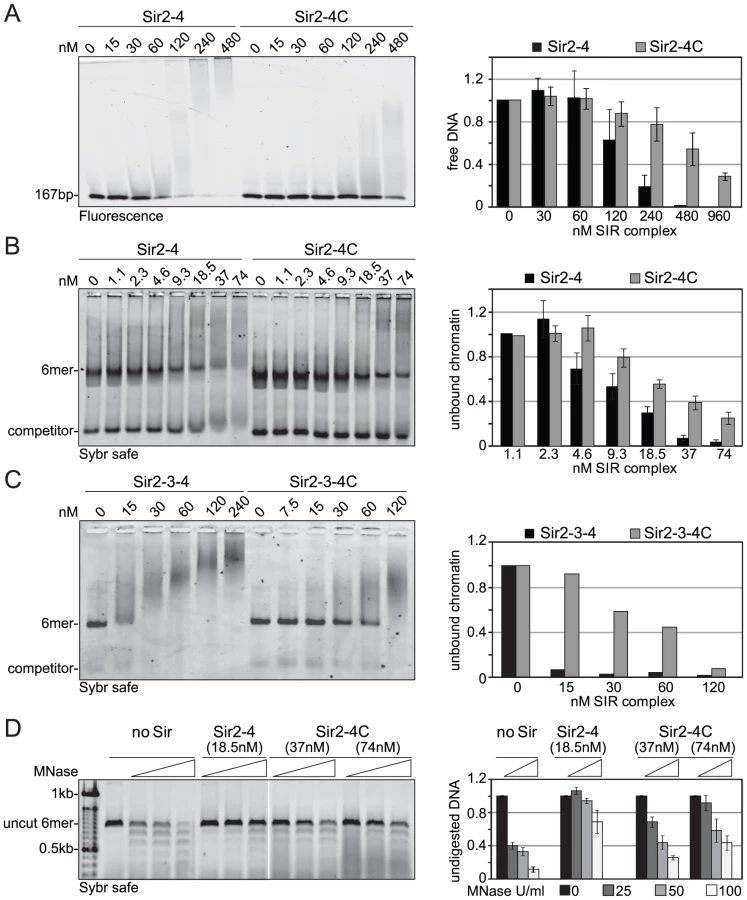
To identify in vivo phosphoacceptor sites in Sir4, we first expressed a functional, epitope-tagged Sir4 from its endogenous locus. The Myc-tagged Sir4 protein was immunoprecipitated either from cycling cells or from cells that were arrested in G2/M phase by repressing CDC20, which encodes an essential anaphase-initiating factor (Figure S4A, S4B). We then used mass spectroscopy to identify the phosphorylated peptides. As predicted by Zill and colleagues [34], most of the phosphorylated amino acid residues were in the N-terminal half of the protein (Figure 7A and 7B, Figure S4C, Table 2). Moreover, ten of the twelve phosphorylated sites we identified contained the minimal consensus sequence for CDK, [S/T*]-P (in bold face in Figure 7A, Table 2), which can also be phosphorylated by the mitogen-activated protein kinase (MAPK). These sites were among those previously predicted to be targets for CDK and MAPK [71], [72].

Five of these [S/T*]-P sites showed at least 1.5-fold higher levels of phosphorylation in G2/M as compared to cycling cells (Figure 7B). One of these was in the Sir4C domain (S1134), which, interestingly, is near a mapped SUMO-acceptor residue (K1128) within the Esc1-binding PAD domain [73]. However, mutation of this C-terminal phosphoacceptor site or the neighboring SUMO acceptor lysine yielded no detectable silencing - or anchoring-related phenotypes (data not shown). We therefore focused on the phosphoacceptor sites in the N-terminus of the protein. Within this domain, serine 63 (S63) was the site we detected most frequently over several experiments, while serine 84 (S84) showed a strict G2/M specificity.
To confirm the presence of CDK target sites within Sir4N, we exposed a recombinant Sir4N–GST fusion protein to a range of purified kinases in vitro. The recombinant protein was modified by CDK and protein kinase C, but not by yeast casein kinase II or the human MAP kinase, ERK (Figure S5A, S5B). To examine whether the sites modified by CDK in vitro corresponded to the sites phosphorylated in vivo, we mutated CDK consensus sites within the Sir4N domain (Figure 7A). We substituted consensus-site threonines 7 and 13 by alanines (A7 and A13), and serines 63 and 84 by glycine residues (Sir4NGG), alone and in combination (Figure 7C). The mutant Sir4N domains were purified and used as substrates for phosphorylation by CDK in the presence of γ32P-ATP. After trypsin digestion, the resulting phosphopeptides were resolved by high-resolution 1D gel electrophoresis (Figure 7C). The identities of the cleavage products were determined both by co-migration with synthesized, digested peptides and by the absence of signal in the mutants in which serine or threonine had been replaced by non-phosphoaccepting amino acids (Figure 7C and data not shown). Unlike the wild-type Sir4N protein, peptides carrying glycine substitutes at S63 and S84 lost almost all CDK-mediated phosphorylation in vitro (Figure 7C, Figure S5B). In contrast, alanine substitutions at T7 or T13 had only minor effects, alone or in combination. Given that S63 and S84 were phosphorylated by CDK both in vitro and in the endogenous protein recovered from mitotic cells, we propose that these two Sir4N residues are the major, physiological targets for CDK.
Mutation of phosphoacceptor sites does not alter Sir4N interaction with Yku80, Sir1, Sif2, or DNA
To test the functional significance of Sir4N phosphorylation at S63 and S84 we analyzed the interactions of the non-phosphorylatable Sir4N mutant (Sir4NGG) and the mutant carrying a mutation that mimics the phosphoserine residues (Sir4NDD) with Sir1, Sif2 and Yku80. In yeast two-hybrid analyses, the interactions between Sir4NGG or Sir4NDD and Yku80, Sif2 or Sir1 were identical to the interactions between wild-type Sir4N and these binding partners (Figure 7D). Thus, at least in this assay, substitution of S63 and S84 by either G or D does not perturb the binding of known ligands to Sir4N.
We next tested mutant and wild-type Sir4N fragments for their ability to bind DNA and protect linker DNA from MNase digestion. Intriguingly, the Sir4NGG mutant showed a higher affinity for DNA than the wild-type protein or the Sir4NDD mutant, suggesting that Sir4N phosphorylation might weaken its interaction with DNA (Figure 7E).The incubation of Sir4N with CDK in vitro prior to DNA binding, increased the affinities of both wild-type Sir4N and the non-phosphorylatable Sir4NGG mutant for DNA, due to nonspecific effects of the kinase (Figure S5C). Indeed, by performing MNase digestion of nucleosomes bound by Sir4NGG, Sir4NDD, or the wild-type protein, we found equal protection of linker DNAs in all cases (Figure 7F). We conclude that point mutations at these two major CDK phosphoacceptor sites in Sir4N do not substantially alter the affinity of the domain for either chromatin or DNA.
Mutation of Sir4N phosphorylation sites affects the stability of gene repression in vivo
Despite the absence of in vitro phenotypes for Sir4N bearing mutated S63 and S84 residues, we checked the effects of these two phosphoacceptor site mutations on silencing in vivo. To test this, we introduced the double mutations S63G–S84G (sir4GG) or S63D–S84D (sir4DD) into the endogenous SIR4 gene in a strain carrying both Tel5R::ADE2 and Tel7L::URA3 telomeric reporter genes. In the course of these experiments we serendipitously created an additional mutation at the Sir4 N-terminus, namely a proline to alanine substitution at residue 2 (sir4P2A). This substitution does not prevent cleavage of the initiator methionine or acetylation of the alanine at position 2 in contrast to the proline, and protein half life is predicted to be the same for either variant [74]. Indeed, we scored no significant effects on the half-life of Sir4 due to any of the mutations described above or below (data not shown).
We first tested the effects of the background P2A mutation alone on ADE2 silencing at Tel5R. We found that most sir4P2A colonies were darker red than wild-type colonies (Figure 8A), which indicates a more stable repression of ADE2. On the other hand, some colonies were completely white, indicating a low frequency of stable Tel5R::ADE2 reporter derepression (Figure 8C–8E). This suggested to us that the P2A mutation stabilizes either “off” or “on” epigenetic states at Tel5R. When the P2A mutation was combined with the non-phosphorylatable sir4P2AGG mutation or the phospho-mimicking mutation (sir4P2ADD), we scored the same dark red color, but also noted that white colonies appeared at higher frequency (Figure 8A, 8B). To quantify this phenomenon, we cultured single white or dark red colonies from each strain in liquid culture and plated them out after 24 h and 48 h to score the status of the ADE2 reporter by red vs white colony color. Whereas less than 1% of the sir4P2A colonies switched from red (ADE2 repressed) to white (ADE2 derepressed), we found that 6–10% of the sir4P2AGG or sir4P2ADD colonies switched color, indicating that these mutations render the repressed state less stable (Figure 8C). Conversely, we found that 0.4–0.6% of sir4P2A colonies switched from a derepressed to a repressed state (white to red), while the sir4P2AGG or sir4P2ADD strains remained completely derepressed, with no red colonies detected after 24 h of culturing of a white colony (Figure 8D). This argues that mutation of S63 and S84 generally destabilizes silencing or impairs re-establishment of a repressed state. The effects are particularly noticeable in combination with the sir4P2A mutation, which alone, for unknown reasons, stabilizes either state.

To test whether the effect of these mutations at Tel5R:ADE2 held true for another telomere, we spotted overnight cultures of single colonies onto uracil-deficient plates, to score for expression of the Tel7L::URA3 reporter gene (Figure 8E). We observed loss of URA3 gene silencing in the sir4P2AGG and sir4P2ADD mutants. Moreover, the colonies growing on uracil-deficient plates were all white (ADE2 derepressed), indicating that Tel7L and Tel5R reporter genes were derepressed simultaneously (Figure 8E). The loss of Tel7L::URA3 repression was far more pronounced for the white colonies of sir4P2AGG and sir4P2ADD strains than for the sir4P2A mutation alone, arguing that alteration of the phosphorylation sites does indeed enhance derepression.
We next investigated whether the Sir4 phospho-site mutants caused general disruption of telomeric silencing when this is scored by growth on rapamycin (Figure 3A). Indeed, the white (derepressed) colonies of the sir4P2ADD and sir4P2AGG strains showed more resistance to rapamycin than the red (repressed) colonies, and were almost as resistant as the sir4Δ strain (Figure 8E). Quantification showed that this effect was far more pronounced in the sir4P2ADD and sir4P2AGG strains, than in the sir4P2A strain (15% for sir4P2A, 49% for sir4P2AGG and 69% for sir4P2ADD). As in the switching assay, the phospho-mimicking mutation sir4P2ADD produced the strongest derepressed state and strongest rapamycin resistance (Figure 8E).
We generalized this observation by scoring mRNA levels at native subtelomeric genes. This was done both by mRNA analysis for genes at two natural telomeres, Tel6R and Tel9R (Figure 3C) and by whole genome tiling arrays, that compared gene expression in wild-type SIR4 cells with either red or white colonies of the sir4P2ADD mutant. The data in Figure 8F confirmed derepression at both natural telomeric genes and at Tel7L::URA3 in white sir4P2ADD colonies, although not to the degree detected in the sir4Δ stain (Figure 8F). We also observed derepression of HML in the white sir4P2ADD cells. On the other hand, when we examine expression in the red sir4P2ADD colonies, we find that subtelomeric genes are as stably repressed in the red sir4P2ADD mutant cells as they are in a SIR4 wild-type strain (Figure 8F). The microarray data confirmed this trend for subtelomeric genes (Figure 8G). By overlaying the transcriptional effects on all genes as a function of their distance from the telomere (red line is lowess smoothed over all genes), we score an increase in expression from genes within the first 5 kb from the telomere in both the sir4Δ and the white sir4P2ADD strains (Figure 8G). We also observed a generally stronger repressed state in the sir4P2ADD red colony for the genes that lie closest to telomeres, as expected. Thus the effects of the mutations on the ADE2 reporter can be confirmed and extended to native subtelomeric genes.
In summary, thanks to the stabilizing effect of the sir4P2A background, we are able to demonstrate a defect in the sir4P2ADD and the sir4P2AGG strains in silencing. The derepressed state suggested by white colony color coincided with general derepression of TPE and with resistance to rapamycin, as reported for the sir4Δ strain. We note that the phenotypes were somewhat stronger for the phospho-mimicking sir4P2ADD mutant than for sir4P2AGG. Taken together, these results indicate that the Sir4 N-terminus helps modulate stable gene repression at telomeres by being phosphorylated on the target sites we have identified.
Discussion
SIR-mediated transcriptional repression in budding yeast has been studied genetically and biochemically for over 20 years, yet we still do not fully understand the functions of its core components nor how it is regulated either during the cell cycle or in response to stress. In this study we addressed the molecular role and regulation of Sir4, the largest and least conserved Sir protein. On the basis of our new findings and a large body of earlier work, we can assign four roles to different domains of Sir4 and describe their functions in SIR-mediated repression. First, as described previously, the C-terminal half of the Sir4 protein has a scaffolding function that is essential for assembling Sir2 and Sir3 into the SIR complex and delivering it to chromatin (by binding to Rap1 and Yku). Second, the N-terminal 270 residues of Sir4 have a recruitment function by binding Sir1 and Yku80. Third, the Sir4N contributes to the tight association of the SIR complex with DNA in vitro and enhances nucleosomal linker protection. This domain is essential in vivo for repression of HM loci under suboptimal conditions (i.e. when the silencers in HMR are compromised or when Yku70 is absent) and contributes significantly to TPE. Finally, we find that both the extreme N-terminus and the adjacent central domain of Sir4 are heavily phosphorylated in vivo and that the mutation of two phosphoacceptor sites in the N-terminus affects the stability of subtelomeric repression. Given that Sir4 residues S63 and S84 are phosphorylated in mitotic cells, we speculate that the phosphorylation of Sir4N regulates the stability of TPE through the cell cycle.
The C-terminus of Sir4 is sufficient to establish a silent chromatin structure
Marshall and colleagues proposed in 1987 [51] that both the N - and C-terminal domains of Sir4 were required for silencing of HML. By using a slightly shorter and significantly more stable C-terminal domain of Sir4 than that used by Marshall and colleagues (residues 747–1358), we show that the Sir4 N-terminus is dispensable for repression of intact HML and HMR loci, although not for the repression of subtelomeric reporter genes. The ability of Sir4C to silence HM reflects, in part, the strong redundancy in Sir factor recruitment pathways at HM loci, i.e. several silencer factors redundantly recruit Sir3 and Sir4 [20].
Consistent with our finding that Sir4C is sufficient to silence intact HMR in the presence of Sir2 and Sir3, we find that recombinant complexes containing Sir2, Sir3, and Sir4C are stable upon isolation, retain full histone H4K16 deacetylation activity and bind nucleosomal arrays in vitro. H4K16 deacetylation by Sir2 has been proposed to provoke a conformational change in the SIR complex that increases its binding to chromatin [28], [52], [75]. We do not know whether in the SIR complex the N-terminal domain of Sir4 contributes to this change in conformation, although we note that Sir4C-containing complexes retain deacetylation activity.
We explain the inability of a longer Sir4C fragment to repress (as described in Marshall et al., 1987) by its instability (Figure S1B). Indeed, this is consistent with an earlier study of the san1-1 mutant, which partially restores mating in a strain expressing only a Sir4 C-terminal domain [76]. San1 is a ubiquitin ligase that targets misfolded proteins for degradation by the proteasome, and one of its targets is Sir4 [77]. Our work and this study suggest that a sufficiently stable Sir4C fragment provides all the essential interactions necessary for formation of silent chromatin at HM loci – most crucially a tight association with Sir2 and Sir3 to create an active, heterotrimeric complex that can interact stably with chromatin [13], [52]. It is not, however, sufficient for TPE.
The Sir4 N-terminus enhances Sir4 binding to HM loci via protein - and DNA-interactions
Although the Sir4 C-terminus is sufficient for HM repression under certain conditions, we also found conditions that render the N-terminus essential for efficient HM silencing, namely when one of the recruiting elements was deleted at the HMR-E silencer (ΔA Orc1-Sir1 site or ΔB Abf1 site), or in the absence of YKU70. This weakens the recruitment of Sir3 or Sir4 to the HM silencers. The requirement for Sir4N under these conditions is consistent with its ability to bind Sir1 and Yku80. Sir1 recruits Sir4 to HM silencers by direct interaction with Orc1 [21]. The Yku70/80 complex can stabilizes silent chromatin by two means, first by recruiting the HM loci to Sir-clusters at the periphery [46], [49]and second by helping form a promoter-silencer interaction at HM loci through a looping mechanism [78]–[80]. Nevertheless, Yku70/80 is only essential for mating in the absence of Sir1 and vice versa [56], [57]. Consistent with this, we observe that Sir4N enhances mating efficiency in the absence of Yku70.
These two interactions, however, are probably not the only functions of the Sir4 N-terminus in HM silencing. First, there are two other sites of contact between Sir4 and the Yku70/80 complex [39], [42], and second, expression of a Sir4N-C fusion protein represses an HMR locus that lacks one of its binding sites for ORC (HMR-EΔA; Figure 2D). This indicates a function for the Sir4 N-terminus that is independent of its interaction with Sir1. We propose that the additional function is its strong non-specific affinity for DNA, which contributes to a tighter interaction of the SIR complex to chromatin and to enhanced linker DNA protection in vitro.
Full-length Sir4 is necessary for full subtelomeric repression
At telomeres, the Sir4 N-terminus is required for TPE. Moreover, while the expression of a Sir4N-C fusion slightly reduces mRNA levels compared to Sir4C, it could not suppress reporter genes when their promoters were induced. Since Sir1 is not required for subtelomeric repression, these effects are independent of Sir1 [15]. Rather, we suggest that the DNA-binding affinity of Sir4N increases binding of the SIR complex to telomeres to enhance the stability of repression. Full TPE, however, appears to require not only the first 270 amino acids of Sir4, but also the unstructured region between 270 and 744. Thus we suggest that another, yet unidentified function may be attributed to this domain.
Besides promoting tight association of the SIR holocomplex with DNA, the N-terminus of Sir4 may also regulate the strength or character of the Sir3–Sir4 interaction. We find that expression of Sir4C enhances the formation Sir3–EGFP foci even when there is no TPE. Similarly, Sir3-EGFP foci in the absence of TPE were observed in a strain overexpressing a non-acetylatable form of Sir3 [62], indicating that Sir protein clustering does not always lead to gene repression. It is possible that the Sir4C protein has a stronger affinity for Sir3 than does full-length Sir4. This is consistent with an earlier hypothesis that the Sir4 N-terminus interferes the binding of Sir3 to Sir4 [43], [81]. If true, the expression of Sir4C alone may lead to the sequestration of Sir3 into foci that antagonize repression. Consistent with this, we note that telomeres are highly sensitive to changes in Sir protein levels [31], and that Sir4C expression is somewhat toxic to cells, while that of the Sir4N–C fusion is not (Figure S2E and data not shown).
The N-terminus is the major site of Sir4 phosphorylation and fine-tunes subtelomeric stress-response genes
In this study we characterize Sir4 as a phosphoprotein and map key phosphoacceptor sites in the N-terminal domain of the protein. We show that two sites that are phosphorylated by CDK in vitro are also phosphorylated in mitotic cells in vivo. Zill and colleagues [34] speculated that the N-terminus of Sir4 may be specialized for fine-tuning or regulating silencing in response to environmental factors. Indeed, we find that the N-terminus of Sir4 is its major site of phosphorylation in vivo.
What functions of Sir4 might be affected by its phosphorylation? As Sir4N is dispensable for HM silencing, but is essential for TPE, we reasoned that the state of Sir4 phosphorylation might affect the repression of subtelomeric genes, many of which are activated only under conditions of nutrient stress [59]. Phosphorylation of the Sir4N terminus by CDK may also destabilize silent chromatin in mitosis. Consistent with this, previous work has shown that SIR complexes are partially released from telomeres in mitotic yeast nuclei [63], [64], and yeast heterochromatin is most accessible to transcription factors and gene activation during G2/M phase [82]. Moreover, passage through mitosis, which is accompanied by CDK-dependent protein phosphorylation and dephosphorylation, is important for the establishment of silencing [83]–[85]. Although it is not clear why, we note that the partial release of factors from chromatin during mitosis is a common feature of eukaryotes. Heterochromatin protein 1 (HP1; [86]) and the Polycomb complex (PcG; [87]) are both partially released during mitosis in Drosophila. The Polycomb protein EZH2 is a direct target of CDK [88], [89], and HP1 release appears to be due to phosphorylation of histone H3 on Ser10 by Aurora B kinase [90], [91].
In yeast, we show that the substitution of the Sir4 phosphoacceptor sites S63 and S84 by acidic amino acids, or by non-phospho-accepting glycines, had only minor effects on the TPE (Figure S6). However, by combining these mutations with a fortuitous mutation of residue 2 (proline to alanine or P2A), we could observe that they indeed tend to derepress TPE. The sir4P2A mutation alone had a strong stabilizing effect on either silent or open epigenetic states, in that ADE2 repression at Tel5R was seen to be either enhanced or abolished. The phospho-acceptor site mutants (sir4P2ADD or sir4P2AGG) has a tendency to derepress TPE and thereafter to retain the derepressed state, which is readily visible thanks to the stabilizing effect of sir4P2A. Indeed, re-establishing a repressed state occurred less frequently in the sir4P2ADD or sir4P2AGG mutant cells. Importantly, we then showed by microarray analysis that this was accompanied by a general derepression of subtelomeric genes, similar - albeit less pronounced - to that observed in sir4Δ cells. Together our analysis suggests that the modification of Sir4N S63 and S84 CDK target sites either directly derepresses TPE or interferes in the re-establishment of a repressed state. We suggest that the Sir4 N-terminal domain regulates repression both during the cell cycle and in response to environmental stress, which is most likely mediated by a MAP kinase cascade. Given the abundance of confirmed CDK phosphoacceptor sites in this domain, and putative MAP kinase sites, this may be one of the main functions of Sir4 N-terminus.
Materials and Methods
Plasmids, strains, and yeast methods
All strains and plasmids are described in Text S1. Standard techniques were used for cloning, yeast strain generation and growth. To obtain Sir4 expression similar to the endogenous levels, the Sir4 locus (1 kb 5′ of start and 250 bp 3′) was cloned into a CEN-ARS plasmid. Introduction of a NcoI site at the Sir4 start codon allowed subcloning of shorter Sir4 fragments and introduced the P2A mutation. To introduce the Sir4 phosphosite mutations into the genome, the respective plasmids were digested with SalI and SacI, purified fragments were integrated into a sir4::KanMX6 (GA5822) strain. Positive clones were selected for growth on SC medium+0.1% 5-FOA and checked by sequencing of the genomic locus. At least two independent transformants were analyzed.
To check Sir4 expression levels, the CEN-ARS plasmids were transformed into a protease-deleted strain (GA73) and cells were grown to OD600<1 prior to lysis by bead-beating. Standard techniques were used for SDS-PAGE and immunoblotting. Mcm2 (yN-19) antibody is from Santa Cruz, anti Myc antibody 9E10. Sir2 and Sir4 antibody have been described previously [7], [92]. Yeast two-hybrid analysis was carried out as described previously but using the GA181 strain [93], [94].
Silencing assays
For quantitative mating assays, plasmid transformed strains and the tester strain were grown overnight in SC medium. 107 cells of the mating-tester strain (GA858) were mixed in 1 ml YPAD medium containing 2×106 cells transformed with a given Sir4 plasmids and grown for 5 h at 30°C (see also [51]). Cells were then grown 3 days at 30°C on SD medium to select for diploids and SC medium – tryptophan/-methionine to normalize cell numbers.
Silencing of indicated reporter genes was performed as described [95], after growth overnight in selective media. Ten-fold dilution series starting at 107 cells/ml were performed in triplicates on appropriate media. Silencing of the ADE2 reporter was scored after 3 days growth on YPAD 30°C and subsequent maintenance at 4°C for 4 days.
Recombinant protein purification
Sir4C-expressing baculovirus was generated using the BaculoGold linearized cDNA (BD Bioscience) and Cellfectin reagent (Invitrogen) according to manufacturer's instructions. SIR complexes were co-expressed in insect sf21 cells and purified as described previously [28], [52]. Buffers contained 10 mM TEA pH8 when the proteins were purified for gradient sedimentation. For gradient sedimentation, 200 µl Calmodulin-column eluate was layered on a 4 ml 5–25% glycerol gradient (10 mM TEA pH8, 150 mM sodium chloride, 0.01% Tween-20) prepared using a Gradient Master (BioComp) in Beckmann 11×61 polyallomer tubes. Gradients were centrifuged for 18 h at 4°C and 30'000 g and fractionated into 100 ul aliquots Samples of 20 µl were analyzed by SDS-PAGE and SyproRuby staining. Sir4N fragments were expressed in E. coli and purified from inclusion bodies using standard Ni-NTA techniques as described previously [52].
In vitro phosphorylation and phosphopeptide mapping
Recombinant Sir4N-GST was purified from E. coli using standard procedures in PBS buffer. For phosphorylation assays, the proteins were incubated with the indicated kinases for 1 h at 37°C (a kind gift of E. Nigg; CDK2 (NM_001798 Proqinase)) and P32-γ-ATP. To analyze phosphorylation, Sir4N was run on a SDS-PAGE and proteins were detected by Coomassie staining and radiography. For phospho-peptide detection, Sir4N-GST was first digested by partial tryptic digest after in vitro phosphorylation. Peptides were lyophilized and analyzed on alkaline peptide gels and radiography according to the method of West and Bonner [96]. Briefly, samples were resuspended in loading buffer containing 0.125 M Tris-HCl pH 6.8 and 6 M urea. Peptides were then separated on 0.5 mm thick gels containing, 40% acrylamide, 0.037% bis-acrylamide, 0.75 M Tris-HCl pH 8.8. Gels were run at 10 mA for approximately 4 h. Peptide size was determined by co-migration with synthesized peptides that were phosphorylated and loaded alongside.
Chromatin and DNA binding, MNase digestion
Chromatin and DNA binding as well as MNase digestions were performed as described previously [28], [52]. Briefly, 6 mer of nucleosomes diluted to 2.5×10−8 M were incubated with increasing amounts of indicated proteins in 10 mM TEA pH7.4, 25 mM sodium chloride for 20 min on ice and analyzed on 0.7% native agarose gels (0.2× TB) run at 4°C. DNA binding assays were performed using a 167 bp 601-Widom sequence DNA fragment Cy5-labeled by Klenow enzyme at an AvaI site. DNA bindings were performed in 150 mM sodium chloride at a DNA concentration of 2.5×10−9 M, using the same conditions as for chromatin binding. Linker DNA protection assays were performed by adding 0.25–1 U MNase and 1 mM calcium chloride to 1 pmol chromatin that had been pre-incubated with indicated amounts of Sir proteins. After 10 min on ice 5 mM EGTA was added and samples were deproteinized by incubation with proteinase K for 30 min at 30°C. Deacetylation, expression and purification of homogeneously H4K16 reactions were carried out as described previously [28]. 10 nM of purified Sir2–Sir3–Sir4 and Sir2–Sir3–Sir4C complex were incubated with 70 nM of H4K16ac histone octamers with or without 150 µM of NAD. The reaction was carried out at 30°C in 50 mM Tris pH 8, 50 mM sodium chloride, 2.7 mM potassium chloride, 1 mM magnesium chloride and 0.005% Tween-20. The reaction was stopped at the indicated time points by addition of Laemmli sample buffer and samples were analyzed by 4–12% SDS PAGE by immuno blotting using H4K16ac antibody (Millipore 07-329) and H3 antibody (Abcam ab1791).
Immunoprecipitation and phosphosite mapping
Cells (GA5691, GA5589, GA1275) were grown overnight to OD600 = 0.6 in YPA-galactose media, then shifted to YPA-glucose for 2 h at 30°C for mitotic arrest (G2/M cells). Cycling cells were grown in the same carbon source, but did not contain the GALp:CDC20 allele that leads to G2/M arrest in glucose. Cells were harvested, washed once in ice-cold PBS and resuspended in one pellet volume of lysis buffer without detergent (50 mM HEPES pH7.5, 500 mM sodium acetate, 5 mM magnesium acetate, 0.1 mM EDTA, 5% glycerol [32]). The resuspended cells were snap frozen in liquid nitrogen and broken using a ball-mill (3×3 min at 30 1/s; Retsch MM4000); the cell powder was stored at −80°C. For immunoprecipitations, the cell powder was mixed with an equal volume of lysis buffer containing protease and phosphatase inhibitors and 1% Triton X-100. After thawing on ice for 5 min, cell extract was cleared by centrifugation and 5 mg of proteins were incubated at 4°C with 50 µl of Affi-prep protein A beads (BioRad) crosslinked to 9E10 antibodies [97]. Beads were washed with lysis buffer and stably bound proteins were eluted twice with 1.5× bead volume of 2 M glycine pH2 which was neutralized afterwards by Tris pH 8.0. For mass spectroscopy analysis, the eluates were processed by reduction and alkylation of the cysteines followed by sequential digestion with AspN and chymotrypsin or with trypsin only. The peptides were separated by nano-HPLC (Agilent 1100 nanoLC system, Agilent Technologies) coupled to an LTQ Orbitrap Velos hybrid mass spectrometer (Thermo Scientific) operated in positive mode using a top 5 DDA method. Inclusion lists were partially added to the method to search for expected peptides and to confirm already identified phosphorylated Sir4 peptides. Phosphorylated peptides and phosphosites were determined searching SwissProt data base restricted to S. cerevisiae using Mascot 2.3 (Matrix Science). Resulting sequences were inspected manually. Relative quantification was performed by integration of LC–MS extracted ion chromatograms. The peak areas of the corresponding phosphorylated peptides were normalized to the average of the peak areas of five non-phosphorylated Sir4 peptides. For prediction of CDK and MAPK sites, GPS2.1 software was used [72].
Microscopy
C-terminally EGFP-tagged Sir3 (GA3128, GA6287, GA6288) was monitored in live cells grown to mid-log phase in SC medium and then embedded in an agarose pad as described [98]. For quantification of Sir3-EGFP foci, all images were taken the same day and treated with the same threshold to quantify foci versus intense foci (above that threshold).
mRNA purification, QPCR, and microarray
Cells of indicated strains/colony color were grown to OD600>0.6 and mRNA was purified using Qiagen mini RNeasy Kit. Reverse Transcriptase (RT) reaction was performed using ProtoScript AMV Kit (NEB#E6550). For QPCR, 0.5 µl of the RT reaction was used in a total volume of 10 µl using the GoTaq qPCR Master Mix (Promega, A6002), sybr green method and the ONE STEP fast cycler (ABI). For primers see Text S1. Values were normalized to ACT1 to account for samples differences and then to Sir4.
For microarrays, 100 ng of total RNA were amplified with the GeneChip WT Double-Stranded Target Assay (Affymetrix) and hybridized to GeneChip S. cerevisiae Tiling 1.0R Arrays following the “GeneChip Whole Transcript (WT) Double-Stranded Target Labeling Assay Manual” (Affymetrix) with a hybridization time of 16 h. The Affymetrix Fluidics protocol FS450_0001 was used for washing. Scanning was performed with Affymetrix GCC Scan Control v. 3.0.0.1214 on a GeneChip Scanner 3000 with autoloader. Raw data CEL files were read into R (version 2.14.1) using the Bioconductor (version 2.9) package Affy and a custom CDF package (available upon request). Probe sets were summarized and probe set-level values normalized with the RMA function. Gene coordinates for S. cerevisiae genes (EF3) were downloaded from Biomart (central.biomart.org) and chromosome length information was retrieved from the chromInfo table of the UCSC genome browser (genom.ucsc.edu) for SacCer_Apr2011/sacCer3. Fold changes were calculated using the lmFit and eBayes functions as implemented in the limma package. Fold changes for telomeric genes were centered around zero and plotted against the distance to the closest chromosome end. Smoothing was performed with the lowess (locally weighted scatterplot smoothing) function and fold changes for each contrast were scaled and centered using the function scale.
Supporting Information
Zdroje
1. BraunsteinMRoseABHolmesSGAllisCDBroachJR 1993 Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev 7 592 604
2. SukaNSukaYCarmenAAWuJGrunsteinM 2001 Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell 8 473 479
3. LooSRineJ 1994 Silencers and domains of generalized repression. Science 264 1768 1771
4. GottschlingDE 1992 Telomere-proximal DNA in Saccharomyces cerevisiae is refractory to methyltransferase activity in vivo. Proc Natl Acad Sci U S A 89 4062 4065
5. SinghJKlarAJ 1992 Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: a novel in vivo probe for chromatin structure of yeast. Genes Dev 6 186 196
6. RaghuramanMKBrewerBJFangmanWL 1997 Cell cycle-dependent establishment of a late replication program. Science 276 806 809
7. GottaMLarocheTFormentonAMailletLScherthanH 1996 The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae. J Cell Biol 134 1349 1363
8. OttavianiAGilsonEMagdinierF 2008 Telomeric position effect: from the yeast paradigm to human pathologies? Biochimie 90 93 107
9. RuscheLNKirchmaierALRineJ 2003 The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72 481 516
10. GasserSMCockellMM 2001 The molecular biology of the SIR proteins. Gene 279 1 16
11. MoazedD 2001 Common themes in mechanisms of gene silencing. Mol Cell 8 489 498
12. BuhlerMGasserSM 2009 Silent chromatin at the middle and ends: lessons from yeasts. Embo J 28 2149 2161
13. CubizollesFMartinoFPerrodSGasserSM 2006 A homotrimer-heterotrimer switch in Sir2 structure differentiates rDNA and telomeric silencing. Mol Cell 21 825 836
14. RineJHerskowitzI 1987 Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116 9 22
15. AparicioOMBillingtonBLGottschlingDE 1991 Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell 66 1279 1287
16. ShoreDStillmanDJBrandAHNasmythKA 1987 Identification of silencer binding proteins from yeast: possible roles in SIR control and DNA replication. Embo J 6 461 467
17. BuchmanARLueNFKornbergRD 1988 Connections between transcriptional activators, silencers, and telomeres as revealed by functional analysis of a yeast DNA-binding protein. Mol Cell Biol 8 5086 5099
18. MorettiPFreemanKCoodlyLShoreD 1994 Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev 8 2257 2269
19. SusselLShoreD 1991 Separation of transcriptional activation and silencing functions of the RAP1-encoded repressor/activator protein 1: isolation of viable mutants affecting both silencing and telomere length. Proc Natl Acad Sci U S A 88 7749 7753
20. BrandAHMicklemGNasmythK 1987 A yeast silencer contains sequences that can promote autonomous plasmid replication and transcriptional activation. Cell 51 709 719
21. TrioloTSternglanzR 1996 Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature 381 251 253
22. TannerKGLandryJSternglanzRDenuJM 2000 Silent information regulator 2 family of NAD - dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A 97 14178 14182
23. ImaiSArmstrongCMKaeberleinMGuarenteL 2000 Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403 795 800
24. ArmstrongCMKaeberleinMImaiSIGuarenteL 2002 Mutations in Saccharomyces cerevisiae gene SIR2 can have differential effects on in vivo silencing phenotypes and in vitro histone deacetylation activity. Mol Biol Cell 13 1427 1438
25. HechtALarocheTStrahl-BolsingerSGasserSMGrunsteinM 1995 Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell 80 583 592
26. GeorgelPTPalacios DeBeerMAPietzGFoxCAHansenJC 2001 Sir3-dependent assembly of supramolecular chromatin structures in vitro. Proc Natl Acad Sci U S A 98 8584 8589
27. JohnsonALiGSikorskiTWBuratowskiSWoodcockCL 2009 Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol Cell 35 769 781
28. OppikoferMKuengSMartinoFSoeroesSHancockS 2011 The dual role of H4K16 acetylation in the establishment of yeast silent chromatin. Embo J 30 2610 2621
29. Strahl-BolsingerSHechtALuoKGrunsteinM 1997 SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11 83 93
30. RuscheLNKirchmaierALRineJ 2002 Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Mol Biol Cell 13 2207 2222
31. CockellMGottaMPalladinoFMartinSGGasserSM 1998 Targeting Sir proteins to sites of action: a general mechanism for regulated repression. Cold Spring Harb Symp Quant Biol 63 401 412
32. RudnerADHallBEEllenbergerTMoazedD 2005 A nonhistone protein-protein interaction required for assembly of the SIR complex and silent chromatin. Mol Cell Biol 25 4514 4528
33. EhrentrautSHasslerMOppikoferMKuengSWeberJM 2011 Structural basis for the role of the Sir3 AAA+ domain in silencing: Interaction with Sir4 and unmethylated histone H3K79. Genes & Development 25 1835 1846
34. ZillOAScannellDTeytelmanLRineJ 2010 Co-evolution of transcriptional silencing proteins and the DNA elements specifying their assembly. PLoS Biol 8 e1000550 doi:10.1371/journal.pbio.1000550
35. TannyJCKirkpatrickDSGerberSAGygiSPMoazedD 2004 Budding yeast silencing complexes and regulation of Sir2 activity by protein-protein interactions. Mol Cell Biol 24 6931 6946
36. ChangJFHallBETannyJCMoazedDFilmanD 2003 Structure of the coiled-coil dimerization motif of Sir4 and its interaction with Sir3. Structure 11 637 649
37. MurphyGASpedaleEJPowellSTPillusLSchultzSC 2003 The Sir4 C-terminal coiled coil is required for telomeric and mating type silencing in Saccharomyces cerevisiae. J Mol Biol 334 769 780
38. MorettiPShoreD 2001 Multiple interactions in Sir protein recruitment by Rap1p at silencers and telomeres in yeast. Mol Cell Biol 21 8082 8094
39. TsukamotoYKatoJIkedaH 1997 Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature 388 900 903
40. LarocheTMartinSGGottaMGorhamHCPrydeFE 1998 Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr Biol 8 653 656
41. MishraKShoreD 1999 Yeast Ku protein plays a direct role in telomeric silencing and counteracts inhibition by rif proteins. Curr Biol 9 1123 1126
42. TaddeiAHedigerFNeumannFRBauerCGasserSM 2004 Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. Embo J 23 1301 1312
43. RoyRMeierBMcAinshADFeldmannHMJacksonSP 2004 Separation-of-function mutants of yeast Ku80 reveal a Yku80p-Sir4p interaction involved in telomeric silencing. J Biol Chem 279 86 94
44. AndrulisEDNeimanAMZappullaDCSternglanzR 1998 Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 394 592 595
45. MailletLBoscheronCGottaMMarcandSGilsonE 1996 Evidence for silencing compartments within the yeast nucleus: a role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes Dev 10 1796 1811
46. HedigerFNeumannFRVan HouweGDubranaKGasserSM 2002 Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol 12 2076 2089
47. AndrulisEDZappullaDCAnsariAPerrodSLaiosaCV 2002 Esc1, a nuclear periphery protein required for Sir4-based plasmid anchoring and partitioning. Mol Cell Biol 22 8292 8301
48. TaddeiAGasserSM 2004 Multiple pathways for telomere tethering: functional implications of subnuclear position for heterochromatin formation. Biochim Biophys Acta 1677 120 128
49. GartenbergMRNeumannFRLarocheTBlaszczykMGasserSM 2004 Sir-mediated repression can occur independently of chromosomal and subnuclear contexts. Cell 119 955 967
50. TaddeiAVan HouweGNagaiSErbIvan NimwegenE 2009 The functional importance of telomere clustering: global changes in gene expression result from SIR factor dispersion. Genome Res 19 611 625
51. MarshallMMahoneyDRoseAHicksJBBroachJR 1987 Functional domains of SIR4, a gene required for position effect regulation in Saccharomyces cerevisiae. Mol Cell Biol 7 4441 4452
52. MartinoFKuengSRobinsonPTsai-PflugfelderMvan LeeuwenF 2009 Reconstitution of yeast silent chromatin: multiple contact sites and O-AADPR binding load SIR complexes onto nucleosomes in vitro. Mol Cell 33 323 334
53. CockellMRenauldHWattPGasserSM 1998 Sif2p interacts with Sir4p amino-terminal domain and antagonizes telomeric silencing in yeast. Curr Biol 8 787 790
54. PijnappelWWSchaftDRoguevAShevchenkoATekotteH 2001 The S. cerevisiae SET3 complex includes two histone deacetylases, Hos2 and Hst1, and is a meiotic-specific repressor of the sporulation gene program. Genes Dev 15 2991 3004
55. PillusLRineJ 1989 Epigenetic inheritance of transcriptional states in S. cerevisiae. Cell 59 637 647
56. PattersonEEFoxCA 2008 The Ku complex in silencing the cryptic mating-type loci of Saccharomyces cerevisiae. Genetics 180 771 783
57. VandreCLKamakakaRTRivierDH 2008 The DNA end-binding protein Ku regulates silencing at the internal HML and HMR loci in Saccharomyces cerevisiae. Genetics 180 1407 1418
58. RenauldHAparicioOMZierathPDBillingtonBLChhablaniSK 1993 Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev 7 1133 1145
59. AiWBertramPGTsangCKChanTFZhengXF 2002 Regulation of subtelomeric silencing during stress response. Mol Cell 10 1295 1305
60. HardyCFSusselLShoreD 1992 A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev 6 801 814
61. LowaryPTWidomJ 1998 New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276 19 42
62. RuaultMDe MeyerALoiodiceITaddeiA 2011 Clustering heterochromatin: Sir3 promotes telomere clustering independently of silencing in yeast. J Cell Biol 192 417 431
63. LarocheTMartinSGTsai-PflugfelderMGasserSM 2000 The dynamics of yeast telomeres and silencing proteins through the cell cycle. J Struct Biol 129 159 174
64. SmithCDSmithDLDeRisiJLBlackburnEH 2003 Telomeric protein distributions and remodeling through the cell cycle in Saccharomyces cerevisiae. Mol Biol Cell 14 556 570
65. MartinSGLarocheTSukaNGrunsteinMGasserSM 1999 Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97 621 633
66. MillsKDSinclairDAGuarenteL 1999 MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97 609 620
67. Radman-LivajaMRubenGWeinerAFriedmanNKamakakaR 2011 Dynamics of Sir3 spreading in budding yeast: secondary recruitment sites and euchromatic localization. Embo J 30 1012 1026
68. RayAHectorRERoyNSongJHBerknerKL 2003 Sir3p phosphorylation by the Slt2p pathway effects redistribution of silencing function and shortened lifespan. Nat Genet 33 522 526
69. HoltLJTuchBBVillenJJohnsonADGygiSP 2009 Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325 1682 1686
70. UbersaxJAWoodburyELQuangPNParazMBlethrowJD 2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
71. MillerMLJensenLJDiellaFJorgensenCTintiM 2008 Linear motif atlas for phosphorylation-dependent signaling. Sci Signal 1 ra2
72. XueYRenJGaoXJinCWenL 2008 GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol Cell Proteomics 7 1598 1608
73. DenisonCRudnerADGerberSABakalarskiCEMoazedD 2005 A proteomic strategy for gaining insights into protein sumoylation in yeast. Mol Cell Proteomics 4 246 254
74. HwangCSShemorryAVarshavskyA 2011 N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327 973 977
75. LiouGGTannyJCKrugerRGWalzTMoazedD 2005 Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell 121 515 527
76. SchnellRD'AriLFossMGoodmanDRineJ 1989 Genetic and molecular characterization of suppressors of SIR4 mutations in Saccharomyces cerevisiae. Genetics 122 29 46
77. DasguptaARamseyKLSmithJSAubleDT 2004 Sir Antagonist 1 (San1) is a ubiquitin ligase. J Biol Chem 279 26830 26838
78. ValenzuelaLDhillonNDubeyRNGartenbergMRKamakakaRT 2008 Long-range communication between the silencers of HMR. Mol Cell Biol 28 1924 1935
79. HofmannJFLarocheTBrandAHGasserSM 1989 RAP-1 factor is necessary for DNA loop formation in vitro at the silent mating type locus HML. Cell 57 725 737
80. WeissKSimpsonRT 1998 High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating type locus HMLalpha. Mol Cell Biol 18 5392 5403
81. MoazedDKistlerAAxelrodARineJJohnsonAD 1997 Silent information regulator protein complexes in Saccharomyces cerevisiae: a SIR2/SIR4 complex and evidence for a regulatory domain in SIR4 that inhibits its interaction with SIR3. Proc Natl Acad Sci U S A 94 2186 2191
82. AparicioOMGottschlingDE 1994 Overcoming telomeric silencing: a trans-activator competes to establish gene expression in a cell cycle-dependent way. Genes Dev 8 1133 1146
83. KirchmaierALRineJ 2006 Cell cycle requirements in assembling silent chromatin in Saccharomyces cerevisiae. Mol Cell Biol 26 852 862
84. MillerAMNasmythKA 1984 Role of DNA replication in the repression of silent mating type loci in yeast. Nature 312 247 251
85. Martins-TaylorKDulaMLHolmesSG 2004 Heterochromatin spreading at yeast telomeres occurs in M phase. Genetics 168 65 75
86. KellumRRaffJWAlbertsBM 1995 Heterochromatin protein 1 distribution during development and during the cell cycle in Drosophila embryos. J Cell Sci 108 Pt 4 1407 1418
87. BuchenauPHodgsonJStruttHArndt-JovinDJ 1998 The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: impact on models for silencing. J Cell Biol 141 469 481
88. WeiYChenYHLiLYLangJYehSP 2011 CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 13 87 94
89. ChenSBohrerLRRaiANPanYGanL 2010 Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 12 1108 1114
90. FischleWTsengBSDormannHLUeberheideBMGarciaBA 2005 Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438 1116 1122
91. HirotaTLippJJTohBHPetersJM 2005 Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature 438 1176 1180
92. PerrodSCockellMMLarocheTRenauldHDucrestAL 2001 A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. Embo J 20 197 209
93. GolemisEASerebriiskiiIFinleyRLJrKoloninMGGyurisJ 2001 Interaction trap/two-hybrid system to identify interacting proteins. Curr Protoc Protein Sci Chapter 19 Unit19 12
94. BjergbaekLCobbJATsai-PflugfelderMGasserSM 2005 Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. Embo J 24 405 417
95. GottaMPalladinoFGasserSM 1998 Functional characterization of the N terminus of Sir3p. Mol Cell Biol 18 6110 6120
96. PantazisPWestMHBonnerWM 1984 Phosphorylation of histones in cells treated with hypertonic and acidic media. Mol Cell Biol 4 1186 1188
97. HerzogFPetersJM 2005 Large-scale purification of the vertebrate anaphase-promoting complex/cyclosome. Methods Enzymol 398 175 195
98. MeisterPGehlenLRVarelaEKalckVGasserSM 2010 Visualizing yeast chromosomes and nuclear architecture. Methods Enzymol 470 535 567
99. GottschlingDEAparicioOMBillingtonBLZakianVA 1990 Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63 751 762
100. MondouxMAZakianVA 2007 Subtelomeric elements influence but do not determine silencing levels at Saccharomyces cerevisiae telomeres. Genetics 177 2541 2546
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 5
Nejčtenější v tomto čísle
- Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice
- Genome-Wide Association of Pericardial Fat Identifies a Unique Locus for Ectopic Fat
- Slowing Replication in Preparation for Reduction
- An Essential Role for Katanin p80 and Microtubule Severing in Male Gamete Production