
The Rim15-Endosulfine-PP2A Signalling Module Regulates Entry into Gametogenesis and Quiescence Distinct Mechanisms in Budding Yeast
The fundamental property of a cell is to sense changes in the environment and then respond in a way that maximizes its chances of survival. When diploid budding yeast cells are subjected to complete nutrient starvation they have two possible fates, namely quiescence and gametogenesis. Quiescent cells have reduced rates of transcription and translation and increased stress tolerance. Gametogenesis results in production of haploid spores that can survive for long periods of time. In this paper, we report a signalling module that regulates entry into both quiescence and gametogenesis in budding yeast. The module consists of three molecular components namely a serine-threonine kinase Rim15, a phosphatase PP2ACdc55 and a conserved protein called as endosulfine. PP2ACdc55 negatively regulates entry into gametogenesis and quiescence. Upon nutrient starvation, Rim15 becomes active and phosphorylates endosulfine. This converts endosulfine to an inhibitor of PP2ACdc55 and thereby leading to entry into quiescence and gametogenesis. Remarkably, an analogous module consisting of Greatwall kinase, PP2A-B55δ and endosulfine regulates entry into mitosis in frog egg extracts and meiotic maturation in flies suggesting that this signalling module is highly conserved and co-opted during evolution to control distinct biological processes in different organisms.
Published in the journal:
. PLoS Genet 10(6): e32767. doi:10.1371/journal.pgen.1004456
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004456
Summary
The fundamental property of a cell is to sense changes in the environment and then respond in a way that maximizes its chances of survival. When diploid budding yeast cells are subjected to complete nutrient starvation they have two possible fates, namely quiescence and gametogenesis. Quiescent cells have reduced rates of transcription and translation and increased stress tolerance. Gametogenesis results in production of haploid spores that can survive for long periods of time. In this paper, we report a signalling module that regulates entry into both quiescence and gametogenesis in budding yeast. The module consists of three molecular components namely a serine-threonine kinase Rim15, a phosphatase PP2ACdc55 and a conserved protein called as endosulfine. PP2ACdc55 negatively regulates entry into gametogenesis and quiescence. Upon nutrient starvation, Rim15 becomes active and phosphorylates endosulfine. This converts endosulfine to an inhibitor of PP2ACdc55 and thereby leading to entry into quiescence and gametogenesis. Remarkably, an analogous module consisting of Greatwall kinase, PP2A-B55δ and endosulfine regulates entry into mitosis in frog egg extracts and meiotic maturation in flies suggesting that this signalling module is highly conserved and co-opted during evolution to control distinct biological processes in different organisms.
Introduction
The ability of cells to sense deleterious changes in environment and mount an appropriate physiological and metabolic response is essential for cellular survival. Response to nutrition starvation in budding yeast has been an extremely powerful model to study this biological trait [1]. Upon complete nutrient starvation, yeast cells enter either gametogenesis or quiescence. Diploid yeast cells undergo gametogenesis when subjected to nitrogen starvation in the absence of glucose and in the presence of a non-fermentable carbon source. They undergo one round of DNA replication followed by two rounds of nuclear divisions to form 4 haploid spores which can stay dormant for long periods of time. Haploid and diploid cells enter quiescence when subjected to nutrient starvation or when treated with a drug called rapamycin, an inhibitor of the TOR (Target of Rapamycin) signalling pathway. Quiescence (-also referred to as G0) is a reversible non-proliferative state characterized by low rates of transcription and translation, increased stress-tolerance, elevated rate of macroautophagy and synthesis of storage carbohydrates (trehalose and glycogen). Many of the G0-features like increased macroautophagy, low rates of transcription and translation are also characteristic of quiescent mammalian cells suggesting that the core features of quiescence program are conserved [2], [3]. Ablation of G0-entry/exit control mechanisms is frequently linked to either reduced life span (especially in unicellular organisms) or cellular transformation (in multi-cellular organisms) [4], [5].
In budding yeast, entry into quiescence is controlled by the master regulator Rim15, a member of the AGC (named after protein kinase A, G and C families) group of serine-threonine kinases [6]. Activity of Rim15 is controlled by two nutrient signalling pathways namely the Ras/Protein Kinase A (Ras/PKA) and the Target of Rapamycin Complex 1 (TORC1) pathways. The TORC1 pathway responds to the availability of nitrogen source in the growth medium [2], [5]. In contrast, the Ras/PKA pathway responds to levels of glucose in the growth medium [2], [5]. Both pathways positively regulate cell proliferation in response to nutrient availability and thereby inhibit entry into G0. PKA phosphorylates Rim15 at five consensus PKA phosphorylation sites to inhibit its kinase activity and promote its retention in the cytosol [6]. Apart from PKA and TORC1 pathways, Rim15 also integrates signalling from Sch9 kinase (ortholog of mammalian Akt/S6 kinase) and Pho85-Pho80 kinase (phosphate-sensing) pathways [7]. Signalling through TORC1, Sch9 and Pho85/Pho80 pathways phosphorylate Rim15 at Thr-1075 and inhibit its nuclear localization. Nutrient deprivation inhibits signalling through these four pathways which results in dephosphorylation of Rim15 at its five PKA sites and Thr-1075 leading to its activation and translocation to the nucleus. Activated Rim15 stimulates stress-responsive transcription factors Msn2/4 and post-diauxic shift transcription factor Gis1 which in turn activate transcription of several genes required for survival in G0. Rim15 phosphorylates endosulfines, a highly conserved family of cAMP regulated phosphoproteins to promote entry into quiescence [8]. Endosulfines, following phosphorylation by Rim15, protect mRNA, which are transcriptionally controlled by Msn2/4 and Gis1, from degradation via the 5′-3′ mRNA decay pathway by inhibiting Dhh1 (decapping activator) and Ccr4 (deadenylation factor) [8].
Entry into gametogenesis in yeast is mainly regulated at the level of transcription of IME1 which encodes the master transcription factor for early-meiosis genes (EMGs) [9]. Like G0, entry into gametogenesis is negatively regulated by TORC1 and Ras/PKA pathways. Ime1 is recruited to promoters of EMGs and activates their transcription [10]. During vegetative growth, a DNA-binding protein Ume6 binds to promoters of EMGs and represses their expression by associating with the Sin3/Rpd3 histone deacetylase and Isw2 chromatin remodelling complexes [11], [12]. Absence of glucose and nitrogen in the medium results in the replacement of Sin3/Rpd3 and Isw2 by Ime1 at the EMG promoter regions [13]. It was proposed that Ime1 activates transcription of EMGs by converting Ume6 from a repressor into an activator [10]. This model was consistent with the observations that Ime1 physically interacts with Ume6 and that cells lacking Ume6 fail to sporulate efficiently [10], [14], [15]. However, this model was disputed by subsequent studies which showed that interaction of Ime1 with Ume6 facilitates Ume6 destruction and meiotic gene induction [16]. Rim15 has been implicated in the removal of histone deacetylase complex from the promoters of EMGs [13] but precisely how this is achieved is not known.
In this paper, we demonstrate that endosulfines are required for entry into gametogenesis and quiescence in budding yeast. Phosphorylation of endosulfine by Rim15 results in its association with the protein phosphatase PP2ACdc55 and inhibition of its phosphatase activity. We show that the Rim15-endosulfine-PP2ACdc55 signalling module regulates entry into quiescence and gametogenesis by distinct mechanisms. We also demonstrate that this signalling module is required for pre-meiotic autophagy which is necessary for gametogenesis in budding yeast. Remarkably a similar signalling module regulates M-phase progression during mitosis and meiosis in higher eukaryotes. In Xenopus egg extracts, the Greatwall kinase phosphorylates α-endosulfine (ENSA) and Arpp19 at a conserved serine residue, which then inhibits PP2A-B55δ to promote entry into mitosis [17], [18]. Depletion of Greatwall kinase and endosulfine in Drosophila leads to mitotic defects suggests that the module regulates entry into mitosis in flies [19], [20]. Inactivation of endosulfine in flies causes a failure in oocyte progression from prophase I to metaphase I indicating that this module regulates entry into M-phase during meiosis [21]. Our results therefore expand the repertoire of functions for this highly conserved signalling module that regulates distinct biological processes in different systems.
Results
Endosulfines are required for entry into gametogenesis
Since Rim15 is required for expression of early meiotic genes [22] we examined the function of endosulfine in gametogenesis. Budding yeast has two endosulfines Igo1 and Igo2. We first assessed the ability of wild type, igo1Δ, igo2Δ and igo1Δ igo2Δ strains to sporulate. While wild type, igo1Δ and igo2Δ strains sporulated with an efficiency of ≥65%, only 3% of igo1Δ igo2Δ cells formed spores (Figure 1A). To determine the precise function of endosulfines in spore formation, we induced wild type and igo1Δ igo2Δ cells to enter meiosis by transferring them to Sporulation medium (SPM). We examined expression of early meiotic proteins Ime1 and Rec8 (by Western blotting), pre-meiotic DNA replication (flow cytometry) and nuclear division (DAPI staining). Wild type cells replicated their DNA after 5 hours into SPM (Figure 1B), expressed Ime1 and Rec8 (Figure 1D), and underwent two rounds of nuclear division to form tetranucleate spores (Figure 1C). However igo1Δ igo2Δ cells failed to express both Rec8 and Ime1, did not undergo pre-meiotic DNA replication and remained mononucleate even after 12 hours into SPM (Figure 1B–1D). These results indicate that endosulfines are required for entry into gametogenesis in budding yeast. Induction of sporulation [23] involves arresting cells in stationary phase by growth in nutrient medium contacting acetate as a carbon source for 16 hours. To rule out the possibility that the failure of endosulfine mutant cells to sporulate was due to their inability to exit from stationary phase, we induced logarithmically growing wild type and igo1Δ igo2Δ cells to enter gametogenesis. Wild type but not igo1Δ igo2Δ cells underwent pre-meiotic DNA replication and spore formation (Figure S1), indicating that endosulfines are required for entry into gametogenesis. The spores formed in igo1Δ igo2Δ cells at a low frequency had viabilities similar to wild type spores (data not shown) suggesting that endosulfines are required for efficient entry into gametogenesis but not for rest of the sporulation program.

Phosphorylation of Igo1 at S64 is required for efficient entry into gametogenesis
Phosphorylation of endosulfine at a conserved serine residue (Figure 1E) by Greatwall kinase is required for entry into mitosis in Xenopus egg extracts [17], [18]. Phosphorylation at the corresponding Serine residue (Serine-64) in budding yeast Igo1 by Rim15 kinase is required for entry into G0 [8]. To test whether phosphorylation at S-64 also regulates entry into gametogenesis, we tested the ability of phospho-inhibitory igo1-S64A mutant to sporulate. About 50% of igo1Δ igo2Δ cells expressing wild type Igo1 sporulated in comparison to just 2% of control igo1Δ igo2Δ cells. In contrast, only 10% of igo1Δ igo2Δ cells expressing Igo1-S64A sporulated (Figure 1F). The sporulation efficiency of igo1Δ igo2Δ cells expressing the phospho-mimetic mutant Igo1-S64D was 1.7 fold more than that of Igo1-S64A expressing igo1Δ igo2Δ cells (Figure 1F). This effect of S64D mutation on sporulation efficiency was independent of Rim15 function (Figure S2). These results suggest that phosphorylation of Igo1 at Serine-64 by Rim15 is required for efficient entry into gametogenesis.
Phosphorylation of Igo1 at Serine-64 occurs at a constant level during the mitotic cell cycle [24]. To examine the phosphorylation of Igo1 at Serine-64 during entry into gametogenesis, we induced IGO1-myc8 and igo1-S64A-myc8 cells to enter gametogenesis by transferring them to SPM. Analysis of DNA content by flow cytometry indicated that pre-meiotic DNA replication was initiated after 3 hours into SPM and completed by 5 hours in both strains (Figure S3B). We prepared whole cell extracts and analysed electrophoretic mobility of Igo1 by Phos-tag affinity gel electrophoresis and SDS-PAGE. Phos-tag specifically retards the mobility of phosphoproteins [25]. We observed a phos-tag dependent mobility shift of wild type Igo1 but not Igo1-S64A. This upshifted band in wild type cells was present before transfer to SPM and was detectable up to 2 hours after transfer (Figure S3A). As expression of early meiotic genes like Ime1 and Rec8 is detectable even after 1 h in SPM (Figure 1C), we conclude that Igo1 is phosphorylated at S-64 during entry into gametogenesis but dephosphorylated subsequently.
Endosulfine contains a conserved protein kinase A site RK/RXS/T at its C-terminus (Figure S4A). Since PKA inhibits entry into gametogenesis, we reasoned that phosphorylation at this site might have an opposite effect to that mediated by Rim15 phosphorylation of S-64. However replacement of the Serine-105 in Igo1 with alanine or aspartate did not affect sporulation (Figure S4B).
Depletion/absence of the PP2A regulatory subunit Cdc55 suppress the gametogenesis - and G0- entry defects of endosulfine mutant cells
Phosphorylated endosulfine promotes entry into mitosis in Xenopus egg extracts by inhibiting the Cdk-antagonizing protein phosphatase PP2A-B55δ [17], [18]. We have demonstrated that PCLB2CDC55 cells which express CDC55 from the mitosis-specific promoter PCLB2, fail to undergo meiotic nuclear divisions and form monads [23]. The meiotic nuclear division defect of PCLB2CDC55 cells can be suppressed by net1-6Cdk, a mutant allele encoding the nucleolar protein Net1 lacking 6 Cdk recognition sites [23]. We also noted that PCLB2CDC55 cells underwent pre-meiotic DNA replication earlier than wild type cells [23] suggesting that PP2ACdc55 might negatively regulate entry into gametogenesis. We therefore investigated whether budding yeast proteins Rim15, endosulfine and PP2ACdc55 regulate entry into gametogenesis and G0. If PP2ACdc55 and Rim15/endosulfines play opposing roles in entry into gametogenesis and endosulfines promote entry into gametogenesis only by antagonising PP2ACdc55 , we reasoned that inactivation of PP2ACdc55 might suppress the sporulation defect of igo1Δ igo2Δ and rim15Δ cells. While 80% of wild type cells formed spores, only about 10% and 18% of igo1Δ igo2Δ and rim15Δ cells respectively, did. Remarkably igo1Δ igo2Δ and rim15Δ cells carrying a meiotic-null allele of CDC55 (PCLB2CDC55) formed monads (75%) like PCLB2CDC55 cells (Figure 2A). Crucially, combining net1-6Cdk with PCLB2CDC55 igo1Δ igo2Δ and PCLB2CDC55 rim15Δ cells resulted in efficient formation of tetrads (Figure 2A). The ability of PCLB2CDC55 to suppress igo1Δ igo2Δ was specific as deletion of a gene encoding an alternative PP2A regulatory subunit Rts1 had no effect on sporulation efficiency of igo1Δ igo2Δ cells (Figure 2A).

To confirm suppression of igo1Δ igo2Δ by PCLB2CDC55 we induced wild type, igo1Δ igo2Δ and igo1Δ igo2Δ PCLB2CDC55 cells to enter meiosis by transferring them to SPM. Wild type cells completed pre-meiotic DNA replication after 4 hours (Figure 2B), and expressed Cdc5 (a marker for mid-meiosis) after 7 hours in SPM (Figure 2C). In contrast, igo1Δ igo2Δ cells did not initiate DNA replication (Figure 2B) and failed to express Cdc5 even after 12 hours in SPM (Figure 2C). Crucially igo1Δ igo2Δ PCLB2CDC55 cells completed pre-meiotic DNA replication (3–4 hours) and expressed Cdc5 (5–6 hours) (Figure 2B–C). These results indicate that PP2ACdc55 and Rim15/endosulfine play opposing roles in regulating entry into gametogenesis.
We then determined whether PP2ACdc55 also negatively regulates entry into quiescence. Wild type, igo1Δ igo2Δ, cdc55Δ and igo1Δ igo2Δ cdc55Δ cells were treated with rapamycin and entry into G0 was monitored by assaying expression of Hsp26, a gene that is specifically induced during entry into G0 [8]. While wild type cells induced expression of Hsp26 after 2 hours following rapamycin treatment, the igo1Δ igo2Δ cells failed to express Hsp26 (Figure 2D). Crucially, both cdc55Δ cells and igo1Δ igo2Δ cdc55Δ cells expressed Hsp26 even in the absence of rapamycin treatment. These results indicate that the Rim15-endosulfine-PP2ACdc55 pathway regulates entry into gametogenesis and quiescence in budding yeast.
Phosphorylation of Igo1 at S64 by Rim15 converts it into an inhibitor of PP2ACdc55
To test whether phosphorylation of Igo1 at S64 results in increased association with PP2ACdc55, we performed an in vitro binding assay. We purified wild type Igo1, Igo1-S64A and Igo1-S64D from bacterial cells by attaching a Maltose Binding Peptide (MBP) to their N-termini. We then incubated endosulfine (and its variants) bound to amylose resin via the MBP with yeast extracts containing Cdc55-TAP (Tandem Affinity Purification). Specifically Igo1-S64D but not WT Igo1/Igo1-S64A physically interacted with Cdc55 in vitro (Figure 3A). We then tested whether phosphorylation of wild type endosulfine by Rim15 results in increased association with Cdc55. For this, we purified either wild type or a Kinase-Dead (kd) version of Rim15 (Rim15-C115A) from yeast cells using a GST affinity tag. We then incubated either wild type or Igo1-S64A or Igo1-S64D bound to amylose resin with Rim15/Rim15-kd for 45 minutes in the presence of ATP. The phospho-mimetic mutant Igo1-S64D interacted with Cdc55 regardless of whether it was incubated with Rim15/Rim15-kd. Wild type Igo1, but not Igo1-S64A, incubated with catalytically active Rim15 interacted with Cdc55 (Figure 3B). These results indicate that phosphorylation of Igo1 at S64 promote its association with PP2ACdc55. We confirmed the phosphorylation of Igo1 at S-64 by Rim15 using a phospho-specific antibody directed against S-64-P which recognized Igo1 incubated with wild-type but not a catalytically dead version of Rim15 (Figure 3B).

To test whether phosphorylation of endosulfine at S64 converts it into an inhibitor of PP2ACdc55, we measured the phosphatase activity of PP2ACdc55 in the presence of Igo1, Igo1-S64A and Igo1-S64D proteins. We purified PP2ACdc55 by attaching a TAP (Tandem Affinity Purification) tag to the C-terminus of Cdc55 (Figure 3C). At first, we measured the phosphatase activity of purified Cdc55 using a phosphorylated peptide as a substrate. Crucially, the TAP eluates from Cdc55-TAP tagged strain but not from an untagged strain, had phosphatase activity (Figure 3D). We then purified MBP-fused versions of Igo1/Igo1-S64D/Igo1-S64A from bacteria and tested their effect on PP2ACdc55 phosphatase activity. Only Igo1-S64D but not Igo1/Igo1-S64A inhibited the phosphatase activity of PP2ACdc55 (Figure 3E). Purified endosulfines had little or no phosphatase activity on their own (Figure S5). These results show that phosphorylation of endosulfine Igo1 at S64 by Rim15 converts it into an inhibitor of PP2ACdc55.
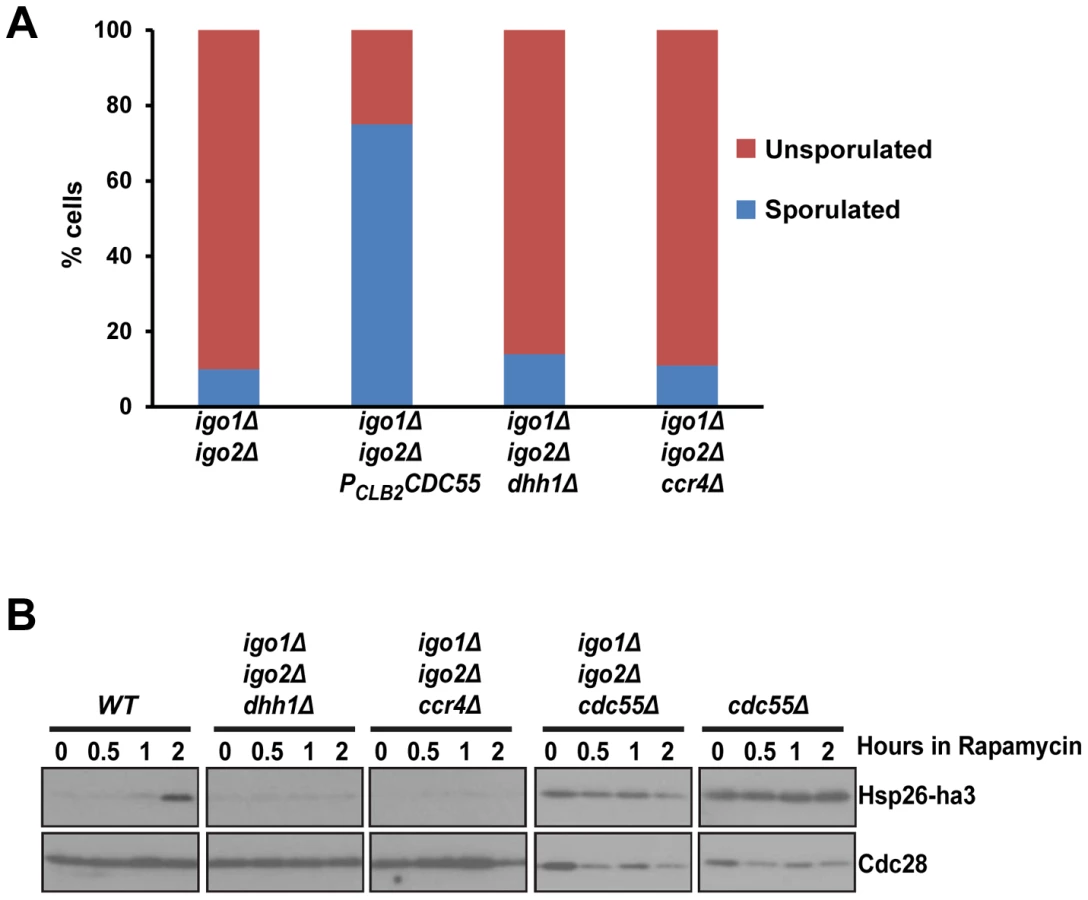
dhh1Δ and ccr4Δ do not suppress the G0 - and gametogenesis - entry defects of igo1Δ igo2Δ cells
Endosulfines activated by Rim15 were proposed to protect mRNA involved in stress response from the 5′ to 3′ mRNA decay pathway by direct inhibition of decapping enzyme Dhh1 [8]. Consistent with this possibility, deletion of genes DHH1 and CCR4, which are required for 5′ to 3′ decay were reported to suppress entry into quiescence defect of igo1Δ igo2Δ cells [8]. We therefore tested whether dhh1Δ and ccr4Δ also suppress the sporulation defect of igo1Δ igo2Δ cells. While PCLB2CDC55 suppressed the sporulation defect of igo1Δ igo2Δ cells, both dhh1Δ and ccr4Δ did not (Figure 4A). We also found that dhh1Δ and ccr4Δ did not suppress the G0 entry defect of igo1Δ igo2Δ cells (Figure 4B) contrary to what was previously reported [8]. We do not know the reason for this discrepancy but differences in the strain background used (BY4741 vs. SK1) for the experiments could be an explanation. However, our data are consistent with a simple model which posits that endosulfines regulate entry into quiescence and gametogenesis only through inhibition of PP2ACdc55.
The Rim15-Endosulfine-PP2ACdc55 pathway regulates entry into gametogenesis independent of G0 transcription factors Msn2, Msn4 and Gis1
Entry into quiescence is mediated by activation of three master transcription factors namely Msn2, Msn4 and Gis1 [5]. If the roles of Rim15-Endosulfine-PP2ACdc55 module during entries into quiescence and gametogenesis were identical then one would predict that cells lacking the three G0–specific transcription factors to be also defective in entry into gametogenesis. However we found that msn2Δ msn4Δ gis1Δ cells formed spores (60%) although with defective spore walls (Figure 5A and data not shown). To confirm that the tetrad formation in msn2Δ msn4Δ gis1Δ cells was dependent on endosulfines, we deleted IGO1 and IGO2 in msn2Δ msn4Δ gis1Δ cells. The quintuple mutant cells were defective in forming tetrads like igo1Δ igo2Δ cells (Figure 5A). We confirmed that msn2Δ msn4Δ gis1Δ cells were defective in entry into quiescence (Figure 5B). These results indicate that Rim15-Endosulfine-PP2ACdc55 pathway regulates entry into gametogenesis independently of activation of G0 transcription factors.

Endosulfines are required for transcriptional induction of IME1 caused by transfer of cells to sporulation medium
Ime1 is a master transcription factor for expression of early meiotic genes [9]. As indicated above (Figure 1D) Ime1 was not strongly expressed in igo1Δ igo2Δ cells after transfer to SPM. In wild type cells, IME1 is not expressed in glucose-containing nutrient medium but is transcribed at low levels in pre-sporulation medium (which contains acetate as a carbon source) and induced further following transfer to SPM [26]–[28]. We tested whether endosulfines are required for this transcriptional induction of IME1 by assaying IME1 mRNA levels by quantitative RT-PCR. In wild type and igo1Δ igo2Δ cells grown in pre-sporulation medium (which contains acetate as the carbon source), the levels of IME1 transcript were around 500-fold higher than in log-phase cells grown in glucose-containing nutrient medium (Figure 6A). However upon transfer to SPM, the IME1 mRNA levels increased further by about 8-fold after 2 hours in wild type but not in igo1Δ igo2Δ cells (Figure 6A). This suggests that endosulfines are required for transcriptional induction of IME1 caused by transfer to SPM.

Expression of Ime1 is not sufficient for suppressing the sporulation defect of endosulfine mutants
If the only role of endosulfines in entry into gametogenesis was to activate transcription of IME1, then ectopic expression of IME1 should bypass the sporulation defect of igo1Δ igo2Δ cells. To test this, we constructed wild type and igo1Δ igo2Δ strains in which IME1 expression can be induced by addition of β-estradiol to the medium using the PGAL/Gal4-ER system [29]. We transferred wild type and igo1Δ igo2Δ cells to SPM in the presence or absence of β-estradiol. While wild type cells sporulated in the presence of β-estradiol, igo1Δ igo2Δ cells failed to do so (Figure 6B). Ime1 was expressed in β-estradiol treated igo1Δ igo2Δ cells although at a lower level compared to wild type cells (Figure 6C). Since endosulfines have been implicated in mRNA stability [8], we tested whether the difference in the Ime1 levels in the two strains was due to difference in the IME1 transcript levels. Quantitative RT-PCR analyses revealed that the IME1 transcript levels were induced to similar extent in wild type and igo1Δ igo2Δ strains and remained relatively unchanged up to 8 hours following induction (Figure 6D). This suggests that endosulfines are not required for regulating IME1 mRNA stability. Decreased Ime1 levels in igo1Δ igo2Δ cells could be caused by either decreased translational efficiency of IME1 mRNA or decreased Ime1 stability. These results indicate that endosulfines promote entry into gametogenesis independently of regulating IME1 expression.
Endosulfines are required for pre-meiotic autophagy
Rim15 is required for autophagy induced by inhibition of PKA and Sch9 but not for autophagy induced by rapamycin treatment [30]. Since autophagy is required for spore formation in yeast [31], we tested whether endosulfines are required for autophagy during entry into gametogenesis. Autophagy can be assayed by following proteolytic cleavage of GFP-Atg8, which is a N-terminal fusion of GFP to Atg8 (a ubiquitin-like protein required for formation of autophagosomal membranes) [32]. We induced wild type, PCLB2CDC55, igo1Δ igo2Δ and igo1Δ igo2Δ PCLB2CDC55 cells to enter meiosis by transferring them to SPM and assayed autophagy. In wild type cells, GFP-Atg8 underwent proteolytic cleavage after 2 hours into SPM (Figure 7A). In contrast, GFP-Atg8 remained intact in igo1Δ igo2Δ cells even after 12 hours in SPM (Figure 7A). Strikingly, GFP-Atg8 was cleaved earlier in PCLB2CDC55 and igo1Δ igo2Δ PCLB2CDC55 cells in comparison to wild type cells. These results are consistent with the hypothesis that PP2ACdc55 inhibits pre-meiotic autophagy and that this inhibition is overcome by endosulfines after transfer to SPM.

We then tested whether endosulfines are required for autophagy induced by rapamycin treatment. We treated wild type, cdc55Δ, igo1Δ igo2Δ and igo1Δ igo2Δ cdc55Δ cells with rapamycin and assayed autophagy by western analysis. While autophagy in cdc55Δ cells was slightly advanced in comparison to wild type cells, endosulfine mutant cells underwent autophagy as efficiently as wild type cells (Figure 7B). We also found that endosulfines were not required for autophagy triggered by nitrogen starvation (Figure S6). Since rapamycin treatment and nitrogen starvation trigger autophagy by inhibiting TORC1, our results indicate that endosulfines are not required for autophagy induced by inhibition of TORC1 signalling. Rapamycin treatment of diploid cells induces sporulation [33]. While rapamycin–treated wild type cells formed tetrads after 24 hours, igo1Δ igo2Δ cells did not (Figure 7C). This suggests that induction of autophagy per se is insufficient for rescuing the sporulation defect of endosulfine mutant cells.
To determine the role of autophagy in sporulation, we induced wild type and atg1Δ cells (ATG1 encodes a serine-threonine kinase required for autophagy) to enter meiosis by transferring them to SPM. Wild type cells completed pre-meiotic DNA replication after 4–5 hours and underwent two rounds of nuclear division to form 45% tetrads (Figure S7A–B). Although the kinetics of Rec8 expression in wild type and atg1Δ cells were similar (Figure S7B), atg1Δ cells were delayed in initiation of pre-meiotic DNA replication by about 1–2 hours in comparison to wild type cells. Expression of Cdc5 (marker for mid-meiosis) in atg1Δ cells was delayed by about 3 hours relative to wild type cells (Figure S7C). However atg1Δ cells failed to undergo nuclear divisions and remained largely mononucleate with prophase I spindles after 10 hours in SPM (Figure S7B and data not shown). Since the phenotype of atg1Δ cells is distinct from that of igo1Δ igo2Δ cells (which fail to enter gametogenesis as indicated in Figure 1), we conclude that endosulfines regulate entry into gametogenesis independently of controlling pre-meiotic autophagy.
Ume6 associates the histone deacetylase Sin3/Rpd3 to negatively regulate entry into gametogenesis [11]. Interestingly, Ume6 is phosphorylated during sporulation in a Rim15-dependent manner [34]. If endosulfines promote entry into gametogenesis through inhibition of Ume6 and Sin3/Rpd3, then ume6Δ and rpd3Δ should suppress igo1Δ igo2Δ. However ume6Δ and rpd3Δ did not suppress the poor sporulation efficiency of igo1Δ igo2Δ cells (Figure S8). Surprisingly, both ume6Δ and rpd3Δ completely abolished the ability of igo1Δ igo2Δ cells to form tetrads (Figure S8). This suggests that endosulfines and Ume6/Rpd3 regulate spore formation via independent pathways.
Discussion
We have shown that a signalling module consisting of a serine-threonine kinase Rim15, endosulfine Igo1/2 and PP2ACdc55 regulates entry into gametogenesis and quiescence in budding yeast (Figure 8). While our manuscript was in preparation another group reported that Rim15-Endosulfine-PP2ACdc55 pathway is required for onset of quiescence in yeast cells [35]. We show that the signalling module also regulates entry into gametogenesis via a mechanism that is independent of entry into quiescence. Remarkably both studies show that Rim15-Endosulfine-PP2ACdc55 signalling module in budding yeast mechanistically works like the Greatwall Kinase-endosulfine-PP2A-B55δ pathway that regulates mitotic entry in Xenopus egg extracts [17], [18].

Entry into quiescence is controlled by activation of the G0 transcription factors Msn2, Msn4 and Gis1. We show that entry into gametogenesis is not dependent on the G0 transcription factors suggesting that the Rim15-Endosulfine-PP2ACdc55 regulates entry into G0 and gametogenesis by distinct mechanisms. Precisely how PP2ACdc55 prevents entry into gametogenesis and G0 remains unknown. Although the stress-responsive transcription factor Gis1 is hyperphosphorylated in cdc55 mutant cells [35], it is not known whether it is a direct substrate of PP2ACdc55. It is possible that PP2ACdc55 inhibits a factor that is a positive regulator of entry into both gametogenesis and G0 (Figure 8). Alternatively, PP2ACdc55 might inhibit entry into gametogenesis and G0 by dephosphorylation of distinct substrates (Figure 8). Comparing the phosphoproteomes of wild type, cdc55 and igo1Δ igo2Δ cells during entry into gametogenesis and quiescence would be illuminating. Contrary to previous observations [8], we did not find any genetic evidence for endosulfine function in controlling 5′ to 3′ mRNA decay pathway. We suggest that endosulfines regulate entry into both gametogenesis and G0 only via inhibition of PP2ACdc55 (Figure 8).
We have shown that the Rim15-Endosulfine-PP2ACdc55 is required for pre-meiotic autophagy. However the inability to undergo autophagy does not account for the meiotic phenotype of igo1Δ igo2Δ cells as atg1Δ cells (defective in autophagy) enter gametogenesis but fail to undergo any nuclear divisions. Induction of autophagy in igo1Δ igo2Δ cells by rapamycin treatment did not rescue the sporulation defect. Expression of Ime1, the master transcription factor for early meiotic genes, also did not rescue the sporulation defect of igo1Δ igo2Δ cells. These results indicate that the Rim15-Endosulfine-PP2ACdc55 module regulates entry into gametogenesis independently of controlling pre-meiotic autophagy and Ime1 expression. Interestingly Cdc55 has been found to physically interact with Atg1 and Atg18, two proteins required for autophagy, in interactome screens [36], [37]. It will be informative to test whether these interactions are altered during entry into gametogenesis.
Precisely how Rim15/Gwl phosphorylated endosulfine inhibits PP2ACdc55 activity is not known. Structural analyses of PP2ACdc55-endosulfine complex would be illuminating in this respect. It is also important to determine whether endosulfine inhibits PP2ACdc55 activity towards all or only a specific subset of its physiological substrates. Hypomorphic mutations in CDC55 suppress the dyad phenotype of spo12Δ strains (Gary William Kerr and Prakash Arumugam, unpublished observations). This is consistent with antagonistic roles of Spo12 and PP2ACdc55 in FEAR pathway and exit from meiosis I [23], [38], [39]. In contrast to cdc55 hypomorphic alleles, igo1-S64D did not suppress the spo12Δ dyad phenotype (data not shown) suggesting that phosphorylated endosulfine inhibits PP2ACdc55 activity towards only some of its cellular substrates.
Testing whether endosulfines are required for quiescence and gametogenesis in mammalian cells would be very interesting. Notably, expression of endosulfines was first noted in brains [40] and was decreased several fold in patients with neurodegenerative diseases [41]. Given the high conservation of this signalling module, deconstructing its mechanism in budding yeast might give insights into regulation of mitosis in human cells, and vice versa.
Materials and Methods
Yeast strains and plasmids
A complete list of yeast strains and their genotypes can be found in Table S1.
Purification of Igo1 and Rim15
The MBP fused wild-type and mutant forms (S64A and S64D respectively) of Igo1 were expressed and purified from bacteria using the amylose resin (NEB) according to the manufacturer's instructions. Briefly, E. coli cells expressing the MBP fusion proteins were grown overnight and sub-cultured in 2× TY medium containing 0.2% glucose and grown at 37°C to an OD600 nm of ∼0.5. IPTG (Isopropyl β-D-1-thiogalactopyranoside) was added to the culture to a final concentration of 0.2 mM and cells were allowed to grow for another 3 hours at 37°C. Cells were harvested at 4000 rpm for 10 minutes at room temperature and resuspended in 5 ml of buffer A (20 mM Tris-Cl, pH 7.5, 250 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, Roche Complete EDTA-free Protease Inhibitors and 100 mM PMSF) and stored at −20°C after freezing it in liquid N2. Cells were thawed in cold water and lysed by sonication (40 Amp, 5×15 seconds, 1–2 minutes interval between each pulse). Cells were centrifuged at 13,200 rpm for 20 minutes at 4°C and supernatant was transferred to separate tubes. Total amount of protein was measured using the Bradford assay [42]. Equal amount of 50% slurry of Amylose resin (pre-equilibrated in buffer A) was added to the cell lysate. The mixture was incubated for 20 minutes on ice. Beads were collected at low speed (2000 rpm, 1 min, 4°C), washed thrice with 1 ml of buffer A. Proteins bound to beads were recovered by elution with maltose (10 mM) or by adding 2× SDS sample buffer to the beads followed by incubation at 95°C for 5 minutes.
GST-tagged wild-type or mutant forms of Rim15 was purified from yeast cells. Briefly, cells carrying the plasmids (encoding either wild-type or mutant Rim15) were grown to log phase in SD –URA medium at 30°C containing 2% raffinose. After allowing the cultures to reach an OD600 nm ∼1.0, YPG (1% Yeast extract, 2% bactopeptone and 2% galactose) was added to the culture and grown for another 4 hours at 30°C. Cells were collected, washed in cold water and frozen in liquid N2 and stored at −80°C. Cells were thawed , resuspended in lysis buffer (50 mM Tris-Cl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF and Roche Complete EDTA-free Protease Inhibitors) and lysed by using glass beads. Total amount of protein was measured; equal amount of protein was mixed with 150 µl of 50% slurry of GST beads (pre-equilibrated in lysis buffer) and rotated at 4°C for 1 hour. Beads were collected and washed once with lysis buffer, twice with lysis buffer +250 mM NaCl and twice with lysis buffer +500 mM NaCl. The GST-fused proteins were eluted using 10 mM reduced glutathione.
In vitro interaction between Igo1 and Cdc55
Yeast cells expressing Cdc55-TAP were grown to log phase at 30°C in YEPD medium, harvested at 4000 rpm for 5 minutes at 4°C. The cell pellets were stored at −80°C after freezing it in liquid N2. The pellet was thawed, resuspended in yeast lysis buffer (50 mM Tris-Cl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF and Roche Complete EDTA-free Protease Inhibitors) and lysed by using glass beads. Protein concentration was measured by Bradford method. MBP fused wild-type and mutant Igo1 proteins were purified as described above. Equal amounts of bead bound proteins were added to equal amounts of total yeast cell extract. The mixture was incubated on ice for 20 minutes. The beads were collected by centrifugation, washed three times with lysis buffer, resuspended in SDS sample buffer, boiled and run on 10% SDS-PAGE.
GST fused Rim15 and Rim15-kd proteins were purified as described above and the purified protein was used to phosphorylate purified MBP-fused Igo1. The reaction was carried out in kinase buffer (50 mM Tris-Cl, pH7.5, 20 mM MgCl2, 1 mM DTT) containing 1 mM ATP at room temperature for 45 minutes. Beads were collected, mixed with equal amount of yeast cell extract containing Cdc55-TAP and incubated on ice for 20 minutes. The beads were washed three times with lysis buffer, resuspended in 2× SDS sample buffer, boiled and analyzed by Western blotting following SDS-PAGE.
Phosphatase assay
Phosphatase assay was carried out using the Ser/Thr phosphatase assay kit containing a phospho-peptide as a substrate (from Millipore). Briefly, strain expressing TAP-tagged Cdc55 was grown in 1 litre of YEPD medium to log-phase. Cells were harvested, resuspended in 5 ml of yeast lysis buffer and soluble extracts were prepared by bead beating. The extract was mixed with 0.2 ml of IgG sepharose beads (pre-equilibrated in lysis buffer) and the mixture was incubated for 2 hours on a rotary wheel at 4°C. The beads were precipitated, washed 4 times with lysis buffer, once with TEV cleavage buffer (10 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1% NP-40 and 1 mM DTT) and resuspended in 350 µl of TEV cleavage buffer. The bound protein was cleaved and eluted from the beads after incubating overnight at 4°C with 15 U of TEV protease (Invitrogen). The eluted protein was then used for phosphatase assay. Purified Cdc55 was mixed with equal amount (25 µg) of MBP-fused Igo1 or Igo1-S64A or Igo1-S64D or MBP alone and incubated for 20 minutes on ice. The mixture was then incubated with 500 µM of phospho-peptide for 1 hour at 30°C. The reaction was terminated by addition of malachite green solution provided with the kit and absorbance was measured at 620 nm.
In situ immunofluorescence
For in situs, cells from 1 ml of yeast culture were fixed for 15 minutes with 3.7% formaldehyde, pelleted and resuspended in 100 mM K-phosphate buffer (pH 6.4) containing 3.7% formaldehyde and kept overnight on ice. Immunostaining was performed as previously described [23]. The following primary antibodies were used: monoclonal rat anti-α-tubulin 1∶500 (Serotec), monoclonal mouse anti-HA 1∶500 (Covance). Secondary antibodies, pre-absorbed against sera from other species used in labeling, were conjugated with Cy3 or Cy5 (Chemicon) and diluted 1∶500 (Cy3) or 1∶50 (Cy5). DNA was visualized by staining with DAPI.
Microscopy
Images were acquired using a Nikon TE-2000 inverted microscope with a 100×1.49 N.A. objective lens equipped with a Photometrics Coolsnap-HQ2 liquid cooled CCD camera (Photometrics, Tucson, AZ). 16 Z-stacks (spacing = 0.2 µm) Exposure times of 1 second were used for both Cy3 and Cy5, and 0.25 seconds for DAPI. Images were analysed using Metamorph (version 7.5.2.0 MAG Biosystems Software).
Immunoblotting
Whole cell extracts were prepared by cell breakage with glass beads in 20% Trichloroacetic acid. Cell pellets were resuspended in 2× SDS sample buffer, neutralized with 2M Tris base and proteins were denatured by heating the samples at 95°C for 5′. After centrifugation, protein samples were electrophoresed on 10% SDS-PAGE gels. The HA epitope was detected by mouse monoclonal antibody 16B12 at 1∶1000. Goat anti-Cdc5 (Santa Cruz SC-6733) antibody, Goat anti-Cdc28 (Santa Cruz-6708) antibody, mouse anti-Pk (Serotec) antibody, mouse anti-GFP (Roche) antibody and rabbit anti-TAP antibody (Pierce) were all used at 1∶1000 dilution. Myc epitope was detected using the 9E10 antibody (Cambridge Biosciences) at 1∶1000 dilution. Phospho-specific antibody was raised against the phosphorylated synthetic peptide KRKYFDpSGDYALC (pS indicates phosphoserine) by Eurogentec.
For phos-tag gels, TCA extracts were prepared as above and analysed as previously described [25] with a few modifications. Briefly 12.5% polyacrylamide gels were prepared and Phos-Tag (Wako) was added at its final concentration of 50 µM to the separating gel mixture before polymerization. Electrophoresis was performed at a constant current of 30 mA at room temp. After electrophoresis, gels were first soaked in Transfer Buffer (25 mM Tris, 192 mM Glycine, 10% methanol) containing 1 mM EDTA for 20 minutes (2×10 minutes) and then in Transfer Buffer for 30 minutes (3×10 minutes). Electrotransfer onto PVDF membrane was done at a constant voltage of 36 V for 16 hours at 4°C.
Silver staining
After running the protein sample on 10% SDS-PAGE, the gel was washed once with water and then fixed with 100 ml of fixative (50% methanol and 5% acetic acid) for 2 hours. The gel was washed once with 100 ml of 20% ethanol and twice with water. The gel was sensitized with 100 ml of 0.02% sodium thiosulfate for 1 minute and washed immediately with water. The gel was incubated with 100 ml of silver nitrate (0.1% in water) solution containing 20 µl of 37% formaldehyde and kept for 20 minutes at 4°C in dark. The gel was then washed again with water and 100 ml of developing solution (2.5% sodium carbonate 0.0185% formaldehyde) was added. After the bands were visible, 5% acetic acid was added to terminate the reaction.
Analysis of mRNA by quantitative RT-PCR
Total RNA was extracted from yeast cell pellets using the MasterPure Yeast RNA purification kit (Epicentre). RNA integrity was confirmed by agarose gel electrophoretic analysis after denaturation with formamide. Reverse transcription reactions were performed on 0.5 µg of DNAase I-treated RNA with Oligo-dT, using the GoScript Reverse Transcription System (Promega). Quantitative real-time PCR primers for analyzing IME1 and ACT1 were designed as previously described [43] and their specificity was confirmed by melt curve analyses. cDNA reactions were diluted 100-fold, and triplicate quantitative real-time PCRs were performed in a Rotor-Gene Q (Qiagen) using the 2× Rotor-Gene SYBR Green PCR kit (Qiagen). Reactions were analyzed using RotorGene Q software by the comparative CT method, normalizing IME1 mRNA levels against the ACT1 reference gene.
Other techniques
Induction of sporulation was carried out as previously described [44]. To measure sporulation efficiency of yeast strains on solid media, cells were streak purified on YEPD plates. Three single colonies were patched onto YEPD plates. After 24 h of growth at 30°C, cells were patched onto Sporulation plates (0.82% Sodium acetate, 0.19% Potassium chloride, 0.035% Magnesium sulphate, 0.12% Sodium chloride and 1.5% Agar) and incubated at 30°C for 24 h. Sporulation efficiency was assayed using a light microscope. To induce GAL1-IME1 expression, β-estradiol was added to the cultures at the final concentration of 1 µM. The DNA content of sporulating cells was measured by flow cytometry as previously described [45].
Supporting Information
Zdroje
1. BroachJR (2012) Nutritional control of growth and development in yeast. Genetics 192 : 73–105.
2. GrayJV, PetskoGA, JohnstonGC, RingeD, SingerRA, et al. (2004) “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 68 : 187–206.
3. KaeberleinM (2010) Lessons on longevity from budding yeast. Nature 464 : 513–519.
4. MalumbresM, BarbacidM (2001) To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1 : 222–231.
5. De VirgilioC (2012) The essence of yeast quiescence. FEMS Microbiol Rev 36 : 306–339.
6. PedruzziI, DuboulozF, CameroniE, WankeV, RoosenJ, et al. (2003) TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell 12 : 1607–1613.
7. SwinnenE, WankeV, RoosenJ, SmetsB, DuboulozF, et al. (2006) Rim15 and the crossroads of nutrient signalling pathways in Saccharomyces cerevisiae. Cell Div 1 : 3.
8. TalarekN, CameroniE, JaquenoudM, LuoX, BontronS, et al. (2010) Initiation of the TORC1-regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5′-3′ mRNA decay pathway. Mol Cell 38 : 345–355.
9. KassirY, AdirN, Boger-NadjarE, RavivNG, Rubin-BejeranoI, et al. (2003) Transcriptional regulation of meiosis in budding yeast. Int Rev Cytol 224 : 111–171.
10. Rubin-BejeranoI, MandelS, RobzykK, KassirY (1996) Induction of meiosis in Saccharomyces cerevisiae depends on conversion of the transcriptional represssor Ume6 to a positive regulator by its regulated association with the transcriptional activator Ime1. Mol Cell Biol 16 : 2518–2526.
11. KadoshD, StruhlK (1997) Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89 : 365–371.
12. GoldmarkJP, FazzioTG, EstepPW, ChurchGM, TsukiyamaT (2000) The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103 : 423–433.
13. PnueliL, EdryI, CohenM, KassirY (2004) Glucose and nitrogen regulate the switch from histone deacetylation to acetylation for expression of early meiosis-specific genes in budding yeast. Mol Cell Biol 24 : 5197–5208.
14. BowdishKS, YuanHE, MitchellAP (1995) Positive control of yeast meiotic genes by the negative regulator UME6. Mol Cell Biol 15 : 2955–2961.
15. SteberCM, EspositoRE (1995) UME6 is a central component of a developmental regulatory switch controlling meiosis-specific gene expression. Proc Natl Acad Sci U S A 92 : 12490–12494.
16. MalloryMJ, CooperKF, StrichR (2007) Meiosis-specific destruction of the Ume6p repressor by the Cdc20-directed APC/C. Mol Cell 27 : 951–961.
17. Gharbi-AyachiA, LabbeJC, BurgessA, VigneronS, StrubJM, et al. (2010) The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330 : 1673–1677.
18. MochidaS, MaslenSL, SkehelM, HuntT (2010) Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330 : 1670–1673.
19. YuJ, FlemingSL, WilliamsB, WilliamsEV, LiZ, et al. (2004) Greatwall kinase: a nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J Cell Biol 164 : 487–492.
20. RangoneH, WegelE, GattMK, YeungE, FlowersA, et al. (2011) Suppression of scant identifies Endos as a substrate of greatwall kinase and a negative regulator of protein phosphatase 2A in mitosis. PLoS Genet 7: e1002225.
21. Von StetinaJR, TranguchS, DeySK, LeeLA, ChaB, et al. (2008) alpha-Endosulfine is a conserved protein required for oocyte meiotic maturation in Drosophila. Development 135 : 3697–3706.
22. VidanS, MitchellAP (1997) Stimulation of yeast meiotic gene expression by the glucose-repressible protein kinase Rim15p. Mol Cell Biol 17 : 2688–2697.
23. KerrGW, SarkarS, TibblesKL, PetronczkiM, MillarJB, et al. (2011) Meiotic nuclear divisions in budding yeast require PP2A(Cdc55)-mediated antagonism of Net1 phosphorylation by Cdk. J Cell Biol 193 : 1157–1166.
24. JuanesMA, KhoueiryR, KupkaT, CastroA, MudrakI, et al. (2013) Budding yeast greatwall and endosulfines control activity and spatial regulation of PP2A(Cdc55) for timely mitotic progression. PLoS Genet 9: e1003575.
25. Kinoshita-KikutaE, AokiY, KinoshitaE, KoikeT (2007) Label-free kinase profiling using phosphate affinity polyacrylamide gel electrophoresis. Mol Cell Proteomics 6 : 356–366.
26. ShahJC, ClancyMJ (1992) IME4, a gene that mediates MAT and nutritional control of meiosis in Saccharomyces cerevisiae. Mol Cell Biol 12 : 1078–1086.
27. ChuS, DeRisiJ, EisenM, MulhollandJ, BotsteinD, et al. (1998) The transcriptional program of sporulation in budding yeast. Science 282 : 699–705.
28. KassirY, GranotD, SimchenG (1988) IME1, a positive regulator gene of meiosis in S. cerevisiae. Cell 52 : 853–862.
29. BenjaminKR, ZhangC, ShokatKM, HerskowitzI (2003) Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev 17 : 1524–1539.
30. YorimitsuT, ZamanS, BroachJR, KlionskyDJ (2007) Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell 18 : 4180–4189.
31. TsukadaM, OhsumiY (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333 : 169–174.
32. KlionskyDJ, CuervoAM, SeglenPO (2007) Methods for monitoring autophagy from yeast to human. Autophagy 3 : 181–206.
33. ZhengXF, SchreiberSL (1997) Target of rapamycin proteins and their kinase activities are required for meiosis. Proc Natl Acad Sci U S A 94 : 3070–3075.
34. XiaoY, MitchellAP (2000) Shared roles of yeast glycogen synthase kinase 3 family members in nitrogen-responsive phosphorylation of meiotic regulator Ume6p. Mol Cell Biol 20 : 5447–5453.
35. BontronS, JaquenoudM, VagaS, TalarekN, BodenmillerB, et al. (2013) Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep 3 : 16–22.
36. PtacekJ, DevganG, MichaudG, ZhuH, ZhuX, et al. (2005) Global analysis of protein phosphorylation in yeast. Nature 438 : 679–684.
37. HoY, GruhlerA, HeilbutA, BaderGD, MooreL, et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415 : 180–183.
38. BizzariF, MarstonAL (2011) Cdc55 coordinates spindle assembly and chromosome disjunction during meiosis. J Cell Biol 193 : 1213–1228.
39. RockJM, AmonA (2009) The FEAR network. Curr Biol 19: R1063–1068.
40. Virsolvy-VergineA, LerayH, KurokiS, LupoB, DufourM, et al. (1992) Endosulfine, an endogenous peptidic ligand for the sulfonylurea receptor: purification and partial characterization from ovine brain. Proc Natl Acad Sci U S A 89 : 6629–6633.
41. KimSH, LubecG (2001) Brain alpha-endosulfine is manifold decreased in brains from patients with Alzheimer's disease: a tentative marker and drug target? Neurosci Lett 310 : 77–80.
42. BradfordMM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72 : 248–254.
43. KahanaS, PnueliL, KainthP, SassiHE, AndrewsB, et al. (2010) Functional dissection of IME1 transcription using quantitative promoter-reporter screening. Genetics 186 : 829–841.
44. KiburzBM, AmonA, MarstonAL (2008) Shugoshin promotes sister kinetochore biorientation in Saccharomyces cerevisiae. Mol Biol Cell 19 : 1199–1209.
45. EpsteinCB, CrossFR (1992) CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev 6 : 1695–1706.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 6
Nejčtenější v tomto čísle
- Early Back-to-Africa Migration into the Horn of Africa
- PINK1-Mediated Phosphorylation of Parkin Boosts Parkin Activity in
- OsHUS1 Facilitates Accurate Meiotic Recombination in Rice
- Ancient DNA Analysis of 8000 B.C. Near Eastern Farmers Supports an Early Neolithic Pioneer Maritime Colonization of Mainland Europe through Cyprus and the Aegean Islands