
Palaeosymbiosis Revealed by Genomic Fossils of in a Strongyloidean Nematode
Bovine lungworms are economically important nematode parasites of cattle. We have sequenced the genome of the bovine lungworm to provide information for drug and vaccine discovery. Within the lungworm genome we found extensive evidence of an ancient association between the lungworm and a bacterium called Wolbachia. The lungworm Wolbachia is now a “fossil” in the genome, but tells of an ancient infection. Association between lungworms, and related nematode worms, and Wolbachia was not known previously. We have used the lungworm Wolbachia sequence to explore the history of nematode-Wolbachia interactions, particularly the jumping of these symbionts between arthropods and nematodes.
Published in the journal:
. PLoS Genet 10(6): e32767. doi:10.1371/journal.pgen.1004397
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004397
Summary
Bovine lungworms are economically important nematode parasites of cattle. We have sequenced the genome of the bovine lungworm to provide information for drug and vaccine discovery. Within the lungworm genome we found extensive evidence of an ancient association between the lungworm and a bacterium called Wolbachia. The lungworm Wolbachia is now a “fossil” in the genome, but tells of an ancient infection. Association between lungworms, and related nematode worms, and Wolbachia was not known previously. We have used the lungworm Wolbachia sequence to explore the history of nematode-Wolbachia interactions, particularly the jumping of these symbionts between arthropods and nematodes.
Introduction
Wolbachia are alphaproteobacterial, intracellular symbionts of many non-vertebrate animal species, related to rickettsia-like intracellular pathogens such as Anaplasma and Ehrlichia [1]. Wolbachia was first detected as a cytoplasmic genetic element causing mating type incompatibilities in Culex pipiens mosquitoes, and subsequently has been found to infect many insect species [2]. In insects, most Wolbachia can be classified as reproductive parasites, as they manipulate their hosts' reproduction to promote their own transmission [3]. This is achieved by induction of mating type incompatibilities, induction of parthenogenesis in females of haplo-diploid species, and killing or feminisation of genetic males. In some insects, Wolbachia infections are apparently “asymptomatic”, in that no reproductive bias has been detected. There is evidence that Wolbachia infection can be beneficial to hosts, particularly in protection from other infectious organisms [4]. Importantly, in most insect systems tested the symbiosis is not essential to the hosts, which can be cured by antibiotic treatment.
Wolbachia strains have been classified into a number of groups using molecular phylogenetic analyses of a small number of marker loci [5], [6]. Insect Wolbachia largely derive from clade A and B. Outside Insecta, arthropod Wolbachia infections have been identified in terrestrial Collembola (Hexapoda), Isopoda (Crustacea), Chelicerata and Myriapoda, and also in marine Amphipoda and Cirripeda (Crustacea). Most non-insect arthropod infections also involve Wolbachia placed in clades A or B. A minority of arthropod infections involves Wolbachia placed in distinct lineages (clades E through N) [5], [7]. In clade A and B symbionts, transmission appears to be essentially vertical (mother to offspring) in ecological time, but phylogenetic analysis reveals that lateral transfer between hosts has been common on longer timescales.
Wolbachia infections have also been identified in nematodes, notably in the animal parasites of the Onchocercidae. These filarial parasites utilise arthropod vectors (dipterans and chelicerates) in transitioning between their definitive vertebrate hosts, but the Wolbachia they carry are not closely related to those of the vector arthropods. The majority of Wolbachia from onchocercid nematodes are placed in two distinct but related clades, C and D [6], [8]. The biology of the interaction between filarial nematodes and their C and D Wolbachia is strikingly different [9]. There is no evidence of reproductive manipulation. Transmission is vertical, as in other Wolbachia, but, unlike the arthropod symbionts, in species with infections all members carry the symbionts, and the phylogeny of hosts and symbionts show remarkable congruence. Treatment with antibiotics both kills onchocercid nematode Wolbachia, and also affects the viability of the nematodes, suggesting a strongly mutualistic, possibly essential interaction [10], [11]. The interaction is not essential on a phylogenetic timescale, as nested within the Wolbachia-infected onchocercids are species that have lost their infections [12]. The biological bases for the mutualism is a topic of significant research interest, and may include manipulation of embryogenesis, metabolic provisioning and modulation of host immune responses [9], [13]–[16].
Not all nematode Wolbachia are placed in clades C and D [17]. Clade F Wolbachia have a distinct host profile compared to the other clades, as they have been found in both onchocercid nematodes (Mansonella, Madathamugadia and Cercopithifilaria species) [18], and arthropods (hexapods and chelicerates). The Wolbachia symbiont from the nematode Dipetalonema gracile is the sole representative of clade J, but is closely related to clade C Wolbachia [19]. A Wolbachia infection has been described in Radopholous similis, a tylenchid plant parasitic nematode distantly related to the Onchocercidae [20]. This symbiont has been placed in a new clade I. The biological role(s) of these nematode Wolbachia have yet to be defined. Wolbachia have been sought in other nematode species, both parasitic and free-living. These searches, carried out using Wolbachia-specific PCR amplification of marker genes, have generally proved negative in individuals sampled across the diversity of Nematoda other than Onchocercidae [21]. In the many ongoing nematode genome and transcriptome projects, Wolbachia-derived sequence has only been described from onchocercid nematodes and R. similis. However, there are two overlapping expressed sequence tags from Ancylostoma caninum (also a member of Strongyloidea) that have high similarity to Wolbachia genes [22], but these have not been verified as derived from a Wolbachia symbiont in this species. (The relationships of the nematode taxa discussed are illustrated in Figure 1 [18], [23], [24].)

Lateral transfer of Wolbachia genome fragments into the host nuclear genome has been detected in arthropods and nematodes that carry live infections [25], [26]. Inserted fragments range from what is likely the whole bacterial genome inserted into an azuki beetle chromosome, to short fragments at the limit of specific detection. These fragments have excited much debate, particularly concerning the Onchocercidae, where it has been hypothesised that they may represent functional gene transfers into the nematode genome and thus play significant roles in host biology [27]–[30]. However most Wolbachia insertions have accumulated many substitutions and insertion-deletion events compared to their functional homologues in extant bacterial genomes. In this they most resemble nuclear insertions of mitochondrial DNA, which are ‘dead on arrival’ and evolve neutrally in the host chromosome [25].
Interestingly, the onchocercid nematodes Onchocerca flexuosa [31], Acanthocheilonema viteae [11], [32] and Loa loa [12] lack Wolbachia despite their placement within the group of Wolbachia-containing species. This suggests that they have lost their live Wolbachia infections. Fragments of Wolbachia-like sequence have been detected in the nuclear genome in these species [31], [33]. Wolbachia nuclear transfers, or nuwts, in nematodes that currently lack live Wolbachia infection can be thought of molecular fossils of the previous symbiosis history of the host. Just as fossil skeletal remains can reveal the past distribution of larger biota, and viral insertions reveal the history of host infection [34], , nuwts can reveal past symbioses, and their divergence from current Wolbachia genomes can be used to estimate the date of the symbiosis.
We are engaged in a phylum-wide survey of genomes within the Nematoda [36]. As part of our analytic procedures we routinely screen raw genomic DNA data for contamination with environmental, commensal and host DNAs with a pipeline that uses read coverage, contig GC% and sequence identity to known protein sequences [37]. This serves to identify, and ease removal of, contaminating genomes, which in turn improves target genome assembly and aids independent assembly of symbiont genomes where present. Here we present an analysis of genome sequence data from the strongyloidean nematode Dictyocaulus viviparus, the bovine lungworm, which reveals molecular fossils of an ancient Wolbachia symbiosis in this economically important species, which is only distantly related to the previously known nematode hosts (Figure 1).
Results
The draft genome sequence of Dictyocaulus viviparus
We generated a draft genome for the strongyloidean nematode D. viviparus based on a single adult male specimen provided from a cow slaughtered at an abattoir in Ngaoundéré, Cameroon. The D. viviparus genome was assembled using Velvet from 16 gigabases of cleaned data from 165 million, 100-base, paired-end reads from a 500 base pair (bp) insert library sequenced on an Illumina HiSeq2500 instrument. The draft assembly spanned 169.4 megabases (Mb) (Table 1). In terms of contiguity, the draft was of moderate quality with an N50 (length of contig at which 50% of the genome is in contigs of this size or larger) of 22 kilobases (kb), and N90 of 5 kb. There were 17,715 contigs above 500 bp. The assembly had a GC content of 34.5% and estimated read coverage of ∼80 fold (Figure 2A). The mitochondrial contigs from the assembly had >99.5% identity to the published mitochondrial genome of D. viviparus. The size of this draft assembly is within the range of published genome sizes from species of the same suborder (Rhabtitina), which range from 80 Mb (Heterorhabditis bacteriophora [38]) to 320 Mb (Haemochus contortus [38]) (Table 2). Given that we used a single library, and had no long-range mapping data, it is likely that this genome size estimate is lower than the true genome as near-identical repeats will have been collapsed or left unassembled. We assessed the completeness of the draft assembly using the Core Eukaryotic Genes Mapping Approach (CEGMA [39]), and identified 90% complete and 93% partial genes. A previous Roche 454 transcriptome assembly for D. viviparus [40] was used to assess the assembly's completeness in terms of representation of known D. viviparus transcripts. Retaining matches where over 70% of the transcript were mapped to the same genome contig, 87% of transcripts were present in the assembly. Many additional transcripts were split between contigs.



Using a MAKER2-Augustus pipeline [41], [42], we predicted 14,306 protein-coding genes, with a median length of 834 bp, median exon length of 168 bp, and a median of 7 exons per gene. We compared this predicted gene set for D. viviparus to those of Caenorhabditis elegans [43], H. bacteriophora [38] and H. contortus [44], [45] using orthoMCL [46]. A majority (75%) of the predicted D. viviparus proteins clustered with proteins from these rhabditine nematodes (Figure 2). The only species which had a low proportion of proteins clustered was H. bacteriophora (∼40%), an observation that has been noted previously [38].
Thus, while the goal of our study was not to produce a high-quality reference genome for D. viviparus, the draft assembly and annotation produced are still of reasonable quality (Table 2). A majority of known D. viviparus genes are present, similarity to related nematode species is high, and most of the genes appear to be present and in full length. The genome assembly and a dedicated BADGER genome exploration environment [47] are available from http://dictyocaulus.nematod.es.
Identification of Wolbachia-like sequences in the nuclear assembly
As part of our standard quality control processes, we generated a taxon-annotated GC-coverage plot (TAGC plot) [37], with the goal of identifying any non-nematode (either bovine host or environmental bacterial) contamination (Figure 3 A). This process allows identification of contaminants by their presence as contigs with differing GC content or estimated read coverage compared to that of assured target genome contigs [48]. The taxonomic annotation, using the NCBI BLAST+ suite, serves to assign contaminant contigs to their possible species of origin. This process identified a total of 193 contigs, spanning 1 Mb, that had best matches to Wolbachia (Figure 3 B). The Wolbachia-like contigs had a GC content very close to the mode for the nematode genome, but they had a wide range of estimated coverages, from approximately equal to the majority of nematode-derived contigs to 3–4 fold higher Figure 3 C). Unusually, the Wolbachia-like contigs were not better assembled than the nuclear genome. The lower complexity of the alphaproteobacterial genome usually results in more contiguous assembly, even at low coverage.

The putative Wolbachia from D. viviparus (wDv) contigs were compared to the complete genomes of Wolbachia from Brugia malayi (wBm) [9] and O. ochengi (wOo) [16]. The average identity of the BLAST hits was 84.5% ±3.2% to both of the other Wolbachia genomes, indicating similar evolutionary distance from these two taxa (Figure 3 D). The matches were distributed across the genomes of other Wolbachia (Figure 3 B). The Wolbachia-like fragments were uploaded to the RAST server [49] for direct annotation, and 1580 coding sequences were predicted, almost double than found in previous nematode Wolbachia genomes (http://rast.nmpdr.org/?page=JobDetails&job=112231; Table 3). This elevated number largely resulted from frameshifts and stop codons in the middle of genes, which fragmented the open reading frames, and overall only 567 different Wolbachia genes (of a usual 800 to 1500) were identified. We also screened the contigs that had Wolbachia matches for other informative similarities, and identified 29 that contained both nematode and Wolbachia matches (examples are illustrated in Figure 3 E). We explored both read coverage and read-pair sanity across these 29 contigs using Tablet [50] to validate the co-assembly of nematode and Wolbachia-like segments, as de Bruijn graph assemblers can create chimaeric contigs. We found the contigs to be valid, contiguous regions of the genome. Even in cases such as scaffold00357 (Figure 3 E) where the nuclear and Wolbachia components had distinct read coverages, manual inspection of the presumed Wolbachia-nuclear junctions revealed no issues of inconsistent read pairing or inferred insert length. Segments with much higher coverage than the nuclear genome may be derived from collapse of dispersed repeat copies of the Wolbachia fragment. From these analyses we conclude that the Wolbachia-like fragments are not from an unsuspected live Wolbachia infection of D. viviparus, but are rather neutrally-evolving insertions of Wolbachia genome fragments into the nematode nuclear genome, and are relics of an ancient symbiosis, now lost. We have called the fragmented Wolbachia wDv, though, obviously, we have no evidence of an extant wDv organism (and in fact regard it as being extinct).

As a preliminary assessment of whether the insertions are restricted to some populations of D. viviparus (and thus that the symbiosis may have been recent and only in part of the species), or are more widespread (and thus likely to derive from more ancient symbiosis), we screened an independent D. viviparus isolate for presence of Wolbachia gene fragments. We performed directed PCR and Sanger sequencing of Wolbachia gene fragments from a D. viviparus isolate maintained at the Moredun Institute, Edinburgh, isolated in Scotland in 2005. Both ftsZ and 16S rRNA fragments were amplified from this strain, and, when sequenced, were closely similar to the whole genome assembly-derived fragments, but differed by several substitutions (Figure 4 A, B). Comparison of the nuclear small subunit ribosomal RNA sequence from the assembly to those from Dictyocaulus species affirmed the species identification (Figure 4 C). We also screened the previous D. viviparus transcriptome assembly [40] for Wolbachia-like fragments and identified six transcribed fragments (Table 4) that were likely to be derived from Wolbachia, confirming presence of symbiont gene fragments in a third isolate.


These transcribed Wolbachia-like fragments might offer evidence for functional integration of the remnants of the wDv genome into the nuclear genome. We thus investigated each fragment for possible function. In four of five fragments deriving from protein-coding genes there were frameshifts and in-frame stop codons. None of the transcribed fragments showed evidence of splicing. One transcript, where the Wolbachia-like sequence was in the likely 3′ UTR of a nematode gene (a homologue of C. elegans FRM-1), showed standard spliceosomal introns in the nematode-gene-like part, but the Wolbachia fragment itself was not spliced. Four of the transcript fragments were very short (500–600 bases, approximately one 454 read length).
Relationships of the Wolbachia of D. viviparus to other Wolbachia
To identify the relationships of wDv, sequences from the Wolbachia-like contigs were added to a five-gene supermatrix (including 16S rDNA, groEL, ftsZ, dnaA and coxA loci) used previously for phylogenetic analyses of Wolbachia [18]. This matrix does not include data from all 14 recognised Wolbachia clades, as sequencing in most has been limited. wDv fragments corresponding to these genes were identified using BLAST and aligned with MUSCLE. We were not able to identify a dnaA gene in the D. viviparus assembly. We added to the alignment data from wOo and available sequences from the Wolbachia from Radopholus similis (wRs). Both RAxML, MrBayes and PhyloBayes analyses suggested that wDv belongs to clade F, with strong branch support (Figure 5). The long terminal branch of wDv compared to other Wolbachia in the same clade is likely to be a consequence of the accumulation of mutations in the wDv regions due to their insertion and subsequent neutral evolution in the nematode genome. wOo was placed robustly within clade C as expected. Placement of wRs was less definite as it clustered as a sister taxon to clade D, but on a long branch with low support. We were unable to recover the published phylogeny [20] with wRs arising basally to other Wolbachia, even when the matrix was analysed with wDv excluded (data not shown), and thus this previous finding may be a methodological artifact.
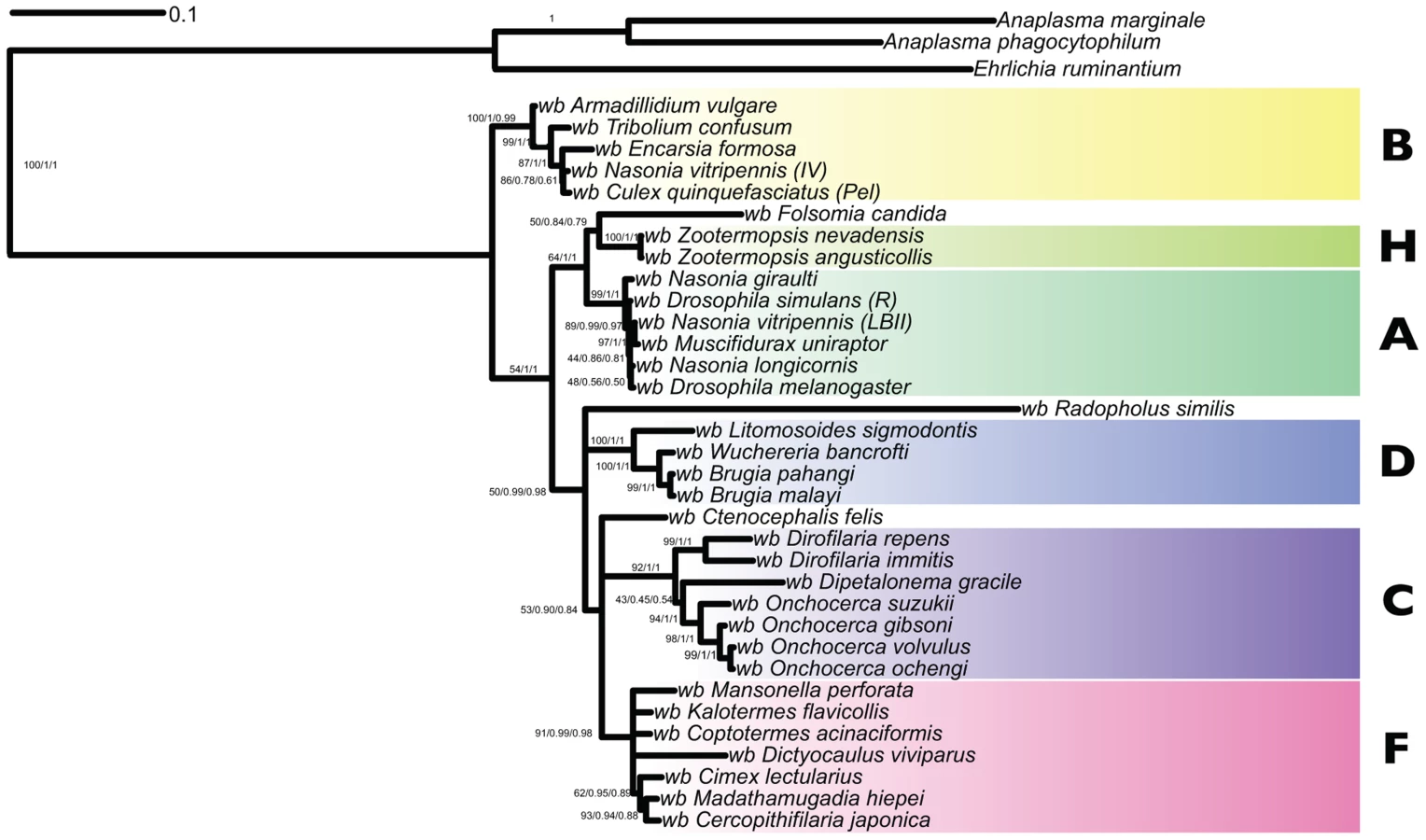
One genomic feature that distinguishes clade C and D Wolbachia is the absence of WO phage. WO phage are active temperate bacteriophage that are present in the sequenced clade A and B genomes, and that may mediate genetic transfer of key symbiosis genes between strains [51]. Using the 1363 protein sequences derived from WO phage available in the NCBI/ENA/DDBJ databases we identified 15 scaffolds in the D. viviparus genome that contained significant (BLAST E-values less than 1e-20) to WO phage proteins. These matches (Table 5) were to a wide range of WO phage genes, including capsid proteins, portal proteins, secretion system components, recombinases and others. In this genomic feature, wDv resembled A and B Wolbachia more than it did C and D.

Discussion
Fossils of Wolbachia infection reveal an unexpected palaeosymbiosis
D. viviparus is the first nematode from the Rhabditina (the group that includes C. elegans and the important animal-parasitic Strongyloidea) that has been shown to have a relationship with Wolbachia. However, the Wolbachia sequences identified in the draft genome sequence do not appear to derive from a living organism, but rather show features suggestive of being ancient laterally transferred fragments of the genome of a clade F-like Wolbachia, which is now extinct. The insertions were not unique to the individual Cameroon nematode sampled, but were identified in another D. viviparus (from Scotland). Published and unpublished transcriptome data for D. viviparus include a very low level of fragments that mapped to Wolbachia-like regions of our assembly. We suggest that the lateral transfers may be found in all D. viviparus, and that it will be exciting to survey additional Dictyocaulinae and related families within Strongyloidea for evidence of (palaeo-) symbiosis, and to better date the origin of the laterally-transferred fragments.
Lateral transfers of Wolbachia DNA into the host nucleus, nuwts, have been identified previously in filarial nematodes and arthropods [26], [52], [53]. The evidence for the D. viviparus Wolbachia-like sequences being ancient lateral transfers include their fragmentation, their interspersion with nematode sequence in robustly-assembled contigs, and their having inactivating mutations. Read coverage of the Wolbachia-like fragments varied greatly. If all the fragments derived from the genome of a live infection, it would be expected that they would have very similar coverage, as seen in other Wolbachia infected nematodes [37], [54]. Fragments with very high read coverage are likely to be repeats (within the nematode genome). While about 1 Mb of contigs had matches to Wolbachia, these did not constitute a complete genome. Only ∼60% of the expected Wolbachia gene content was present (for example the dnaA gene was missing) and many genes and gene fragments were duplicated. Genome fragmentation and gene inactivation is suggestive of a long period of residence in the D. viviparus nuclear genome [25].
Do these Wolbachia-like but nuclear-encoded sequences have a current expressed function in D. viviparus? The majority of the potential protein-coding genes in the Wolbachia-like fragments contain insertions, deletions, frameshift mutations or nonsense codons compared to their homologues from living Wolbachia genomes. We identified only six Wolbachia-like transcript fragments in 61,134 transcripts assembled from 3 million D. viviparus transcriptome sequences [40]. Four of the transcript fragments were very short, about one 454 read length, and one Wolbachia-like match was in the 3′ untranslated region of a bona fide nematode gene. Four of five fragments from protein-coding genes had frameshift and in-frame stop codon mutations, while the 16S rRNA fragment had a large deletion compared to 16S from living Wolbachia. On these bases it is unlikely these Wolbachia-derived sequences play roles in D. viviparus biology.
This discovery suggests that all three suborders of the nematode order Rhabditida (Rhabditina, Tylenchina and Spirurina) have members whose genomes and biology have been shaped by symbioses with Wolbachia. In the well-studied clade C and D Wolbachia the relationship has features of mutualism [14]. The Wolbachia observed in R. similis is apparently live, as bacterial cells can be seen within host cells by microscopy [20], but there are currently no data on the nature of the symbiosis: its genome sequence is awaited with interest. In D. viviparus we have no positive evidence for live infection. Our analyses placed both wDv and wRs close to clade F Wolbachia, and showed that clades C, D and F form a group distinct from clades A and B. From these and previous [18] analyses Clade F appears more “promiscuous” in its host relationships (its known hosts include both nematodes and arthropods). The symbiont biology of clade F is not well known: in Cimex, the clade F symbiont may be essential for fertility and nymphal development [55] but symbiont-host interactions remain unexplored elsewhere. We note that the presence of Wolbachia (albeit now extinct) in D. viviparus, a nematode that does not use an arthropod intermediate vector host, suggests that a simple model of nematode acquisition of Wolbachia from their vector arthropods is less likely. Clade F-like Wolbachia emerge as a credible source of the clade C and D Wolbachia of filarial nematode species. The wDv genome was likely to have contained WO phage [51], a mobile element present in clade A and B genomes but strikingly absent from clade C and D genomes.
In this scenario, the genomic fossils of Wolbachia found in D. viviparus are evidence of infection of an F-like Wolbachia in a dictyocauline ancestor. We identified insertions in independent isolates of the parasite suggesting that the association was not limited to one subpopulation of D. viviparus. We note that there are Wolbachia-like sequences in transcriptome data from A. caninum, another strongyloidean nematode, and thus it is possible that Wolbachia infections may have been widespread in this group. While reports of Wolbachia in the strongyloidean Angiostrongylus have been discounted [56], [57], we are excited by the possibility that other palaeosymbioses, now extinct, may be revealed in forthcoming genome projects across the Nematoda and Metazoa.
Finally, we provide a first draft assembly and annotation of the important nematode parasite D. viviparus. The identification of our specimen as D. viviparus is based on close identity of sequenced loci and the complete mitochondrial genome between our specimen and previously published D. viviparus data. As the specimen was destroyed during DNA extraction we no longer have a voucher for the individual. We note that there are very few records of D. viviparus in sub-Saharan Africa, and it is typically described as a temperate species [58]. A very large abattoir survey in the Democratic Republic of Congo found only 3 infected carcasses from 571 examined, and all of these were from cattle reared above 1,500 m (Ngaoundéré is at 1,200 m) [59]. The genome and annotation can be used as a springboard for further analysis both investigating the Wolbachia-nematode interaction and also potential gene identification for drug and vaccine development.
Materials and Methods
Nematode isolation and genome sequencing
A single Dictyocaulus viviparus male was isolated from Bos indicus (an individual of the local Gudali breed) in Ngaoundéré abattoir, Adamawa Region in Cameroon by David Ekale and Vincent Tanya during the ongoing Enhancing Protective Immunity Against Filariasis EU-Africa programme. The nematode was frozen at −80°C and shipped to Liverpool, UK, where DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen). Genomic sequencing was carried out by the Edinburgh Genomics Facility, using Illumina TruSeq library preparation reagents and a HiSeq 2500 instrument. A single 300 bp insert library was constructed, and 100 base paired-end data generated. Raw data have been submitted to the International Nucleotide Sequence Database Consortium under the project accession PRJEB5116 (study ERP004482).
Genome assembly and annotation
All software tools used (including versioning and command line options used) are summarised in Table 6. The quality of Illumina reads was checked with FASTQC [60]. Raw reads were quality trimmed (base quality of 20), and paired reads were discarded if either pair was below 51 bases using fastq-mcf [61]. The trimmed reads were digitally normalised to ∼20X coverage with khmer [62]. A draft assembly was generated using the normalised reads with Velvet [63] and gaps within scaffolds were filled using GapFiller [64]. Scaffold coverage was obtained by mapping all the reads back to the assembly using the clc-bio toolkit (CLC-Bio Ltd). Taxon-annotated GC%-coverage plots (TAGC plots) [37] were used to identify potential bovine and other contamination. Bovine contamination, which was minimal, was removed.

A MAKER2-Augustus annotation pipeline was used to predict protein-coding genes from the genome [41]. The MAKER2 program combines multiple ab initio and evidence-based gene predictors and predicts the most likely gene model. MAKER2 was run in a SGE cluster using the SNAP ab initio gene finder trained by CEGMA [39] output models, GeneMark-ES ab initio finder, D. viviparus transcripts and SwissProt proteins. We used the MAKER2 predictions to train Augustus [42] and create a gene finder profile for D. viviparus. Using the gene finder profile, the assembled transcriptome [40] and available expressed sequence tag data [65], Augustus was used alone to predict the final gene set, which was used for downstream analysis. Protein sets from selected nematode species, downloaded from Wormbase [66], were clustered using orthoMCL [46].
Analysis of Wolbachia-like fragments
The Dictycaulus viviparus draft assembly was broken into 500 bp fragments and each fragment was compared to Brugia malayi and Onchocerca ochengi Wolbachia endosymbiont genomes using BLAST+ [67]. Similarity hits with lengths above 100 bases were considered for downstream analysis. Contigs with Wolbachia-like sequences were annotated using the RAST server, which provided both gene finding and gene functional annotation. Junction fragments between putative Wolbachia insertions and D. viviparus nuclear genomic DNA were identified using BLAST+. Putative phage WO fragments were identified through tBLASTn comparison of the 1353 phage WO proteins available in NCBI nr to the D. viviparus assembly, using an E-value cutoff of 1e-20.
The phylogenetic relationships of Wolbachia from D. viviparus were assessed by identifying orthologues of 16S rDNA, groEL, ftsZ, dnaA, and coxA genes, and aligning these to orthologues from other Wolbachia. The five-gene supermatrix was analysed using RAxML [68], MrBayes [69] and PhyloBayes [70] (see Table 6 for specific parameters used). Trees were visualised in iTol [71] and FigTree [72].
Identification of Wolbachia insertions in other D. viviparus
D. viviparus genomic DNA from the Moredun, Scotland, isolate was provided by Prof. Jacqui matthews, Moredun Institute [73]. The Moredun strain has no known connection with Cameroon. Caenorhabditis elegans (free-living rhabditid nematode, which does not carry Wolbachia) and Litomosoides sigmodontis (a filarial nematode that carries a clade D Wolbachia [11]) genomic DNAs were used as negative and positive controls, respectively. PCR primers designed to amplify Wolbachia 16S, Wolbachia ftsZ [8], nematode nuclear small subunit rRNA (nSSU) [74] and mitochondrial cytochrome oxidase I (cox1) [75] were used in PCR with Phusion enzyme (NEB) to identify similar fragments in each nematode genomic DNA. A list of primers used and PCR conditions are given in Table 7. Positive PCR fragments were directly sequenced in both directions using BigDye v3 reagents in the Edinburgh Genomics facility. D. viviparus Roche 454 transcriptome data (Bioproject PRJNA20439) were downloaded from ENA and screened using BLAST for sequences corresponding to the Wolbachia insertions in our assembly.

Zdroje
1. WerrenJH (1997) Biology of Wolbachia. Annual Reviews of Entomology 42 : 587–609.
2. ZugR, HammersteinP (2012) Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PloS one 7: e38544.
3. WerrenJH, BaldoL, ClarkME (2008) Wolbachia: master manipulators of invertebrate biology. Nature reviews Microbiology 6 : 741–751.
4. WalkerT, JohnsonPH, MoreiraLA, Iturbe-OrmaetxeI, FrentiuFD, et al. (2011) The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476 : 450–453.
5. LoN, ParaskevopoulosC, BourtzisK, O'NeillSL, WerrenJH, et al. (2007) Taxonomic status of the intracellular bacterium Wolbachia pipientis. International journal of systematic and evolutionary microbiology 57 : 654–657.
6. Comandatore F, Sassera D, Montagna M, Kumar S, Koutsovoulos G, et al. (2013) Phylogenomics and analysis of shared genes suggest a single transition to mutualism in Wolbachia of nematodes. Genome biology and evolution.
7. AugustinosAA, Santos-GarciaD, DionyssopoulouE, MoreiraM, PapapanagiotouA, et al. (2011) Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PloS one 6: e28695.
8. BandiC, AndersonTJ, GenchiC, BlaxterML (1998) Phylogeny of Wolbachia in filarial nematodes. Proc Biol Sci 265 : 2407–2413.
9. FosterJ, GanatraM, KamalI, WareJ, MakarovaK, et al. (2005) The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biology 3: e121.
10. GenchiC, SacchiL, BandiC, VencoL (1998) Preliminary results on the effect of tetracycline on the embryogenesis and symbiotic bacteria (Wolbachia) of Dirofilaria immitis. An update and discussion. Parassitologia 40 : 247–249.
11. HoeraufA, Nissen-PahleK, SchmetzC, Henkle-DuhrsenK, BlaxterML, et al. (1999) Tetracycline therapy targets intracellular bacteria in the filarial nematode Litomosoides sigmodontis and results in filarial infertility. J Clin Invest 103 : 11–18.
12. McGarryHF, PfarrK, EgertonG, HoeraufA, AkueJP, et al. (2003) Evidence against Wolbachia symbiosis in Loa loa. Filaria journal 2 : 9.
13. TaylorMJ, CrossHF, BiloK (2000) Inflammatory responses induced by the filarial nematode Brugia malayi are mediated by lipopolysaccharide-like activity from endosymbiotic Wolbachia bacteria. The Journal of experimental medicine 191 : 1429–1436.
14. FennK, BlaxterM (2004) Are filarial nematode Wolbachia obligate mutualist symbionts? Trends in Ecology and Evolution 19 : 163–166.
15. FennK, BlaxterM (2006) Wolbachia genomes: revealing the biology of parasitism and mutualism. Trends Parasitol 22 : 60–65.
16. DarbyAC, ArmstrongSD, BahGS, KaurG, HughesMA, et al. (2012) Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res 22 : 2467–2477.
17. CasiraghiM, BordensteinSR, BaldoL, LoN, BeninatiT, et al. (2005) Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology 151 : 4015–4022.
18. LefoulonE, GavotteL, JunkerK, BarbutoM, UniS, et al. (2012) A new type F Wolbachia from Splendidofilariinae (Onchocercidae) supports the recent emergence of this supergroup. Int J Parasitol 42 : 1025–1036.
19. CasiraghiM, BainO, GuerreroR, MartinC, PocacquaV, et al. (2004) Mapping the presence of Wolbachia pipientis on the phylogeny of filarial nematodes: evidence for symbiont loss during evolution. International journal for parasitology 34 : 191–203.
20. HaegemanA, VanholmeB, JacobJ, VandekerckhoveTT, ClaeysM, et al. (2009) An endosymbiotic bacterium in a plant-parasitic nematode: member of a new Wolbachia supergroup. International journal for parasitology 39 : 1045–1054.
21. BordensteinSR, FitchDH, WerrenJH (2003) Absence of wolbachia in nonfilariid nematodes. Journal of nematology 35 : 266–270.
22. ElsworthB, WasmuthJ, BlaxterM (2011) NEMBASE4: the nematode transcriptome resource. International journal for parasitology 41 : 881–894.
23. BlaxterM (2011) Nematodes: the worm and its relatives. PLoS biology 9: e1001050.
24. FerriE, BainO, BarbutoM, MartinC, LoN, et al. (2011) New insights into the evolution of Wolbachia infections in filarial nematodes inferred from a large range of screened species. PloS one 6: e20843.
25. BlaxterM (2007) Symbiont Genes in Host Genomes: Fragments with a Future? Cell Host and Microbe 2 : 211–213.
26. FennK, ConlonC, JonesM, QuailMA, HolroydNE, et al. (2006) Phylogenetic relationships of the Wolbachia of nematodes and arthropods. PLoS Pathog 2: e94.
27. Dunning Hotopp JC, Clark ME, Oliveira DCSG, Foster JM, Fischer P, et al. (2007) Widespread Lateral Gene Transfer from Intracellular Bacteria to Multicellular Eukaryotes. Science Epub ahead of print on ScienceExpress Aug 30, 2007.
28. McNultySN, AbubuckerS, SimonGM, MitrevaM, McNultyNP, et al. (2012) Transcriptomic and proteomic analyses of a Wolbachia-free filarial parasite provide evidence of trans-kingdom horizontal gene transfer. PloS one 7: e45777.
29. McNultySN, FischerK, CurtisKC, WeilGJ, BrattigNW, et al. (2013) Localization of Wolbachia-like gene transcripts and peptides in adult Onchocerca flexuosa worms indicates tissue specific expression. Parasites & vectors 6 : 2.
30. IoannidisP, JohnstonKL, RileyDR, KumarN, WhiteJR, et al. (2013) Extensively duplicated and transcriptionally active recent lateral gene transfer from a bacterial Wolbachia endosymbiont to its host filarial nematode Brugia malayi. BMC Genomics 14 : 639.
31. McNultySN, FosterJM, MitrevaM, Dunning HotoppJC, MartinJ, et al. (2010) Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS One 5: e11029.
32. CasiraghiM, AndersonTJ, BandiC, BazzocchiC, GenchiC (2001) A phylogenetic analysis of filarial nematodes: comparison with the phylogeny of Wolbachia endosymbionts. Parasitology 122 Pt 1 : 93–103.
33. DesjardinsCA, CerqueiraGC, GoldbergJM, Dunning HotoppJC, HaasBJ, et al. (2013) Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nature genetics 45 : 495–500.
34. KatzourakisA (2013) Paleovirology: inferring viral evolution from host genome sequence data. Philos Trans R Soc Lond B Biol Sci 368 : 20120493.
35. KatzourakisA, GiffordRJ (2010) Endogenous viral elements in animal genomes. PLoS Genet 6: e1001191.
36. KumarS, KoutsovoulosG, KaurG, BlaxterM (2012) Toward 959 nematode genomes. Worm 1 : 1–9.
37. KumarS, JonesM, KoutsovoulosG, ClarkeM, BlaxterM (2013) Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Frontiers in Genetics 4 : 237.
38. BaiX, AdamsBJ, CicheTA, CliftonS, GauglerR, et al. (2013) A lover and a fighter: the genome sequence of an entomopathogenic nematode Heterorhabditis bacteriophora. PloS one 8: e69618.
39. ParraG, BradnamK, KorfI (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23 : 1061–1067.
40. CantacessiC, GasserRB, StrubeC, SchniederT, JexAR, et al. (2011) Deep insights into Dictyocaulus viviparus transcriptomes provides unique prospects for new drug targets and disease intervention. Biotechnology advances 29 : 261–271.
41. CantarelBL, KorfI, RobbSM, ParraG, RossE, et al. (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome research 18 : 188–196.
42. StankeM, KellerO, GunduzI, HayesA, WaackS, et al. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34: W435–439.
43. The C. elegans Genome Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282 : 2012–2018.
44. SchwarzEM, KorhonenPK, CampbellBE, YoungND, JexAR, et al. (2013) The genome and developmental transcriptome of the strongylid nematode Haemonchus contortus. Genome biology 14: R89.
45. LaingR, KikuchiT, MartinelliA, TsaiIJ, BeechRN, et al. (2013) The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome biology 14: R88.
46. LiL, StoeckertCJJr, RoosDS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13 : 2178–2189.
47. ElsworthB, JonesM, BlaxterM (2013) Badger–an accessible genome exploration environment. Bioinformatics 29 : 2788–2789.
48. KumarS, BlaxterML (2011) Simultaneous genome sequencing of symbionts and their hosts. Symbiosis 55 : 119–126.
49. AzizRK, BartelsD, BestAA, DeJonghM, DiszT, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC genomics 9 : 75.
50. MilneI, BayerM, CardleL, ShawP, StephenG, et al. (2010) Tablet–next generation sequence assembly visualization. Bioinformatics 26 : 401–402.
51. KentBN, BordensteinSR (2010) Phage WO of Wolbachia: lambda of the endosymbiont world. Trends Microbiol 18 : 173–181.
52. KondoN, NikohN, IjichiN, ShimadaM, FukatsuT (2002) Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc Natl Acad Sci U S A 99 : 14280–14285.
53. Dunning-HotoppJC, ClarkME, OliveiraDC, FosterJM, FischerP, et al. (2007) Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317 : 1753–1756.
54. GodelC, KumarS, KoutsovoulosG, LudinP, NilssonD, et al. (2012) The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 26 : 4650–4661.
55. HosokawaT, KogaR, KikuchiY, MengXY, FukatsuT (2010) Wolbachia as a bacteriocyte-associated nutritional mutualist. Proceedings of the National Academy of Sciences of the United States of America 107 : 769–774.
56. TsaiKH, HuangCG, WangLC, YuYW, WuWJ, et al. (2007) Molecular evidence for the endosymbiont Wolbachia in a non-filaroid nematode, Angiostrongylus cantonensis. Journal of biomedical science 14 : 607–615.
57. FosterJM, KumarS, FordL, JohnstonKL, BenR, et al. (2008) Absence of Wolbachia endobacteria in the non-filariid nematodes Angiostrongylus cantonensis and A. costaricensis. Parasites & vectors 1 : 31.
58. ThamsborgSM, BoaME, MakundiAE, KassukuAA (1998) Lungworm infection (Dictyocaulus viviparus) on dairy cattle farms in tropical highlands of Tanzania. Tropical animal health and production 30 : 93–96.
59. ChartierC (1990) [Dominant characteristics of helminth parasitism in cattle in Ituri (Haut-Zaire). I. The helminth fauna]. Rev Elev Med Vet Pays Trop 43 : 75–84.
60. Andrews S (2013) fastqc. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
61. Aronesty E (2011) ea-utils: Command-line tools for processing biological sequencing data.
62. Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH (2012) A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. arXiv q-bio: http://arxiv.org/abs/1203.4802.
63. ZerbinoDR, BirneyE (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18 : 821–829.
64. NadalinF, VezziF, PolicritiA (2012) GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13 Suppl 14: S8.
65. RanganathanS, NagarajSH, HuM, StrubeC, SchniederT, et al. (2007) A transcriptomic analysis of the adult stage of the bovine lungworm, Dictyocaulus viviparus. BMC genomics 8 : 311.
66. HarrisTW, BaranJ, BieriT, CabunocA, ChanJ, et al. (2013) WormBase 2014: new views of curated biology. Nucleic Acids Research 42: D789–793.
67. BoratynGM, CamachoC, CooperPS, CoulourisG, FongA, et al. (2013) BLAST: a more efficient report with usability improvements. Nucleic Acids Research 41: W29–33.
68. StamatakisA (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 : 2688–2690.
69. RonquistF, TeslenkoM, van der MarkP, AyresDL, DarlingA, et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic biology 61 : 539–542.
70. LartillotN, LepageT, BlanquartS (2009) PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25 : 2286–2288.
71. LetunicI, BorkP (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Research 39: W475–478.
72. Rambaut A (2012) FigTree version 1.4. http://tree.bio.ed.ac.uk/software/figtree/.
73. PezzementiL, KrejciE, ChatonnetA, SelkirkME, MatthewsJB (2012) A tetrameric acetylcholinesterase from the parasitic nematode Dictyocaulus viviparus associates with the vertebrate tail proteins PRiMA and ColQ. Molecular and biochemical parasitology 181 : 40–48.
74. BlaxterML, De LeyP, GareyJR, LiuLX, ScheldemanP, et al. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature 392 : 71–75.
75. FolmerO, BlackM, HoehW, LutzR, VrijenhoekR (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular marine biology and biotechnology 3 : 294–299.
76. KorfI (2004) Gene finding in novel genomes. BMC Bioinformatics 5 : 59.
77. LomsadzeA, Ter-HovhannisyanV, ChernoffYO, BorodovskyM (2005) Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research 33 : 6494–6506.
78. EdgarRC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5 : 113.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 6
Nejčtenější v tomto čísle
- Early Back-to-Africa Migration into the Horn of Africa
- PINK1-Mediated Phosphorylation of Parkin Boosts Parkin Activity in
- OsHUS1 Facilitates Accurate Meiotic Recombination in Rice
- Ancient DNA Analysis of 8000 B.C. Near Eastern Farmers Supports an Early Neolithic Pioneer Maritime Colonization of Mainland Europe through Cyprus and the Aegean Islands