
Characterization of Inducible Models of Tay-Sachs and Related Disease
Tay-Sachs and Sandhoff diseases are lethal inborn errors of acid β-N-acetylhexosaminidase activity, characterized by lysosomal storage of GM2 ganglioside and related glycoconjugates in the nervous system. The molecular events that lead to irreversible neuronal injury accompanied by gliosis are unknown; but gene transfer, when undertaken before neurological signs are manifest, effectively rescues the acute neurodegenerative illness in Hexb−/− (Sandhoff) mice that lack β-hexosaminidases A and B. To define determinants of therapeutic efficacy and establish a dynamic experimental platform to systematically investigate cellular pathogenesis of GM2 gangliosidosis, we generated two inducible experimental models. Reversible transgenic expression of β-hexosaminidase directed by two promoters, mouse Hexb and human Synapsin 1 promoters, permitted progression of GM2 gangliosidosis in Sandhoff mice to be modified at pre-defined ages. A single auto-regulatory tetracycline-sensitive expression cassette controlled expression of transgenic Hexb in the brain of Hexb−/− mice and provided long-term rescue from the acute neuronopathic disorder, as well as the accompanying pathological storage of glycoconjugates and gliosis in most parts of the brain. Ultimately, late-onset brainstem and ventral spinal cord pathology occurred and was associated with increased tone in the limbs. Silencing transgenic Hexb expression in five-week-old mice induced stereotypic signs and progression of Sandhoff disease, including tremor, bradykinesia, and hind-limb paralysis. As in germline Hexb−/− mice, these neurodegenerative manifestations advanced rapidly, indicating that the pathogenesis and progression of GM2 gangliosidosis is not influenced by developmental events in the maturing nervous system.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002943
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002943
Summary
Tay-Sachs and Sandhoff diseases are lethal inborn errors of acid β-N-acetylhexosaminidase activity, characterized by lysosomal storage of GM2 ganglioside and related glycoconjugates in the nervous system. The molecular events that lead to irreversible neuronal injury accompanied by gliosis are unknown; but gene transfer, when undertaken before neurological signs are manifest, effectively rescues the acute neurodegenerative illness in Hexb−/− (Sandhoff) mice that lack β-hexosaminidases A and B. To define determinants of therapeutic efficacy and establish a dynamic experimental platform to systematically investigate cellular pathogenesis of GM2 gangliosidosis, we generated two inducible experimental models. Reversible transgenic expression of β-hexosaminidase directed by two promoters, mouse Hexb and human Synapsin 1 promoters, permitted progression of GM2 gangliosidosis in Sandhoff mice to be modified at pre-defined ages. A single auto-regulatory tetracycline-sensitive expression cassette controlled expression of transgenic Hexb in the brain of Hexb−/− mice and provided long-term rescue from the acute neuronopathic disorder, as well as the accompanying pathological storage of glycoconjugates and gliosis in most parts of the brain. Ultimately, late-onset brainstem and ventral spinal cord pathology occurred and was associated with increased tone in the limbs. Silencing transgenic Hexb expression in five-week-old mice induced stereotypic signs and progression of Sandhoff disease, including tremor, bradykinesia, and hind-limb paralysis. As in germline Hexb−/− mice, these neurodegenerative manifestations advanced rapidly, indicating that the pathogenesis and progression of GM2 gangliosidosis is not influenced by developmental events in the maturing nervous system.
Introduction
Two-thirds of the seventy or so inborn errors of lysosomal function affect the nervous system. Tay-Sachs disease [1], [2] and Sandhoff disease [3] are GM2 gangliosidoses arising from deficiency of the lysosomal acid hydrolase, β-N-acetylhexosaminidase; they are characterized by neuronal accumulation of GM2 ganglioside and related glycoconjugates [4]–[6]. Infantile GM2 gangliosidosis is a relentless neurodegenerative disorder with developmental regression, dystonia, blindness and seizures causing death in childhood [7], [8]. Characteristically, infants with GM2 gangliosidoses are healthy at birth and during the neonatal period but loss of motor function and cognition, with regression of acquired skills, becomes apparent after the first few months of life [9] - suggesting that disease onset is influenced by developmental processes involved in post-natal organization of the brain.
Development of genetically coherent models of Sandhoff disease generated by disruption of the Hexb gene in embryonic stem cells in mice [10], [11] provides a platform for pathological and therapeutic investigation of GM2 gangliosidoses. However, questions as to the pathogenesis, mechanisms inducing progression of disease, and the true extent of therapeutic reversibility remain. Ascertaining how the lysosomal defect contributes to widespread neuronal injury and other cardinal features of this condition, mandates the need for an authentic model of the disease which allows temporal and spatial dissection of the neuropathology to be analysed during its evolution. To accomplish this, we developed a reversible transgenic murine counterpart of human Sandhoff disease which utilizes the tetracycline-inducible gene expression system.
Mouse models employing the tetracycline-inducible system [12] have been created for the investigation of other neurogenetic diseases such as Huntington's disease [13] and Alzheimer's disease [14]. While these models used the tetracycline-inducible system to deliver a single deleterious gene product, creation of an informative experimental model to study diffuse neurodegeneration in a recessively transmitted disorder of lysosomal function, requires global rescue of the nervous system. Inherent challenges to this stratagem relate particularly to the extent of functional restitution and robustness with which long-term expression can be obtained in the neuraxis [15].
Here we characterize two novel inducible strains of transgenic Sandhoff disease mice: one expresses a construct harbouring proximal elements of the mouse Hexb promoter, its counterpart is under the control of the human Synapsin 1 gene (SYN1) promoter. Phenotypic rescue of Sandhoff mice with autoregulatory expression cassettes based on the tet-off system led to a threefold extension of lifespan with sparing of most of the central nervous system from lysosomal storage and accompanying injurious effects. Unexpectedly, doxycycline-induced suppression of β-hexosaminidase expression in the adult animal caused acute neurodegeneration with the stereotypical murine simulacrum of Sandhoff disease and a course indistinguishable from the unmodified germline Hexb−/− background strain. Accordingly, neuro-developmental processes probably have little influence on the lysosomal metabolism of gangliosides or on the cellular ontogeny of Tay-Sachs and Sandhoff diseases. These findings may inform the timing and clinical stage at which therapeutic interventions such as gene therapy are considered for patients with GM2 gangliosidoses.
Methods
Ethics Statement
Mice were handled in accordance with the Animals (Scientific Procedures) Act 1986.
Transgene Construction
Bovine growth hormone polyadenylation (BGH-polyA) sequence from the pcDNA3 plasmid (Invitrogen) was subcloned into the SphI site of pSP73 plasmid (P2221, Promega) to make plasmid P1. Coding sequence for the tet-transactivator from the pTet-off advanced plasmid (tTA2s, 630934, Clontech) was digested with EcoRI and BamHI and subcloned into the pSP73 plasmid upstream of the BGH-polyA sequence (P2). The proximal murine Hexb promoter (330 bp) [16] was amplified with primers: forward 5′-CTC CTG GGA ATT CTG ACT CG-3′ and reverse: 5′-TCC GCG AGT CTG GCT AGG-3′. The human Synapsin 1 promoter [17] (SYN1, 585 bp) containing the neuron-restrictive silencing element [18] was amplified by PCR using primers: forward 5′-AGT CTT GTA CAC CCT CTG TGA GGG GGT TAT T-3′ and reverse 5′-AGT GTG AAT TCC TCT CAG GCA CGA CAC GAC-3′. Both promoter fragments were cloned using the TOPO 2.1 TA cloning vector (Invitrogen). Either one of these promoters was sequenced and subcloned into the EcoRI site upstream of the tet-transactivator in plasmid P2 after dephosphorylation of the vector with shrimp alkaline phosphatase (plasmids P3Hex or P3SYN).
The TRE-Tight promoter from pTRE-Tight (631059, Clontech) was digested with ZraI and EcoRI and subcloned into the EcoRV and EcoRI restriction sites upstream of the BGH-polyA sequence in P1 to make plasmid P4. The full murine Hexb cDNA was amplified from a plasmid (Image ID: 100015010, GenBank accession: BC146503) using primers: forward 5′-ATG CAG AAT TCA GCA GAA GGG CCG TCA AG-3′ and reverse 5′-CGA ACC GAA CAG GCT TAT GT-3′. This amplification product was cloned into a TOPO 2.1 plasmid and digested with EcoRI and StuI and subcloned into the EcoRI and EcoRV restriction sites of pcDNA3 for expression analysis. Hexb coding sequence was then digested with EcoRI and PvuII and subcloned into EcoRI and PvuII restriction sites downstream of the TRE-Tight promoter in plasmid P4 to make P5.
A BglII/BsrBI fragment containing the Hexb promoter/tTA-cDNA/BGH-PolyA construct from P3Hex and a BglII/HpaI fragment containing the SYN1 promoter/tTA-cDNA/BGH-PolyA construct from P3SYN were subcloned into ZraI and BglII restriction sites in P5 such that they were in opposite ‘back-to-back’ orientation to the TRE-Tight/Hexb-cDNA/BGH-PolyA construct on the same vector. This created the auto-regulatory constructs P6Hex and P6SYN (Figure S1). The BGH-PolyA sequence downstream of the Hexb coding sequence in P6Hex and P6SYN was replaced with a NotI/PciI fragment from the pTRE-Tight plasmid containing SV40-PolyA sequence, creating plasmids P7Hex and P7SYN (Figure S1).
cHS4 sequence, reported to be a good insulating element [19], was amplified from genomic DNA isolated from commercially available chicken liver using primers: forward 5′-ACG TAG ATC TTC CTG GAA GGT CCT GGA AG-3′ and reverse 5′-TCA AAC ATG CAG GCT CAG AC-3′ and sequenced. Cloned cHS4 sequence was found to contain mutations when compared to published sequence. Consequently, this sequence was re-amplified using the forward primer 5′-ATA CGG AGA TCT GAG CTC ATG GGG ACA GCC CCC CCC CAA AGC CCC CAG GGA TGT AAT TAC-3′ in order to change the sequence of the second element of cHS4 so it was identical to sequence previously published. cHS4 was subcloned into a BglII site, in between the two expression constructs in each of the two P7 plasmids, to create four new plasmids where the cHS4 element was placed in either of two orientations – P8Hex (+), P8Hex (−), P8SYN (+) and P8SYN (−). P6Hex, P8Hex (+) and P8SYN (+) constructs were chosen for standard pronuclear injection techniques (Figure S1) [20].
Generation of Transgenic Animals
P6Hex, P8Hex (+) and P8SYN (+) plasmids, digested with DrdI and PvuII to produce 3.8, 4.6, and 4.8 kbp fragments, respectively, were injected into fertilized oocytes produced from matings of B6CBA F1 mice. HexTg or SYNTg transgenic founder animals, where Hexb cDNA was under the control of the Hexb or SYN1 promoters, respectively, were genotyped by PCR. For routine genotyping, the presence of either the Hex or the SYN inducible constructs were detected using primers: 5′-AGC TCA CTC AAA GGC GGT AA-3′ and 5′-GGG AGG ATT GGG AAG ACA AT-3′ to amplify sequence across the tail to head junctions of the integrated tandem repeats.
Transgenic animals were crossed with germline Hexb−/− (Sandhoff) mice (strain: B6; 129S-Hexbtm1Rlp, Jackson Laboratory [10]). Crossing transgenic founder HexTg and SYNTg mice and Sandhoff mice was performed until Hexb−/−HexTg or Hexb−/−SYNTg mice were generated. The Hex and SYN transgenes were maintained in a hemizygous state. The presence or absence of the Hexb knockout allele was detected using a three primer PCR reaction: MNEO1 : 5′-ATC TGG ACG AAG AGC ATC AG-3′; MHexb18 : 5′-TAG ACT GCT TTG GAA ACT GC-3′; MHexb19 : 5′-TCA GGA AGG AAG TGT CTC AC-3′.
Animal Experimentation
Mice were allowed access to food ad libitum and were supplied with transgel (Charles River Laboratories) and mashed food pellets on the cage floor when they displayed signs of Sandhoff disease. For disease induction, mice were exposed continuously to doxycycline in food pellets at a dose of 1 g doxycycline/kg of food (S3949, Bio-Serv, NJ). Motor performance was assessed by the inverted screen test as previously described [21]. For biochemical studies, animals were culled by asphyxiation with CO2 and tissues rapidly dissected out and frozen on dry ice. For histological analysis, using fresh frozen tissues, animals were asphyxiated with CO2 and tissues embedded in OCT and frozen on dry ice. For histological analysis using perfuse-fixed tissue, animals were terminally anaesthetised with an overdose of sodium pentobarbital and transcardially perfused with phosphate-buffered saline (PBS) containing 4% w/v paraformaldehyde. Brains were postfixed for two hours in paraformaldehyde and cryopreserved in PBS containing 30% w/v sucrose.
Histological Processing
OCT embedded tissue was sectioned at 15 or 30 µm thickness and mounted onto superfrost plus microscope slides, and stored at −80°C. For immunostaining, primary antibodies used were mouse anti-GFAP (1∶50, sc-6171, Santa-Cruz Biotechnology), rat anti-CD68 (1∶50, MCA-1957, Serotec), or rabbit anti-PAX2 (1∶100, 71–6000, Invitrogen). Secondary antibodies used were goat anti-mouse Ig-HRP (1∶100, P0477, DAKO), biotinylated anti-rat (1∶100, BA-4001, Vector Laboratories), or goat anti-rabbit Ig-HRP (1∶100, P0488, DAKO).
Slides were thawed, dried and fixed in PBS containing 4% paraformaldehyde for 10 minutes. Tissue was permeabilized in PBS containing 0.1% v/v Triton-X100 for 15 minutes and endogenous peroxidase activity was quenched by incubation in PBS containing 3% H2O2 for 5 minutes. Sections were blocked in PBS containing 2% bovine serum albumin for twenty minutes and were then incubated with blocking solution containing primary antibody overnight at 4°C. If the avidin/biotin complex system was used for staining, endogenous biotin was blocked at this stage using an avidin/biotin blocking kit (SP-2001, Vector Laboratories). Sections were washed in PBS three times for five minutes each and incubated in blocking solution containing secondary antibody for one hour. Sections that were incubated with biotinylated anti-rat antibody were washed in PBS as above and incubated with avidin-HRP (Vectastain ABC kit, PK-4000, Vector Laboratories) according to manufacturer's directions. Slides were washed in PBS as above and developed with diaminobenzidine (DAB, SK-4100, Vector Laboratories), dehydrated, cleared and mounted in DPX.
Detection of β-hexosaminidase activity in tissues was carried out as previously described [22] by incubating sections in the substrate naphthol AS-BI N-acetyl-β-glucosaminide (Sigma, N4006-1G). Hex-azotized pararosaniline, prepared by treatment of pararosaniline (P3750-5G, Sigma) with sodium nitrite and hydrochloric acid, was used to visualise the enzymatic reaction-product. For co-localization with the neuron-specific marker, NeuN, sections stained for β-hexosaminidase activity were permeabilized and blocked as above, and then subsequently incubated with mouse anti-NeuN-Alexa 488 conjugate (1∶100, MAB377X, Millipore) for one hour. Slides were washed in PBS and mounted in Prolong Gold anti-fade mounting medium (P36931, Invitrogen). Periodic acid-Schiff (PAS) staining was carried out with Schiff reagent (3803800E, Leica Microsystems) and periodic acid (P0430-100G, Sigma) as instructed by the manufacturer. PAS-stained sections were counterstained with haematoxylin, dehydrated, cleared and mounted in DPX.
In situ hybridization was performed as previously described [23]. Expression of tet-transactivator mRNA was assessed by hybridization with an antisense probe generated using the T7 promoter on plasmid P2 that was cut with EcoRI (1.1 kb).
Tissue Analysis
Fluorometric analysis of enzyme activity using the substrate 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside (MUG, M-5504, Biosynth), was performed as previously described [23]. Briefly, tissue samples were homogenized in 10 mM sodium phosphate (pH 6.0) containing 100 mM NaCl and 0.1% Triton X-100, centrifuged at 13,000 RPM for 10 minutes and the supernatant taken for enzyme analysis. Activities were standardized by determination of protein concentration using the bicinchoninic acid protein assay (23227, Thermo Scientific). Anion-exchange chromatography, used to resolve the activity of the different β-hexosaminidase isoforms (HexA, HexB and HexS), was carried out as described previously [23].
Lipids were quantified using high performance thin–layer silica chromatography as previously described [21]. Relative quantification by densitometry was performed using ImageJ software (NIH) and sphingolipids were standardized to ganglioside GM1.
Gene expression was quantified by real-time PCR. Mouse tissue was homogenised and total RNA extracted using an RNeasy Mini Kit (74104, Qiagen). Total RNA was reverse-transcribed with a Quantitect Reverse Transcriptase Kit (205311, Qiagen). Real-time PCR was performed with Power SYBR Green PCR Master Mix (4367659, Applied Biosystems) on a 7500 Fast Real-Time PCR System (Applied Biosystems). Gene expression was standardized by comparison with the housekeeping gene for β-actin, using the commercially available primer set Quantitect Primer Assay Mm_Actb_2_SG (QT01136772, Qiagen). Transgenic mouse Hexb cDNA was amplified using the forward and reverse primers: 5′ - AAT GGT CAG CCG TGG AAT AG-3′ and 5′ - CAA ATG TGG TAT GGC TGA TTA TG-3′. Transgenic tet-transactivator (tTA22) cDNA sequence was amplified using the forward and reverse primers: 5′ - AGA GCA CAG CGG AAT GAC TT-3′ and 5′ - CCT GTA CTG GCA CGT GAA GA-3′. For all amplification reactions, the following cycle conditions were used: 95°C for 10 min×1; 95°C for 15 s, 58°C for 1 min×35.
Image Manipulation, Counting, and Statistical Analysis
All drawing was performed using Inkscape (version 0.47). Manipulation of photomicrographs was carried out with ImageJ (NIH) and arranged using Inkscape.
PAX2-positive neurons in the ventral spinal cord were quantified using ImageJ (NIH) software. For each of the four regions of the spinal cord counted, three non-consecutive 15 µm thick sections were quantified per animal. PAX2-stained neurons ventral to the central canal were counted. Counts were standardized by area to give a relative density per mm2. For statistical analysis, ANOVA was used with Bonferroni's post hoc analysis for comparison (GraphPad Prism v5.0, GraphPad Software).
Results
Assessment of Transgenic β-Hexosaminidase Expression
Inducible expression constructs were tested by transient transfection of HEK 293T cells, with and without exposure to various concentrations of doxycycline. Two main types of inducible cassette were tested – one expressing from the proximal mouse Hexb promoter (Hex), the other from the human SYN1 promoter (SYN). Both inducible constructs showed strong expression of β-hexosaminidase activity, and brisk silencing in response to low concentrations of doxycycline added to the media of transiently transfected cells (Figures S1 and Figure 1A). Constructs were then microinjected into fertilized oocytes to generate transgenic founder mice (Table S1). Constructs driven from both the Hexb and SYN1 promoters were used to create transgenic lines that were intended to compare body-wide rescue from Sandhoff disease with rescue in nervous tissues only.

Transgenic founder mice were identified using PCR and crossed over two successive generations onto a Hexb−/− background. Two transgenic lines, one bearing a Hex cassette (Hexb−/−HexTg) and one bearing a SYN cassette (Hexb−/−SYNTg) (from P8Hex (+) and P8SYN (+), respectively), expressed transgenic β-hexosaminidase in the brain, as shown by a fluorometric MUG assay performed on homogenized brain tissue (Figure 1B). As anticipated, transgenic β-hexosaminidase expression from the SYN1 promoter was found only in nervous tissues. Contrary to expectation, the Hex cassette did not express in the wide variety of tissues as the endogenous mouse Hexb promoter does. Instead, expression was found only in the nervous tissue, skin and skeletal muscle.
Anion-exchange chromatography on tissue lysate from the cerebrum showed the presence of all three β-hexosaminidase isozymes, HexB, HexA and HexS, in the brains of both lines; the presence of HexB and HexA rely on expression of transgenic Hexb coding sequence (Figure S2C and S2E). Control heterozygous (Hexb+/−) and germline mutant (Hexb−/−) animals showed the predicted pattern of β-hexosaminidase expression. Importantly, the relative amounts of the three isozymes in the transgenic lines and heterozygous mice were similar.
Throughout the neuraxis, transgenic β-hexosaminidase expression, assessed by staining for β-hexosaminidase activity on cryo-sections, appeared to be more widespread in the Hexb−/−SYNTg line than in the Hexb−/−HexTg strain. The SYN transgenic mouse expressed β-hexosaminidase activity throughout the forebrain, including the diencephalon where activity was largely weak or absent in the Hex line (Figure 1C iii–v). In both lines transgenic β-hexosaminidase activity was present in the cerebellum and absent in the brainstem, with a few exceptions in the Hex line; the inferior colliculi and the inferior olivary complex did show enzyme activity expression. Interestingly, strong transgene expression was associated with neurons, rather than with glia in both lines (Figure 1D).
The pattern of β-hexosaminidase staining throughout the neuraxis showed that expression appeared restricted to particular neuronal populations. β-hexosaminidase can be secreted and recaptured by neighbouring cells [24]. To define the pattern of neuronal cell populations expressing the transgene and potential relevance to disease manifestations, in situ hybridization was used against the tet-transactivator. Strong expression of tet-transactivator transcript was observed in the brains of mice carrying SYNTg or HexTg transgenes, with a few notable differences. In the SYNTg cerebral cortex, tet-transactivator transcript was strongly expressed in layer 6b neurons, but not as strongly expressed in other layers. This contrasted with the Hex line, where expression was more evenly distributed through the cortex. Furthermore, strong hybridization signal found in the olfactory bulbs and the thalamic reticular nucleus of the Hex transgenic animal was absent in the SYN line (Figure S3). It is noteworthy that even though no in situ hybridization signal could be seen in the SYNTg strain olfactory bulbs, there was strong transgenic β-hexosaminidase activity. However, in most cases expression of tet-transactivator transcript was seen in the same populations of cells that strongly expressed β-hexosaminidase activity.
Inducible Hexb Expressing Transgenes Rescue the Sandhoff Mouse
Expression of β-hexosaminidase activity from the Hex and SYN transgenic cassettes was sufficient to rescue the Sandhoff mouse from stereotypic features of acute Sandhoff disease. By the time the Hexb−/− Sandhoff animals reach four to five months of age, they show marked bradykinesia and tremor when compared with heterozygous mice of the same age (Videos S1 and S2). By this stage the Sandhoff mouse loses weight and reaches its humane endpoint (defined as losing 10–20% of its highest body weight) at an average age of 127 days. In contrast, Sandhoff mice that carry either the Hex or SYN transgenic constructs show unrestricted movement and no overt disease up to six months of age (Videos S3 and S4, respectively). However, the transgenic constructs do not provide complete rescue, and mice deteriorate between six months of age and their humane endpoints that are on average 373 days and 404 days for Hexb−/−HexTg and Hexb−/−SYNTg mice, respectively (Figure 2A).

Hexb−/−HexTg and Hexb−/−SYNTg mice show progressive but comparatively milder tremor than their germline counterparts that is accompanied by progressive limb hypertonia, evident when mice are held by their tails (Videos S5 and S6). By approximately one year of age, limb hypertonicity restricted movement of the mouse around its home cage; during walking the hindlimbs appeared un-coordinated (particularly evident in the mouse shown in Video S6). Hypertonicity in the limbs is a major contributing factor to deterioration in motor performance as measured with the inverted screen test, between six months and one year of age (Figure 2B).
Transgenic Hexb expression is still present in animals at their humane endpoint and is comparable with mice that are six months of age (Figure 3B–3C and 3E–3F). This is also reflected by the fact that when transgenic animals are culled, areas that strongly express transgenic Hexb, such as the cerebral cortex and the cerebellum, remain free of glycolipid storage as determined by PAS stained cryo-sections (Figure S4, Table S2). Overall, Hexb−/−HexTg and Hexb−/−SYNTg mice show no storage in the cerebral cortex (including the motor corticies, Figure 4A) and most other structures in the cerebrum, such as the striatum (Figure 4B), beyond one year of age (humane endpoint). However, Hexb−/−SYNTg mice did show less neuronal storage in the hypothalamic, midbrain and brainstem structures than Hexb−/−HexTg mouse line at humane endpoint (Table S2).


In Sandhoff animals rescued with a transgenic expression construct, nuclei of the hindbrain such as the gigantocellular reticular nucleus showed neurons that had substantial glycoconjugate storage (Figure 4C). Both Hexb−/−HexTg and Hexb−/−SYNTg mice accumulated extensive amounts of storage material in the hindbrain and ventral spinal cord by one year of age (Figure 4C and 4D), accompanied by activated microglia (Figure 4G and 4H). Injury to these lower motor nuclei may account for the marked hypertonicity seen in the transgenic animals older than six months (Videos S5 and S6). To further characterize pathological processes in the ventral spinal cord of the Hex line at humane endpoint where abundant amoeboid microglia were seen, ventral PAX2-positive interneurons were counted (Figure 4I, 4J and 4K). Compared with one year old Hexb+/− mice, Hexb−/− and Hexb−/−HexTg mice at their respective humane endpoints had decreased PAX2-positive interneuron density in their ventral horns. Interestingly, loss of PAX2-positive interneuron density in Hexb−/− and Hexb−/−HexTg mice did not differ significantly in regions above the sacral spinal cord and also did not co-vary with degrees of hindlimb spasticity observed at the humane endpoint (Videos S2 and S5).
Variegation of transgene expression occurred in some regions of the brain such as the dorsal root ganglia (Figure 1D vii–viii). In some cases, neurons with abundant intense PAS-stained material were present in apposition to similar neurons without any apparent storage (not shown). Similar examples indicating a lack of complete cross-complementation were detected in numerous other nuclei of the mid and hindbrain in Hexb−/−HexTg and Hexb−/−SYNTg mice.
Transgenic Hexb Expression Is Reversibly Inducible In Vivo
Transgene expression in mouse cerebrum was assessed with quantitative real-time PCR. Expression of both transgenic Hexb transcript and tet-transactivator transcript from the single autoregulatory cassette was standardized by comparison to endogenous β-actin transcript. In the absence of doxycycline, Hexb expression was greater than tet-transactivator expression. After only 24 hours of exposure to doxycycline, transgenic Hexb expression in the cerebrum had decreased by 96% and 98% in HexTg and SYNTg mice respectively (Figure 5A and 5B). This reduction was stable over the next six days of doxycycline exposure.

To determine whether transgenic Hexb expression could return after being repressed, animals were fed doxycycline laced food (1 g doxycycline/kg food) for six days and sacrificed six, 12 and 18 days post doxycycline withdrawal (Figure 5A and 5B). HexTg mice showed rapid recovery of transgenic Hexb expression, within six days. Transgenic Hexb expression post doxycycline withdrawal in the forebrain was approximately equal to pre-doxycycline exposure levels and did not change between six and 18 days post doxycycline withdrawal (Figure 5A). In contrast, transgenic Hexb expression in SYNTg mice barely increased at six days after doxycycline withdrawal. Transgenic Hexb expression recovered progressively over the next 12 days but did not reach pre-doxycycline exposure levels after 18 days withdrawal.
In line with a reduction in transgenic Hexb mRNA on exposure to doxycycline, β-hexosaminidase activity also decreased after transgenic animals were exposed to doxycycline. β-hexosaminidase activity was measured in brain lysates (Hexb−/−SYNTg mice) using the MUG assay after 0, 7, 14 and 21 days of exposure to doxycycline. Half life of Hexb protein was approximately four days in the mouse brain and activity had dropped to its minimum within 14 days of doxycycline exposure (Figure 5C).
Induction of Sandhoff Disease in the Adult Mouse Results in Stereotypic Sandhoff Disease
Once we established that the Hex and SYN transgenic cassettes provide rescue from acute Sandhoff disease, and were sensitive to doxycycline treatment in vivo, mice were fed doxycycline (1 g/kg of food) to continuously suppress β-hexosaminidase expression from five weeks of age until they reached their humane endpoint. This experiment tested whether suppression of transgenic Hexb expression permitted the accumulation of glycosphingolipids and at the same time, addressed the question as to whether developmental factors interacted with the course and signs of disease.
Analysis of motor performance using the inverted screen test showed that the doxycycline treatment regimen did not impact on the motor performance of Hexb+/− animals (Figure 6A), nor did it alter the disease course of Hexb−/− animals (Figure 6B). As shown in Figure 6C and 6D, Hexb−/−HexTg and Hexb−/−SYNTg animals that were not exposed to doxycycline showed stable motor performance over the time tested. However, animals of the same age and genotype that were fed doxycycline from five weeks of age showed a dramatic decrease in motor performance starting at age 20 weeks. Reduced motor performance of Hexb−/− animals began at 15 weeks.

Hexb−/−HexTg and Hexb−/−SYNTg mice that were fed doxycycline from five weeks of age onwards developed progressive tremor from 17–19 weeks of age. Mouse weight, on average, had reached a plateau by 20 weeks of age and thereafter started to decrease (Figure S5). Mice reached their humane endpoints with stereotypic acute Sandhoff disease on average 172.5 and 175 days of age, respectively (Figure 6E). Survival of Hexb−/−HexTg and Hexb−/−SYNTg animals under exposure to doxycycline from five weeks of age was similar to total survival of germline Hexb−/− animals. We conclude that induction of acute Sandhoff disease in the adult mouse does not modify signs and disease course significantly. At the humane endpoint, these mice showed typical features of acute Sandhoff disease, such as bradykinesia and tremor (Videos S7 and S8), unlike mice of the same genotype and age that had not been exposed to doxycycline (Videos S3 and S4).
Staining for β-hexosaminidase activity in the brains of Hexb−/−HexTg and Hexb−/−SYNTg mice that were exposed to doxycycline until their humane endpoint (Figure 3H and 3I), showed complete suppression of Hexb expression throughout the entire neuraxis. Similarly, anion-exchange chromatography performed on samples of cerebrum showed loss of HexB and HexA β-hexosaminidase isoforms upon doxycycline exposure (Figure S2D and S2F).
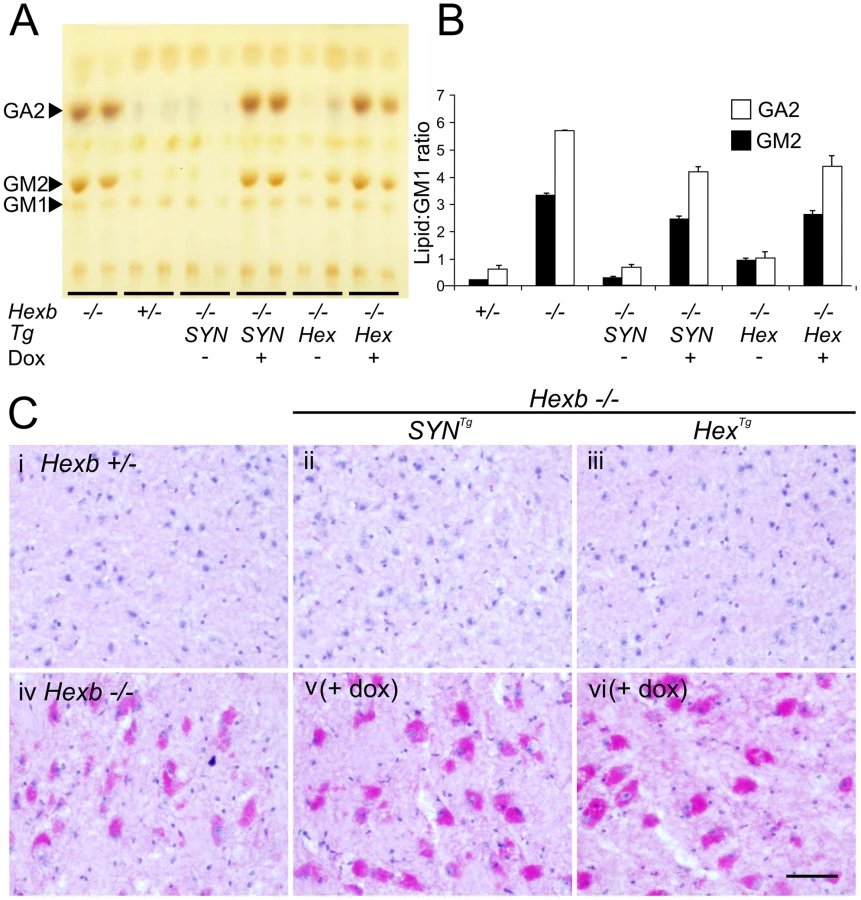
After four to five months of exposure to doxycycline, amounts of storage material in Hexb−/−HexTg and Hexb−/−SYNTg mouse brain were similar to germline Hexb−/− mice at their humane endpoint as shown by TLC analysis (Figure 7A and 7B). PAS staining also revealed storage neurons in doxycycline exposed Hexb−/−HexTg and Hexb−/−SYNTg mice where there was no trace of PAS staining in animals not exposed to doxycycline (Figure 7C). This showed that doxycycline mediated silencing of Hexb expression was sufficient to cause accumulation of glycosphingolipids. Similarly, another histological hallmark of acute Sandhoff disease was also apparent in the same tissue. Staining for the neuroinflammatory markers glial fibrillary acidic protein (GFAP) and CD68 (showing activated astroglia and microglia, respectively) was markedly increased in the cerebrum and cerebellum at the humane endpoint in animals that had been exposed to doxycycline (Figure 8). Staining for neuroinflammatory markers in doxycycline exposed Hexb−/−HexTg and Hexb−/−SYNTg animals appeared similar to Hexb−/− animals at their respective humane endpoints. Furthermore, doxycycline itself had no affect on neuroinflammation in either Hexb+/− or Hexb−/− animals (Figure 8).

Discussion
Using autoregulatory constructs, we report the generation of inducible mouse models of Sandhoff disease. The single inducible constructs used here showed widespread expression of β-hexosaminidase in the mouse brain and rescued acute Sandhoff disease. Our inducible models displayed near-total gene silencing in the presence of doxycycline: administration of the agent induced Sandhoff disease with all its stereotypic features in the adult mouse. Moreover, expression from the transgenic constructs proved to be reversible on withdrawing the doxycycline in vivo.
The use of a single genetic construct carrying both elements (the tet-transactivator and coding sequence expressed from a tet-responsive element bearing promoter) of the tet-inducible system is not in common use for generating transgenic animals by pronuclear microinjection. This stratagem has been used to deliver autoregulatory cassettes by viral vectors [25]–[27] and to generate targeted ‘knock-ins’ in stem cells utilizing the Rosa26 locus [28], [29]. Here we show the utility of this approach is feasible for creating functional inducible transgenic mice by pronuclear injection into fertilized oocytes. The obvious advantage of using a single genetic cassette is that breeding schedules are simplified and reduced numbers of animals are required when an inducible system is bred onto a knockout background.
When crossed onto a Hexb−/− background, both the Hex and SYN inducible cassettes rescued the mouse from acute Sandhoff disease. However, there were differences in expression pattern between the two constructs. Although the Hexb promoter used to drive the Hex inducible cassette was intended to provide systemic expression of Hexb based on its ‘housekeeping function’, expression of β-hexosaminidase activity outside the brain was only found in the skin and skeletal muscle. This precludes assessment of the role of β-hexosaminidase activity in organs such as the liver and kidneys; however it was surprising that for each promoter, expression throughout the central nervous system was similar, since, even for the construct driven by the Hexb promoter, expression appeared to be confined to neurons. Lack of expression from the Hex transgene may have been due to the absence of expression elements in the construct used in this study. Alternatively, this phenomenon could be explained by position effects [19].
Although expression of transgenic β-hexosaminidase throughout the central nervous system rescued the Hexb−/− mouse from acute Sandhoff disease, rescue was incomplete and residual neurodegenerative disease became apparent beyond six months of age (Videos S5 and S6). At the humane endpoint, Hexb−/−HexTg and Hexb−/−SYNTg mice had a mild tremor, and sporadic glycoconjugate storage was seen in Purkinje cells in the cerebellum as revealed by PAS staining; indeed, both strains showed abundant storage of glycoconjugate in lobe ten of the cerebellum. Storage was also detected in regions that interact with the cerebellum such as the pons and the red nucleus. Of note, no pathological storage was found in the substantia nigra at the delayed humane endpoint in these animals.
The most obvious aspect of residual neurological disease in the transgenic animals was increased limb tone (spasticity) - observed as clasping of the limbs when the mice were lifted by their tails. This disease feature was associated with a reproducible pattern of storage in the brain at the humane endpoint (Figure S4, Table S2). Most cerebral structures in the forebrain were free of storage material. Importantly, the motor cortex (origin of the cortico-spinal tract) showed no storage of glycolipid and the striatum (except for the globus pallidus) was also free of PAS-staining material. Accumulation of glycoconjugate was found in neurons, accompanied by CD68-positive amoeboid microglia, in the reticular nuclei in the pons and medulla and throughout the ventral spinal cord (Figure 4). Although storage did occur in other centres of the brain, such as the septum (both strains) and the hypothalamus (Hexb−/−HexTg), pathological changes in reticular nuclei of the medulla and pons and in ventral spinal interneurons are thought to contribute to spasticity through modulation of lower motor neurons [30]–[34]. The ventral spinal cord in Hexb−/−HexTg mice at the humane endpoint showed loss of PAX2-positive ventral interneuron density. Loss of PAX2-staining ventral interneurons is supported by similar results obtained by staining for the neural cell marker NeuN [35]. It is noteworthy that this result was comparable to loss of ventral interneurons in a study of a spastic mouse model [36], suggesting that loss of interneurons in the ventral spinal cord might be responsible for limb spasticity in long-surviving Hexb−/−HexTg mice. Similar loss of PAX2-positive interneuron density was also observed in the Hexb−/− animal at its endpoint, and although limb clasping was less marked, this probably reflects increasing spastic paralysis.
Declining motor function (Figure 2B) may contribute to the final deterioration of the animals, which reach the humane endpoint (Figure 2A) past one year of age. Although we cannot exclude the presence of disease in peripheral organs playing a role in weight loss, other studies from this laboratory [21], [23] showed that correction in the central nervous system was sufficient to rescue mice from acute Sandhoff disease for up to two years.
Efficient silencing of transgenic Hexb mRNA expression was seen in both inducible strains of mice when exposed to doxycycline (Figure 5). In the brain, transcript disappeared within 24 hours of doxycycline exposure and half life of enzyme activity was about four days, similar to human HEXB activity in fibroblasts that had a half life of six days [37]. In contrast, recovery of transgene expression upon withdrawal of doxycycline differed greatly between the two strains. In the frontal cortex of the Hex line, Hexb transgene recovered to expression levels comparable with pre-doxycycline exposure, within six days. The frontal cortex of the SYN line recovered its transgene expression much more slowly, such that 18 days after doxycycline withdrawal β-hexosaminidase activity had only recovered to approximately one third of pre-doxycycline exposure levels. Rapid recovery of transgene expression in the Hex line after doxycycline withdrawal means that it was not slow clearance of doxycycline that was causing sluggish recovery of transgene expression in the SYN line; this was considered because the skeleton can act as a reservoir of doxycycline [38]. Incomplete recovery of transgene expression in tet-inducible systems has been reported in other models [39], [40] and this is largely ascribed to changes in chromatin configuration during silencing that influence the accessibility and expression of transgenes [41], [42].
Suppression of β-hexosaminidase expression in the adult mouse induced an acute phenotype of Sandhoff disease. This was comparable in progression and phenotype to the disease seen in the germline Hexb−/− animal, where pathogenesis takes place alongside dynamic developmental processes. During rodent brain development, ganglioside synthesis changes dramatically. Gangliosides GM3 and GD3, which are abundant in the early fetal brain, decrease in expression and give way to increasing synthesis of GM1 and GD1a as neurons differentiate [43], [44]. Additionally, studies on developing postnatal cerebral cortex show a large temporary induction of GM2 expression that correlates tightly with strict developmental patterns of dendritogenesis [45]–[47]. Based on this observation, we predicted that after initiation in the adult mouse, progression of disease may take much longer than in the conventional germline model of Sandhoff disease. The fact that Sandhoff disease progressed largely unmodified when initiated in the adult mouse suggests that either developmental changes in ganglioside synthesis are insignificant compared with adult neuronal output of ganglioside, or that lysosomal disorder disease processes are resilient to fluctuations in the absolute amount of storage material. Absence of a developmental component of neurological pathogenesis has also been observed in a conditional model of Niemann-Pick type C disease [48] and may be a general feature of neuronopathic lysosomal disorders.
In the present study, a high dose of doxycycline was used to completely abolish transgene expression. However, it may be possible to titrate the dose of doxycycline to give partial correction of the phenotype - as has been reported in studies of GDNF expression induced by adeno-associated viral vectors under the control of the tet system in the substantia nigra [49]. By allowing partial rescue of the Sandhoff phenotype mediated by either of the transgenes, this approach might be used to generate attenuated models of GM2 gangliosidosis.
We contend that the inducible models of Sandhoff disease reported here will facilitate exploration of the pathogenesis in GM2 gangliosidosis. In mice that were allowed to develop residual elements of Sandhoff disease, regionalized storage of glycolipids and related storage molecules in the brain showed that limb hypertonicity can arise independently from glycolipid storage in the motor cortex and could originate in the hindbrain and spinal cord. Tetracycline-inducible models of acute Sandhoff disease were responsive to doxycycline and showed that the pathogenesis of acute Sandhoff disease is not dependent or significantly modified by processes that occur in the developing brain. Future research will focus on the effects of increasing transgene dosage; in addition, the inducible models will be useful for identifying early cellular events in the evolution of experimental GM2 gangliosidosis in vivo.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. TayW (1881) Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Opthalmol Soc 1 : 55–57.
2. SachsB (1887) On arrested cerebral development with special reference to cortical pathology. J Nerv Ment Dis 14 : 541–554.
3. SandhoffK, AndreaeU, JatzkewitzH (1968) Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Life Sci 7 : 283–288.
4. SvennerholmL (1962) The chemical structure of normal human brain and Tay-Sachs gangliosides. Biochem Biophys Res Commun 9 : 436–441.
5. MakitaA, YamakawaT (1963) The glycolipids of the brain of Tay-Sachs' disease. The chemical structures of globoside and main ganglioside. Jpn J Exp Med 33 : 361–368.
6. LedeenR, SalsmanK (1965) Structure of the Tay-Sachs' ganglioside. Biochemistry 4 : 2225–2233.
7. BleyAE, GiannikopoulosOA, HaydenD, KubilusK, TifftCJ, et al. (2011) Natural history of infantile G(M2) gangliosidosis. Pediatrics 128: e1233–1241.
8. SmithNJ, WinstoneAM, StellitanoL, CoxTM, VerityCM (2012) GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol 54 : 176–182.
9. Schneck L (1964) The clinical aspects of Tay-Sachs disease. In: BWVolk, editor. Tay-Sachs' Disease. New York: Grune and Stratton. pp. 16–35.
10. SangoK, YamanakaS, HoffmannA, OkudaY, GrinbergA, et al. (1995) Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet 11 : 170–176.
11. PhaneufD, WakamatsuN, HuangJQ, BorowskiA, PetersonAC, et al. (1996) Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet 5 : 1–14.
12. GossenM, BujardH (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89 : 5547–5551.
13. YamamotoA, LucasJJ, HenR (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101 : 57–66.
14. SantacruzK, LewisJ, SpiresT, PaulsonJ, KotilinekL, et al. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309 : 476–481.
15. LopezME, KleinAD, DimbilUJ, ScottMP (2011) Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci 31 : 4367–4378.
16. NorflusF, YamanakaS, ProiaRL (1996) Promoters for the human beta-hexosaminidase genes, HEXA and HEXB. DNA Cell Biol 15 : 89–97.
17. RalphGS, BienemannA, HardingTC, HoptonM, HenleyJ, et al. (2000) Targeting of tetracycline-regulatable transgene expression specifically to neuronal and glial cell populations using adenoviral vectors. Neuroreport 11 : 2051–5.
18. SchochS, CibelliG, ThielG (1996) Neuron-specific gene expression of synapsin I. Major role of a negative regulatory mechanism. J Biol Chem 271 : 3317–3323.
19. ChungJH, BellAC, FelsenfeldG (1997) Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci U S A 94 : 575–580.
20. Hogan B, Beddington R, Costantini F, Lacy E (1994) Manipulating the mouse embryo, a laboratory manual (second edition). New York: Cold Spring Harbor Laboratory Press.
21. Cachón-GonzálezMB, WangSZ, LynchA, ZieglerR, ChengSH, CoxTM (2006) Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci U S A 103 : 10373–10378.
22. LacorazzaHD, JendoubiM (1995) In situ assessment of beta-hexosaminidase activity. Biotechniques 19 : 434–440.
23. Cachón-GonzálezMB, WangSZ, McNairR, BradleyJ, LunnD, et al. (2012) Gene Transfer Corrects Acute GM2 Gangliosidosis-Potential Therapeutic Contribution of Perivascular Enzyme Flow. Mol Ther 27 March [E-pub ahead of print].
24. HickmanS, ShapiroLJ, NeufeldEF (1974) A recognition marker required for uptake of a lysosomal enzyme by cultured fibroblasts. Biochem Biophys Res Commun 57 : 55–61.
25. ChtartoA, BenderHU, HanemannCO, KempT, LehtonenE, et al. (2003) Tetracycline-inducible transgene expression mediated by a single AAV vector. Gene Ther 10 : 84–94.
26. MizuguchiH, HayakawaT (2002) The tet-off system is more effective than the tet-on system for regulating transgene expression in a single adenovirus vector. J Gene Med 4 : 240–247.
27. HofmannA, NolanGP, BlauHM (1996) Rapid retroviral delivery of tetracycline-inducible genes in a single autoregulatory cassette. Proc Natl Acad Sci U S A 93 : 5185–5190.
28. MiyazakiS, MiyazakiT, TashiroF, YamatoE, MiyazakiJ (2005) Development of a single-cassette system for spatiotemporal gene regulation in mice. Biochem Biophys Res Commun 338 : 1083–1088.
29. MasuiS, ShimosatoD, ToyookaY, YagiR, TakahashiK, et al. (2005) An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res 33: e43.
30. ShapovalovAI, GurevitchNR (1970) Monosynaptic and disynaptic reticulospinal actions on lumbar motoneurons of the rat. Brain Res 21 : 249–263.
31. PetersonBW, PittsNG, FukushimaK (1979) Reticulospinal connections with limb and axial motoneurons. Exp Brain Res 36 : 1–20.
32. RiddleCN, EdgleySA, BakerSN (2009) Direct and indirect connections with upper limb motoneurons from the primate reticulospinal tract. J Neurosci 29 : 4993–4999.
33. CroneC, PetersenNT, NielsenJE, HansenNL, NielsenJB (2004) Reciprocal inhibition and corticospinal transmission in the arm and leg in patients with autosomal dominant pure spastic paraparesis (ADPSP). Brain 127 : 2693–2702.
34. NielsenJB, CroneC, HultbornH (2007) The spinal pathophysiology of spasticity–from a basic science point of view. Acta Physiol (Oxf) 189 : 171–180.
35. SargeantTJ, WangS, BradleyJ, SmithNJ, RahaAA, et al. (2011) Adeno-associated virus-mediated expression of β-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum Mol Genet 20 : 4371–4380.
36. MolonA, Di GiovanniS, HathoutY, NataleJ, HoffmanEP (2006) Functional recovery of glycine receptors in spastic murine model of startle disease. Neurobiol Dis 21 : 291–304.
37. HalleyDJ, de Wit-VerbeekHA, ReuserAJ, GaljaardH (1978) The distribution of hydrolytic enzyme activities in human fibroblast cultures and their intercellular transfer. Biochem Biophys Res Commun 82 : 1176–1182.
38. AndersK, BuschowC, CharoJ, BlankensteinT (2011) Depot formation of doxycycline impairs Tet-regulated gene expression in vivo. Transgenic Res In press.
39. KrestelHE, ShimshekDR, JensenV, NevianT, KimJ, et al. (2004) A Genetic Switch for Epilepsy in Adult Mice. J Neurosci 24 : 10568–10578.
40. BejarR, YasudaR, KrugersH, HoodK, MayfordM (2002) Transgenic calmodulin-dependent protein kinase II activation: dose-dependent effects on synaptic plasticity, learning, and memory. J Neurosci 22 : 5719–5726.
41. ZhuP, AllerMI, BaronU, CambridgeS, BausenM, et al. (2007) Silencing and un-silencing of tetracycline-controlled genes in neurons. PLoS ONE 2: e533 doi:10.1371/journal.pone.0000533.
42. PankiewiczR, KarlenY, ImhofMO, MermodN (2005) Reversal of the silencing of tetracycline-controlled genes requires the coordinate action of distinctly acting transcription factors. J Gene Med 7 : 117–132.
43. NgamukoteS, YanagisawaM, ArigaT, AndoS, YuRK (2007) Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem 103 : 2327–2341.
44. YuRK, MacalaLJ, TakiT, WeinfieldHM, YuFS (1988) Developmental changes in ganglioside composition and synthesis in embryonic rat brain. J Neurochem 50 : 1825–1829.
45. ZervasM, WalkleySU (1999) Ferret pyramidal cell dendritogenesis: changes in morphology and ganglioside expression during cortical development. J Comp Neurol 413 : 429–448.
46. GoodmanLA, WalkleySU (1996) Elevated GM2 ganglioside is associated with dendritic proliferation in normal developing neocortex. Brain Res Dev Brain Res 93 : 162–171.
47. WalkleySU, SiegelDA, DobrenisK (1995) GM2 ganglioside and pyramidal neuron dendritogenesis. Neurochem Res 20 : 1287–1299.
48. YuT, ShakkottaiVG, ChungC, LiebermanAP (2011) Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum Mol Genet 20 : 4440–4451.
49. ManfredssonFP, BurgerC, RisingAC, Zuobi-HasonaK, SullivanLF, et al. (2009) Tight Long-term dynamic doxycycline responsive nigrostriatal GDNF using a single rAAV vector. Mol Ther 17 : 1857–1867.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese