
Identification of Novel Genetic Loci Associated with Thyroid Peroxidase Antibodies and Clinical Thyroid Disease
Autoimmune thyroid diseases (AITD) are common, affecting 2-5% of the general population. Individuals with positive thyroid peroxidase antibodies (TPOAbs) have an increased risk of autoimmune hypothyroidism (Hashimoto's thyroiditis), as well as autoimmune hyperthyroidism (Graves' disease). As the possible causative genes of TPOAbs and AITD remain largely unknown, we performed GWAS meta-analyses in 18,297 individuals for TPOAb-positivity (1769 TPOAb-positives and 16,528 TPOAb-negatives) and in 12,353 individuals for TPOAb serum levels, with replication in 8,990 individuals. Significant associations (P<5×10−8) were detected at TPO-rs11675434, ATXN2-rs653178, and BACH2-rs10944479 for TPOAb-positivity, and at TPO-rs11675434, MAGI3-rs1230666, and KALRN-rs2010099 for TPOAb levels. Individual and combined effects (genetic risk scores) of these variants on (subclinical) hypo - and hyperthyroidism, goiter and thyroid cancer were studied. Individuals with a high genetic risk score had, besides an increased risk of TPOAb-positivity (OR: 2.18, 95% CI 1.68–2.81, P = 8.1×10−8), a higher risk of increased thyroid-stimulating hormone levels (OR: 1.51, 95% CI 1.26–1.82, P = 2.9×10−6), as well as a decreased risk of goiter (OR: 0.77, 95% CI 0.66–0.89, P = 6.5×10−4). The MAGI3 and BACH2 variants were associated with an increased risk of hyperthyroidism, which was replicated in an independent cohort of patients with Graves' disease (OR: 1.37, 95% CI 1.22–1.54, P = 1.2×10−7 and OR: 1.25, 95% CI 1.12–1.39, P = 6.2×10−5). The MAGI3 variant was also associated with an increased risk of hypothyroidism (OR: 1.57, 95% CI 1.18–2.10, P = 1.9×10−3). This first GWAS meta-analysis for TPOAbs identified five newly associated loci, three of which were also associated with clinical thyroid disease. With these markers we identified a large subgroup in the general population with a substantially increased risk of TPOAbs. The results provide insight into why individuals with thyroid autoimmunity do or do not eventually develop thyroid disease, and these markers may therefore predict which TPOAb-positives are particularly at risk of developing clinical thyroid dysfunction.
Published in the journal:
. PLoS Genet 10(2): e32767. doi:10.1371/journal.pgen.1004123
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004123
Summary
Autoimmune thyroid diseases (AITD) are common, affecting 2-5% of the general population. Individuals with positive thyroid peroxidase antibodies (TPOAbs) have an increased risk of autoimmune hypothyroidism (Hashimoto's thyroiditis), as well as autoimmune hyperthyroidism (Graves' disease). As the possible causative genes of TPOAbs and AITD remain largely unknown, we performed GWAS meta-analyses in 18,297 individuals for TPOAb-positivity (1769 TPOAb-positives and 16,528 TPOAb-negatives) and in 12,353 individuals for TPOAb serum levels, with replication in 8,990 individuals. Significant associations (P<5×10−8) were detected at TPO-rs11675434, ATXN2-rs653178, and BACH2-rs10944479 for TPOAb-positivity, and at TPO-rs11675434, MAGI3-rs1230666, and KALRN-rs2010099 for TPOAb levels. Individual and combined effects (genetic risk scores) of these variants on (subclinical) hypo - and hyperthyroidism, goiter and thyroid cancer were studied. Individuals with a high genetic risk score had, besides an increased risk of TPOAb-positivity (OR: 2.18, 95% CI 1.68–2.81, P = 8.1×10−8), a higher risk of increased thyroid-stimulating hormone levels (OR: 1.51, 95% CI 1.26–1.82, P = 2.9×10−6), as well as a decreased risk of goiter (OR: 0.77, 95% CI 0.66–0.89, P = 6.5×10−4). The MAGI3 and BACH2 variants were associated with an increased risk of hyperthyroidism, which was replicated in an independent cohort of patients with Graves' disease (OR: 1.37, 95% CI 1.22–1.54, P = 1.2×10−7 and OR: 1.25, 95% CI 1.12–1.39, P = 6.2×10−5). The MAGI3 variant was also associated with an increased risk of hypothyroidism (OR: 1.57, 95% CI 1.18–2.10, P = 1.9×10−3). This first GWAS meta-analysis for TPOAbs identified five newly associated loci, three of which were also associated with clinical thyroid disease. With these markers we identified a large subgroup in the general population with a substantially increased risk of TPOAbs. The results provide insight into why individuals with thyroid autoimmunity do or do not eventually develop thyroid disease, and these markers may therefore predict which TPOAb-positives are particularly at risk of developing clinical thyroid dysfunction.
Introduction
Autoimmune thyroid disease (AITD), including Hashimoto's thyroiditis and Graves' disease, is one of the most common autoimmune diseases, affecting 2–5% of the general population [1], [2], [3]. Thyroid dysfunction has been associated with osteoporosis, depression, atrial fibrillation, heart failure, metabolic syndrome, and mortality [4], [5], [6], [7], [8], [9], [10], [11]. High serum antibodies against the enzyme thyroid peroxidase (TPO), which is located in the thyroid and plays a key role in thyroid hormone synthesis, are present in 90% of patients with Hashimoto's thyroiditis [12], [13], the most frequent cause of hypothyroidism and goiter. Although TPO antibodies (TPOAbs) are a useful clinical marker for the detection of early AITD, it remains controversial if these antibodies play a causative role in the pathogenesis of Hashimoto's thyroiditis [14], [15], [16].
Interestingly, TPOAb-positive persons also have an increased risk of developing autoimmune hyperthyroidism (Graves' disease) [17], [18], which is caused by stimulating antibodies against the thyroid stimulating hormone (TSH) receptor [19]. Numerous studies have shown that Graves' hyperthyroidism and Hashimoto's thyroiditis show co-inheritance [17], [20], [21]. Finally, thyroid autoimmunity is the most common autoimmune disorder in women of childbearing age, and TPOAb-positive women have an increased risk of developing pregnancy complications such as miscarriage and pre-term delivery [17], [18], [22], [23], [24], [25], [26].
The prevalence of TPOAb-positivity in the general population ranges from 5–24%, but it is currently unknown why these people develop TPOAbs, nor is it known why not all individuals with thyroid autoimmunity develop clinical thyroid disease [27], [28]. It is estimated that around 70% of the susceptibility to develop thyroid autoantibodies is due to genetic factors [29]. In this context it is remarkable to note that little is known about the genetic factors that determine TPOAb-positivity and the risk of AITD.
We therefore performed a genome wide association study (GWAS) meta-analysis for TPOAbs in the general population in 18,297 individuals from 11 populations. Newly identified genetic variants were studied in relation to subclinical and overt hypo - and hyperthyroidism, goiter, thyroid autoimmunity during pregnancy and thyroid cancer risk.
Results
Characteristics of the studied populations are shown in Table 1 and the Supplementary Material S1. Heritability estimates in the family-based cohorts SardiNIA, TwinsUK and Val Borbera were, respectively, 0.65, 0.66, and 0.54 for TPOAb-positivity, and 0.43, 0.66, and 0.30 for TPOAb levels.

Loci associated with TPOAb-positivity and TPOAb levels
See Table 1 and Supplementary Figure S1 for TPOAb measurements and Supplementary Table S1 for genotyping procedures. In most autoimmune diseases, both the presence and the level of autoantibodies are relevant for the disease onset [18], [30], [31]. Furthermore, different pathophysiological processes may be involved in the initiation and severity of the autoimmune response. We therefore performed a GWAS on TPOAb-positivity (including 1769 TPOAb-positives and 16,528 TPOAb–negatives), as well as a GWAS on continuous TPOAb levels (including 12,353 individuals) in stage 1. See Supplementary Figures S2 and S3 for QQ (quantile-quantile) and Manhattan plots.
In stage 2, we followed-up 20 stage 1 SNPs (P<5×10−6; 13 TPOAb-positivity and 10 TPOAb level SNPs, with 3 SNPs overlapping) in 5 populations, including up to 8,990 individuals for TPOAb-positivity (922 TPOAb-positives and 8068 TPOAb–negatives) and 8,159 individuals for TPOAb level analyses (see Supplementary Material S1). Results of the combined stage 1 and 2 meta-analyses, including heterogeneity analyses, are shown in Supplementary Tables S2 and S3. Regional association plots are shown in Supplementary Figures S4 and S5. In the combined stage 1 and 2 meta-analyses GWAS significant associations (P<5×10−8) were observed near TPO (Chr 2p25; rs11675434), at ATXN2 (Chr 12q24.1; rs653178), and BACH2 (Chr 6q15; rs10944479) for TPOAb-positivity, and near TPO (rs11675434), at MAGI3 (Chr 6q15; rs1230666), and KALRN (Chr 3q21; rs2010099) for TPOAb levels (Table 2 and Figure 1). The TPOAb level meta-analysis P-values for the 3 GWAS significant TPOAb-positivity loci were: TPO-rs11675434: P = 7.4×10−13, ATXN2-rs653178: P = 1.3×10−7, and BACH2-rs10944479: P = 2.0×10−4.

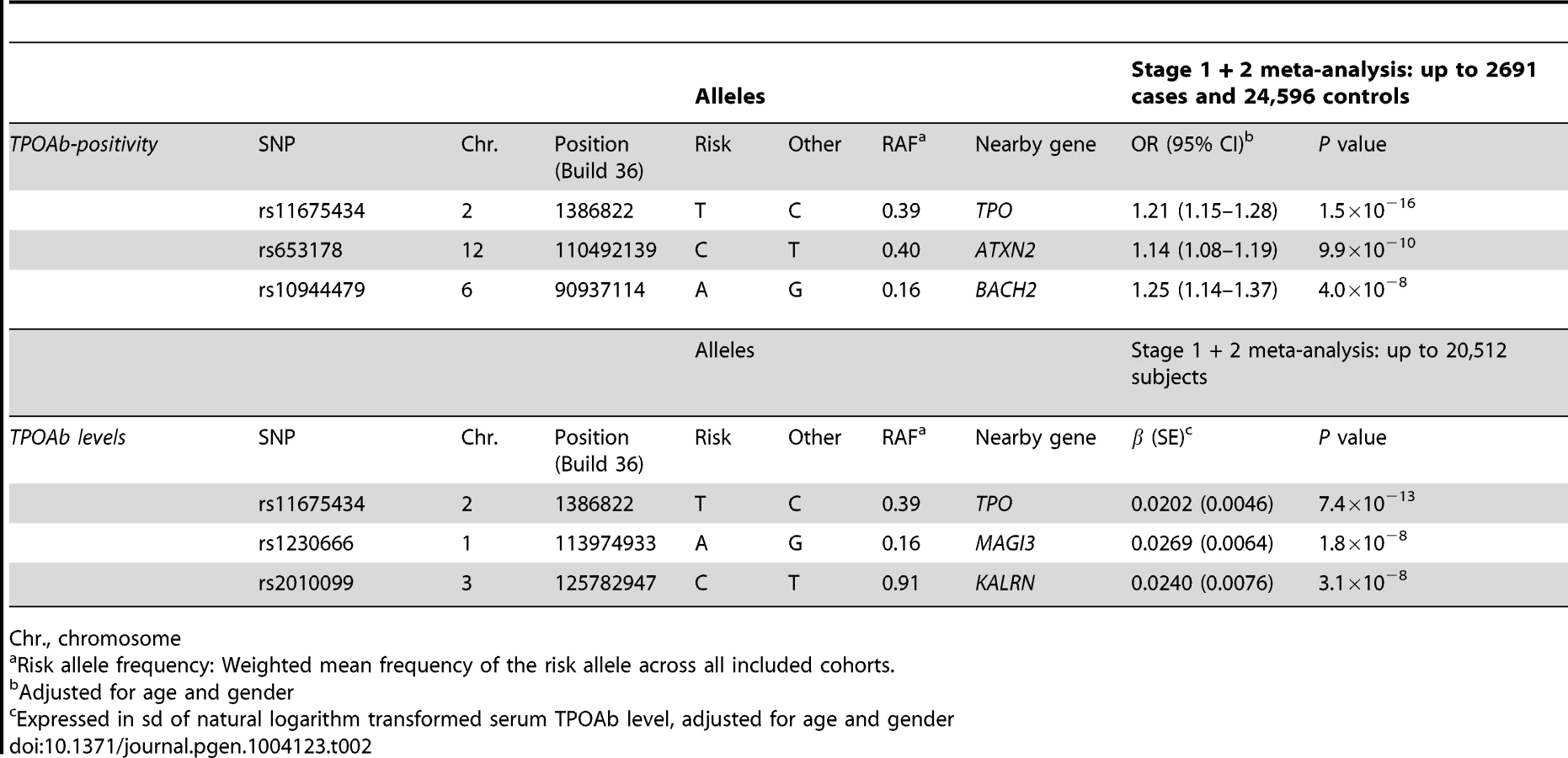
As the 3 GWAS significant loci for TPOAb levels also showed associations with TPOAb-positivity (TPO-rs11675434: OR, 1.21 [95% CI, 1.15–1.28)], P = 1.5×10−16; MAGI3-rs1230666: OR, 1.23 [95% CI, 1.14–1.33], P = 1.5×10−6; KALRN-rs2010099: OR, 1.24 [95% CI, 1.12–1.37], P = 7.4×10−5), we subsequently studied the (combined) effects of these 5 SNPs on clinical thyroid disease. Genetic risk scores were calculated as described in the Supplementary Material. The variance explained by these 5 SNPs was 3.1% for TPOAb-positivity and 3.2% for TPOAb levels. Subjects with a high genetic risk score had a 2.2 times increased risk of TPOAb-positivity compared to subjects with a low genetic risk score (P = 8.1×10−8) (Table 3).

Table S4 shows the stage 1 TPOAb-positivity and TPOAb level meta-analyses results for GWAS significant SNPs reported in previous GWAS on thyroid related phenotypes.
Associations with hypo - and hyperthyroidism
The associations between the 5 GWAS significant SNPs and the risk of abnormal thyroid function tests are shown in Table 4. MAGI3 - rs1230666 was associated with an increased risk of overt hypothyroidism and increased TSH levels below the Bonferroni threshold (i.e., P = 0.05/5 = 0.01). Borderline significant signals were observed at BACH2 - rs10944479 with a higher risk of increased TSH levels as well as overt hyperthyroidism (P = 0.011 and P = 0.012), and at the KALRN-rs2010099 SNP with a lower risk of decreased TSH levels (P = 0.010).

Furthermore, a higher genetic risk score was associated with a higher risk of increased TSH levels (Supplementary Table S5). No effects of the genetic risk score on the risk of overt hypothyroidism, hyperthyroidism or decreased TSH levels were observed.
Associations with goiter
Individuals with a high genetic risk score had a 30.4% risk of sonographically-proven goiter, compared to 35.2% in subjects with a low score (P = 6.5×10−4) (Table 5). None of the individual SNPs was significantly associated with goiter risk.

Thyroid autoimmunity during pregnancy
As autoimmunity significantly changes during pregnancy [25], we additionally studied these effects in an independent pregnant population. Pregnant women with a high genetic risk score had a 2.4 times increased risk of TPOAb-positivity compared to women with a low score (10.3% vs 4.8%, P = 0.03). These women did not have a higher risk of increased TSH levels. However, a borderline significant signal with a lower risk of increased TSH levels was observed at ATXN2 - rs653178 (OR, 0.54 [95% CI, 0.34–0.87], P = 0.012).
Associations with thyroid disease in independent populations
a) Graves' disease
As MAGI3 - rs1230666 and BACH2 - rs10944479 showed promising associations (i.e., P≤0.05) with hyperthyroidism in our meta-analyses, we tested these SNPs in an independent population of 2478 patients with Graves' disease and 2682 controls (see Supplementary Material for further details). Both were associated with an increased risk of Graves' disease (MAGI3 - rs1230666: OR, 1.37 [95% CI, 1.22–1.54]; P = 1.2×10−7; BACH2 - rs10944479: OR, 1.25 [1.12–1.39]; P = 6.2×10−5).
b) Thyroid cancer
Supplementary Table S6 shows the associations of the 5 GWAS significant SNPs with thyroid cancer. No statistically significant associations were detected, but a borderline significant signal with an increased risk of thyroid cancer was observed at ATXN2 - rs653178 (OR, 1.32 [95% CI, 1.02–1.70], P = 0.03).
Pathway analyses
Ingenuity Pathway Analyses (IPA; Ingenuity Systems, Ca, USA) and GRAIL analyses [32] were performed to identify potential pathways involved in AITD, the results of which are shown in Supplementary Tables S7 and S8, and Figure S6. The identified top pathways involved cell death, survival, movement, and OX40 signalling.
Discussion
This is the first GWAS meta-analysis investigating the genetics of TPOAbs in the normal population in up to 18,297 individuals from 11 populations with replication in up to 8,990 individuals from 5 populations. We identified 5 GWAS significant loci associated with TPOAb-positivity and/or levels.
The most significant hit for both TPOAb-positivity and TPOAb levels was located near the TPO gene itself. TPO is a membrane-bound protein located on the apical membranes of the thyroid follicular cell, catalyzing key reactions in thyroid hormone synthesis [33]. Mutations in TPO have been found in patients with congenital hypothyroidism [34], [35]. Although TPOAbs are valid clinical biomarkers of AITD, they are generally considered to be secondary to the thyroid damage inflicted by T-cells.
The FOXE1 gene has been previously associated with hypothyroidism [36], [37] and is known to regulate transcription of TPO [38]. In this context it is interesting to note that we did not find any associations of the variant near TPO with hypothyroidism. Most genes that have been associated with AITD (predominantly Graves' disease) by candidate gene and GWAS studies so far are located in the HLA class I and II regions, or in genes involved in T-cell (i.e., CTLA-4, PTPN22) or other autoimmune responses [28], [39]. Until now, the TPO gene itself had not been associated with AITD, except in one recent candidate gene analysis in a small cohort (n = 188) without replication [40]. A variant near TPO (rs11694732), which is in LD with rs11675434 (r2 = 0.97 in HapMap2), has previously been associated with TSH levels by Gudmundsson et al [41]. However, various other GWAS on serum TSH and FT4 levels have not found any significant associations in or near this locus, including a recent similar sized GWAS by Porcu et al [42].
Three of the other four loci identified here are located in or are in linkage disequilibrium (LD) with genes previously associated with other autoimmune diseases. Rs1230666 is located in intron 9 of MAGI3, encoding a protein that modulates activity of AKT/PKB. AKT/PKB is expressed in the thyroid and regulates apoptosis [43], which seems to play an important role in the development of AITD [44], [45]. In addition, rs1230666 is in LD with rs2476601 (r2 = 0.70 in HapMap2), a variant causing a R620W substitution in PTPN22. PTPN22 is a lymphoid-specific intracellular phosphatase involved in the T-cell receptor signaling pathway. Variations in PTPN22, and specifically R620W, are associated with various autoimmune disorders including type 1 diabetes, rheumatoid arthritis, systemic lupus erythematosus and Graves' disease [46], [47], [48], [49]. The associations of the MAGI3 locus with TPOAb-positivity and Graves' disease may therefore also be explained by linkage with disease-associated variants in PTPN22 [50]. Of note, the association signal at rs2476601 is one order weaker than that of the top variant rs1230666.
The BACH2 locus has been implicated in the susceptibility to several autoimmune diseases, including celiac disease, type 1 diabetes, vitiligo, Crohn's disease, and multiple sclerosis [46], [51], [52], [53], [54]. A recent candidate gene analysis associated the BACH2 locus with an increased risk of AITD, including Hashimoto's thyroiditis and Graves' disease [55]. However, the associations were not significant when Hashimoto's thyroiditis and Graves' disease were studied separately. BACH2 is specifically expressed in early stages of B-cell differentiation and represses different immunoglobulin genes [56]. Interestingly, BACH2 can bind to the co-repressor SMRT (silencing mediator of retinoid and thyroid receptor), which may suggest a more direct effect on thyroid hormone secretion and action as well.
Polymorphisms in ATXN2 have been associated with multiple neurodegenerative diseases, including spinocerebellar ataxia and Parkinson's disease [57], [58], [59]. Different epidemiological studies have associated thyroid dysfunction with cerebellar ataxia [60], [61]. Furthermore, the identified SNP in ATXN2 has been previously associated with renal function, serum urate levels and blood pressure [62], [63], [64]. However, this SNP is in high LD with rs3184504 (r2 = 0.873), a variant causing a Trp262Arg substitution of SH2B adaptor protein 3 (SH2B3). SH2B3 encodes the adaptor protein LNK, a key negative regulator of cytokine signaling playing a critical role in hematopoiesis. This variant is associated with susceptibility to several autoimmune diseases, including celiac disease, type 1 diabetes, vitiligo, and rheumatoid arthritis [46], [51], [53], [65], suggesting more relevance for TPOAb levels than ATXN2. This is supported by a recent study which showed that variants in LD with SH2B3, BACH2, and PTPN22 are associated with TPOAb levels in patients with type 1 diabetes [66].
Whereas the above four loci are located in genes involved in the immune response or the autoantigen, the KALRN (Kalirin) gene encodes a multi-domain guanine nucleotide exchange factor for GTP-binding proteins of the Rho family. The relation of KALRN with levels of TPOAbs is unclear. This gene has recently been found to be associated with megakaryopoiesis and platelet formation [67], which may suggest a function in the immune system [68]. We furthermore performed pathway analyses on the stage 1 TPOAb-positivity and TPOAb level lead SNPs, and identified the cell death, survival and movement pathway as an important pathway for TPOAbs. This finding is supported by previous studies, which show an important role for apoptosis in the development of AITD [44], [45]. Another top pathway involved was the OX40 signalling pathway, and it is of interest to note that OX40 is a T-cell activator promoting the survival of CD4+ T-cells at sites of inflammation [69].
Our results have potential clinical relevance for several reasons. Genetic risk scores based on these novel common (risk allele frequencies: 9–40%) TPOAb-associated SNPs enabled us to identify a large subgroup in the general population with a two-fold increased risk of TPOAb-positivity (10.4% vs 5.4%). These individuals also have a higher risk of increased TSH levels and a lower risk of goiter, suggesting an advanced stage of destruction of the thyroid due to autoimmune processes. Furthermore, pregnant women with high genetic risk scores had a 2.4 times increased risk of TPOAb-positivity during pregnancy. In this context it is interesting to note that TPOAb-positive pregnant women have an increased risk of miscarriages and preterm births independent of thyroid function [70].
Associations with thyroid disease were also found on an individual SNP level. The MAGI3 SNP was associated with a substantially increased risk of hypothyroidism, and the BACH2 SNP showed a borderline significant association (P = 0.011) with a higher risk of increased TSH levels, which includes subjects with subclinical and overt hypothyroidism. Furthermore, both loci were significantly associated with an increased risk of Graves' hyperthyroidism in an independent population. To predict which patients with first or second degree relatives with documented Hashimoto's or Graves' disease will develop clinical thyroid disease, a clinical algorithm has been developed (i.e., the THEA score) [18]. Future studies should analyze if these genetic markers increase the sensitivity of the THEA score. Graves' hyperthyroidism and Hashimoto's thyroiditis co-segregate in families and subjects with TPOAbs have an increased risk of both diseases [17], [18], [20], [21], [22], [26]. The current study provides insight into this phenomenon by showing that specific loci associated with TPOAbs and (subclinical) hypothyroidism, i.e. MAGI3 and BACH2, are also associated with Graves' hyperthyroidism in an independent case-control study.
The prevalence of TPOAb-positivity in the general population is high (5–24%), but it is currently unknown why part of the individuals with thyroid autoimmunity develop clinical thyroid disease whereas others do not [27], [28]. In this context it is interesting to note that the TPOAb-associated SNPs located in TPO and ATXN2 were not associated with clinical thyroid disease. This suggests that the TPOAbs in these individuals may be of less clinical relevance, providing insight into why TPOAb-positive individuals do or do not eventually develop clinical thyroid disease.
Our study has some limitations. The validity of the results is restricted to individuals from populations of European ancestry. Future GWASs in populations from non-European descent will be required to determine to which extent our results can be generalized to other ethnic groups. Secondly, we did not perform conditional analyses to further identify secondary association signals within the identified loci, nor did we perform functional studies for the identified variants. Further research is therefore needed to unravel the exact biological mechanism behind the observed associations. The fact that various TPOAb assays were used across the participating cohorts could lead to bias. We therefore used TPOAb-positivity cut-off values as provided by the respective assay manufacturer, instead of using one fixed cut-off value. This is also of clinical importance as in clinical practice most institutions rely on the TPOAb-positivity cut-off as provided by the assay manufacturer. Furthermore, we did not detect heterogeneity in our results, supporting the fact that results obtained with different assays can be combined across cohorts using the z-score based meta-analysis. Finally, as AITD coincides with other autoimmune diseases, our results could be driven by indirect associations with other autoimmune diseases. However, AITD is the most common autoimmune disease in the general population. We furthermore show that carriage of multiple risk alleles is associated with an increased risk of thyroid dysfunction, which underlines the clinical importance of our findings.
In conclusion, this first GWAS for TPOAbs identified five newly associated loci, three of which were also associated with clinical thyroid disease. Furthermore, we show that carriage of multiple risk variants is not only associated with a substantial increased risk of TPOAb-positivity, but also with a higher risk of increased TSH levels (including subclinical and overt hypothyroidism) and a lower risk of goiter. These genetic markers not only help to identify large groups in the general population with an increased risk of TPOAb-positivity, but may also predict which TPOAb-positive persons are particularly at risk of developing clinical thyroid disease.
Materials and Methods
Study cohorts
For the TPOAb GWAS stage 1 and 2 analyses, and the hypothyroidism, hyperthyroidism and goiter analyses, individuals were recruited from 16 independent community-based and family studies. For the Graves' disease analyses, cases were recruited from the United Kingdom Graves' disease cohort and controls from the British 1958 Birth Cohort. Thyroid cancer cases and controls were recruited from the Nijmegen and Ohio thyroid cancer cohorts. A detailed description of the original cohorts contributing samples is provided in Table 1 and in the Supplementary Material. All participants provided written informed consent and protocols were approved by the institutional review boards or research ethics committees at the respective institutions, and conducted according to the Declaration of Helsinki.
Phenotype definitions
Serum TPOAb levels were determined with a range of assays. TPOAb-positives were defined as subjects with TPOAb levels above the assay-specific TPOAb-positivity cut-off, as defined by the manufacturer (Table 1). Serum TSH and free thyroxine (FT4) levels were determined using a range of assays (Table 1). Assay-specific TSH and FT4 reference ranges were used, as provided by the manufacturer (Table 1). Overt hypothyroidism was defined as a high TSH (i.e., a TSH level above the TSH reference range) and a low FT4. Increased TSH was defined as a high TSH, including persons with overt hypothyroidism or subclinical hypothyroidism (i.e., high TSH with a normal FT4). Overt hyperthyroidism was defined as a low TSH and a high FT4. Decreased TSH was defined as a low TSH, including persons with subclinical or overt hyperthyroidism.
The diagnosis of goiter is described in the Supplementary Material, and the diagnosis of Graves' disease and thyroid cancer in the respective cohorts have been described previously [41].
Genotyping
Samples were genotyped with a range of GWAS genotyping arrays (Supplementary Table S1). Sample and SNP quality control procedures were undertaken within each study. For each GWAS, over 2.5 million SNPs were imputed using CEU samples from Phase 2 of the International HapMap project (www.hapmap.org). Genotyping procedures in the stage 2, Graves' disease and thyroid cancer populations are described in the Supplementary Material.
Association analyses
The heritabilities of TPOAb-positivity and serum TPOAb levels were estimated, as described in the Supplementary Material.
In stage 1, we performed a GWAS on TPOAb-positivity as well as a GWAS on continuous TPOAb levels. Persons taking thyroid medication were excluded. Each SNP was tested for association with TPOAb-positivity using logistic regression analyses, adjusting for age and sex. For cohorts with family structure, we approximated the probability of being affected with a linear mixed model adjusting for age and sex. The produced model was used to predict the expected proportion of “risk” (effective) alleles in cases and controls, hence giving the means to estimate odds ratios. Only unrelated individuals were considered for the SardiNIA cohort. For the GWAS of continuous TPOAb levels, samples with a TPOAb level lower than the minimum TPOAb assay detection limit (Table 1) were excluded. TPOAb levels were natural log-transformed, and sex-specific, age adjusted standardized residuals were calculated. Each SNP was tested for association with these TPOAb level residuals using linear regression analyses (additive model), correcting for relatedness in studies with family structure. See Supplementary Table S1 for the software used for these analyses.
Before meta-analysis, SNPs with a minor allele frequency (MAF) <1% or a low imputation quality were excluded (Supplementary Material), after which the results of each GWAS were combined in a population size weighted z-score based meta-analysis using METAL [71]. Genomic control was applied to individual studies if λ>1.0.
In stage 2, we followed-up stage 1 GWAS significant SNPs, as well as promising SNPs not reaching GWAS significance, in an attempt to reach GWAS significant associations by increasing sample size (Supplementary Material). Results from stage 1 and 2 were combined in a population size weighted z-score based meta-analysis using METAL [71]. A z-score based meta-analysis was used to reduce bias that might be induced by different assays. As this method does not provide betas, and we wanted to provide a rough estimate of the actual effect sizes for convenience, we calculated betas using the fixed effects (inverse variance based) meta-analysis method. Heterogeneity was tested, applying bonferroni based P-value thresholds of P = 0.004 for the TPOAb-positivity analyses and P = 0.005 for the TPOAb level analyses.
All studies assessed and, if present, corrected for population stratification using principal-component analysis (PCA) and/or multidimensional-scaling (MDS), with the exception of SardiNIA and ValBorbera where the high isolation substantiates a lack of stratification (Table S1) [72], [73]. Lambda values were all ∼1, indicating that population stratification was overall properly accounted for (Table S1). To fully remove residual effects, we applied genomic correction to studies were lambda was >1. The final meta-analyses reported a lambda of 1.01 for both the TPOAb-positivity and the TPOAb level GWAS, thus no genomic correction was applied.
The variances explained by the GWAS significant SNPs were calculated. We subsequently studied the individual as well as the combined effects of the GWAS significant SNPs on the risk of clinical thyroid disease, as specified in the Supplementary Material. In short, to study combined effects, a genetic risk score was calculated for every person as the weighted sum of TPOAb risk alleles. The associations between the individual SNPs, genetic risk scores and the risk of abnormal thyroid function tests were studied using logistic regression analyses. Logistic regression analyses were used to study the associations with goiter, Graves' disease and thyroid cancer (Supplementary Material). The results of each study were combined in a population size weighted z-score based meta-analysis using METAL [71].
Various bioinformatic tools were searched for evidence for functional relevance of the GWAS significant SNPs and pathway analyses were performed on the Stage 1 lead SNPs (see Supplementary Material).
Supporting Information
Zdroje
1. GoughSC (2000) The genetics of Graves' disease. Endocrinol Metab Clin North Am 29 : 255–266.
2. SimmondsMJ, GoughSC (2004) Unravelling the genetic complexity of autoimmune thyroid disease: HLA, CTLA-4 and beyond. Clin Exp Immunol 136 : 1–10.
3. TunbridgeWM, EveredDC, HallR, AppletonD, BrewisM, et al. (1977) The spectrum of thyroid disease in a community: the Whickham survey. Clin Endocrinol (Oxf) 7 : 481–493.
4. BiondiB (2012) Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur J Endocrinol 167 : 609–618.
5. ColletTH, GusseklooJ, BauerDC, den ElzenWP, CappolaAR, et al. (2012) Subclinical hyperthyroidism and the risk of coronary heart disease and mortality. Arch Intern Med 172 : 799–809.
6. DavisJD, TremontG (2007) Neuropsychiatric aspects of hypothyroidism and treatment reversibility. Minerva Endocrinol 32 : 49–65.
7. GencerB, ColletTH, VirginiV, BauerDC, GusseklooJ, et al. (2012) Subclinical thyroid dysfunction and the risk of heart failure events: an individual participant data analysis from 6 prospective cohorts. Circulation 126 : 1040–1049.
8. NichollsJJ, BrassillMJ, WilliamsGR, BassettJH (2012) The skeletal consequences of thyrotoxicosis. J Endocrinol 213 : 209–221.
9. RodondiN, den ElzenWP, BauerDC, CappolaAR, RazviS, et al. ((201)) Subclinical hypothyroidism and the risk of coronary heart disease and mortality. Jama 304 : 1365–1374.
10. RuhlaS, WeickertMO, ArafatAM, OsterhoffM, IskenF, et al. (2010) A high normal TSH is associated with the metabolic syndrome. Clin Endocrinol (Oxf) 72 : 696–701.
11. SelmerC, OlesenJB, HansenML, LindhardsenJ, OlsenAM, et al. (2012) The spectrum of thyroid disease and risk of new onset atrial fibrillation: a large population cohort study. Bmj 345: e7895.
12. PearceEN, FarwellAP, BravermanLE (2003) Thyroiditis. N Engl J Med 348 : 2646–2655.
13. SchweizerU, ChiuJ, KohrleJ (2008) Peroxides and peroxide-degrading enzymes in the thyroid. Antioxid Redox Signal 10 : 1577–1592.
14. BrixTH, HegedusL, GardasA, BangaJP, NielsenCH (2011) Monozygotic twin pairs discordant for Hashimoto's thyroiditis share a high proportion of thyroid peroxidase autoantibodies to the immunodominant region A. Further evidence for genetic transmission of epitopic “fingerprints”. Autoimmunity 44 : 188–194.
15. HuberG, StaubJJ, MeierC, MitracheC, GuglielmettiM, et al. (2002) Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab 87 : 3221–3226.
16. NielsenCH, BrixTH, LeslieRG, HegedusL (2009) A role for autoantibodies in enhancement of pro-inflammatory cytokine responses to a self-antigen, thyroid peroxidase. Clin Immunol 133 : 218–227.
17. StriederTG, PrummelMF, TijssenJG, EndertE, WiersingaWM (2003) Risk factors for and prevalence of thyroid disorders in a cross-sectional study among healthy female relatives of patients with autoimmune thyroid disease. Clin Endocrinol (Oxf) 59 : 396–401.
18. StriederTG, TijssenJG, WenzelBE, EndertE, WiersingaWM (2008) Prediction of progression to overt hypothyroidism or hyperthyroidism in female relatives of patients with autoimmune thyroid disease using the Thyroid Events Amsterdam (THEA) score. Arch Intern Med 168 : 1657–1663.
19. WeetmanAP (2000) Graves' disease. N Engl J Med 343 : 1236–1248.
20. BrixTH, HegedusL (2011) Twins as a tool for evaluating the influence of genetic susceptibility in thyroid autoimmunity. Ann Endocrinol (Paris) 72 : 103–107.
21. TomerY, DaviesTF (2003) Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev 24 : 694–717.
22. KordonouriO, DeissD, DanneT, DorowA, BassirC, et al. (2002) Predictivity of thyroid autoantibodies for the development of thyroid disorders in children and adolescents with Type 1 diabetes. Diabet Med 19 : 518–521.
23. MediciM, de RijkeYB, PeetersRP, VisserW, de Muinck Keizer-SchramaSM, et al. (2012) Maternal early pregnancy and newborn thyroid hormone parameters: the Generation R study. J Clin Endocrinol Metab 97 : 646–652.
24. NegroR, MestmanJH (2011) Thyroid disease in pregnancy. Best Pract Res Clin Endocrinol Metab 25 : 927–943.
25. PoppeK, VelkeniersB, GlinoerD (2008) The role of thyroid autoimmunity in fertility and pregnancy. Nat Clin Pract Endocrinol Metab 4 : 394–405.
26. VanderpumpMP, TunbridgeWM, FrenchJM, AppletonD, BatesD, et al. (1995) The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham Survey. Clin Endocrinol (Oxf) 43 : 55–68.
27. HollowellJG, StaehlingNW, FlandersWD, HannonWH, GunterEW, et al. (2002) Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab 87 : 489–499.
28. WeetmanAP (2011) Diseases associated with thyroid autoimmunity: explanations for the expanding spectrum. Clin Endocrinol (Oxf) 74 : 411–418.
29. HansenPS, BrixTH, IachineI, KyvikKO, HegedusL (2006) The relative importance of genetic and environmental effects for the early stages of thyroid autoimmunity: a study of healthy Danish twins. Eur J Endocrinol 154 : 29–38.
30. LinnikMD, HuJZ, HeilbrunnKR, StrandV, HurleyFL, et al. (2005) Relationship between anti-double-stranded DNA antibodies and exacerbation of renal disease in patients with systemic lupus erythematosus. Arthritis Rheum 52 : 1129–1137.
31. NielsenSF, BojesenSE, SchnohrP, NordestgaardBG (2012) Elevated rheumatoid factor and long term risk of rheumatoid arthritis: a prospective cohort study. Bmj 345: e5244.
32. RaychaudhuriS, PlengeRM, RossinEJ, NgAC, PurcellSM, et al. (2009) Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet 5: e1000534.
33. RufJ, CarayonP (2006) Structural and functional aspects of thyroid peroxidase. Arch Biochem Biophys 445 : 269–277.
34. BakkerB, BikkerH, VulsmaT, de RandamieJS, WiedijkBM, et al. (2000) Two decades of screening for congenital hypothyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update). J Clin Endocrinol Metab 85 : 3708–3712.
35. BikkerH, BaasF, De VijlderJJ (1997) Molecular analysis of mutated thyroid peroxidase detected in patients with total iodide organification defects. J Clin Endocrinol Metab 82 : 649–653.
36. DennyJC, CrawfordDC, RitchieMD, BielinskiSJ, BasfordMA, et al. (2011) Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome - and phenome-wide studies. Am J Hum Genet 89 : 529–542.
37. ErikssonN, TungJY, KieferAK, HindsDA, FranckeU, et al. (2012) Novel associations for hypothyroidism include known autoimmune risk loci. PLoS One 7: e34442.
38. OrtizL, Aza-BlancP, ZanniniM, CatoAC, SantistebanP (1999) The interaction between the forkhead thyroid transcription factor TTF-2 and the constitutive factor CTF/NF-1 is required for efficient hormonal regulation of the thyroperoxidase gene transcription. J Biol Chem 274 : 15213–15221.
39. SimmondsMJ (2013) GWAS in autoimmune thyroid disease: redefining our understanding of pathogenesis. Nat Rev Endocrinol 9 : 277–287.
40. FaamB, DaneshpourMS, AziziF, SalehiM, HedayatiM (2012) Association between TPO gene polymorphisms and Anti-TPO level in Tehranian population: TLGS. Gene 498 : 116–119.
41. GudmundssonJ, SulemP, GudbjartssonDF, JonassonJG, MassonG, et al. (2012) Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat Genet 44 : 319–322.
42. PorcuE, MediciM, PistisG, VolpatoCB, WilsonSG, et al. (2013) A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS Genet 9: e1003266.
43. WuY, DowbenkoD, SpencerS, LauraR, LeeJ, et al. (2000) Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem 275 : 21477–21485.
44. BossowskiA, CzarnockaB, BardadinK, Stasiak-BarmutaA, UrbanM, et al. (2008) Identification of apoptotic proteins in thyroid gland from patients with Graves' disease and Hashimoto's thyroiditis. Autoimmunity 41 : 163–173.
45. WangSH, BakerJR (2007) The role of apoptosis in thyroid autoimmunity. Thyroid 17 : 975–979.
46. BarrettJC, ClaytonDG, ConcannonP, AkolkarB, CooperJD, et al. (2009) Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 41 : 703–707.
47. OrruV, TsaiSJ, RuedaB, FiorilloE, StanfordSM, et al. (2009) A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet 18 : 569–579.
48. RaychaudhuriS, RemmersEF, LeeAT, HackettR, GuiducciC, et al. (2008) Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet 40 : 1216–1223.
49. VelagaMR, WilsonV, JenningsCE, OwenCJ, HeringtonS, et al. (2004) The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves' disease. J Clin Endocrinol Metab 89 : 5862–5865.
50. SteerS, AbkevichV, GutinA, CordellHJ, GendallKL, et al. (2007) Genomic DNA pooling for whole-genome association scans in complex disease: empirical demonstration of efficacy in rheumatoid arthritis. Genes Immun 8 : 57–68.
51. DuboisPC, TrynkaG, FrankeL, HuntKA, RomanosJ, et al. (2010) Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 42 : 295–302.
52. FrankeA, McGovernDP, BarrettJC, WangK, Radford-SmithGL, et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 42 : 1118–1125.
53. JinY, BirleaSA, FainPR, FerraraTM, BenS, et al. (2012) Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet 44 : 676–680.
54. SawcerS, HellenthalG, PirinenM, SpencerCC, PatsopoulosNA, et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476 : 214–219.
55. CooperJD, SimmondsMJ, WalkerNM, BurrenO, BrandOJ, et al. (2012) Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet 21 : 5202–5208.
56. MutoA, HoshinoH, MadisenL, YanaiN, ObinataM, et al. (1998) Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3' enhancer. Embo J 17 : 5734–5743.
57. HouldenH, SingletonAB (2012) The genetics and neuropathology of Parkinson's disease. Acta Neuropathol 124 : 325–338.
58. LiuX, LuM, TangL, ZhangN, ChuiD, et al. (2013) ATXN2 CAG repeat expansions increase the risk for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging 34 : 2236.e5–8.
59. MaganaJJ, Velazquez-PerezL, CisnerosB (2013) Spinocerebellar ataxia type 2: clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol Neurobiol 47 : 90–104.
60. BonuccelliU, NutiA, MonzaniF, De NegriF, MuratorioA (1991) Familial occurrence of hypothyroidism and cerebellar ataxia. Funct Neurol 6 : 171–175.
61. TandeterH, LevyA, GutmanG, ShvartzmanP (2001) Subclinical thyroid disease in patients with Parkinson's disease. Arch Gerontol Geriatr 33 : 295–300.
62. KottgenA, AlbrechtE, TeumerA, VitartV, KrumsiekJ, et al. (2013) Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet 45 : 145–154.
63. KottgenA, PattaroC, BogerCA, FuchsbergerC, OldenM, et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat Genet 42 : 376–384.
64. WainLV, VerwoertGC, O'ReillyPF, ShiG, JohnsonT, et al. (2011) Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet 43 : 1005–1011.
65. StahlEA, RaychaudhuriS, RemmersEF, XieG, EyreS, et al. (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42 : 508–514.
66. PlagnolV, HowsonJM, SmythDJ, WalkerN, HaflerJP, et al. (2011) Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS Genet 7: e1002216.
67. GiegerC, RadhakrishnanA, CvejicA, TangW, PorcuE, et al. (2011) New gene functions in megakaryopoiesis and platelet formation. Nature 480 : 201–208.
68. KnausUG (2000) Rho GTPase signaling in inflammation and transformation. Immunol Res 21 : 103–109.
69. GramagliaI, WeinbergAD, LemonM, CroftM (1998) Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol 161 : 6510–6517.
70. ThangaratinamS, TanA, KnoxE, KilbyMD, FranklynJ, et al. (2011) Association between thyroid autoantibodies and miscarriage and preterm birth: meta-analysis of evidence. Bmj 342: d2616.
71. WillerCJ, LiY, AbecasisGR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26 : 2190–2191.
72. MilaniG, MasciulloC, SalaC, BellazziR, BuettiI, et al. (2011) Computer-based genealogy reconstruction in founder populations. J Biomed Inform 44 : 997–1003.
73. PiliaG, ChenWM, ScuteriA, OrruM, AlbaiG, et al. (2006) Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet 2: e132.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 2
Nejčtenější v tomto čísle
- Genome-Wide Association Study of Metabolic Traits Reveals Novel Gene-Metabolite-Disease Links
- A Cohesin-Independent Role for NIPBL at Promoters Provides Insights in CdLS
- Classic Selective Sweeps Revealed by Massive Sequencing in Cattle
- Arf4 Is Required for Mammalian Development but Dispensable for Ciliary Assembly