
Stress-Induced Nuclear RNA Degradation Pathways Regulate Yeast Bromodomain Factor 2 to Promote Cell Survival
Cells adapt to changes in the environment through modulating gene expression at both the RNA and protein levels. RNA degradation plays a central role in this adaption response, by controlling the stability of specific mRNAs to optimize protein production in different conditions. In this study, we show that the gene encoding Bromodomain Factor 2 (BDF2) is tightly regulated according to environmental conditions by two distinct RNA degradation mechanisms. We show that these RNA degradation pathways are critical for cell growth in specific conditions. Our study suggests that environmental modulation of nuclear RNA degradation pathways is a previously unappreciated aspect of gene expression control.
Published in the journal:
. PLoS Genet 10(9): e32767. doi:10.1371/journal.pgen.1004661
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004661
Summary
Cells adapt to changes in the environment through modulating gene expression at both the RNA and protein levels. RNA degradation plays a central role in this adaption response, by controlling the stability of specific mRNAs to optimize protein production in different conditions. In this study, we show that the gene encoding Bromodomain Factor 2 (BDF2) is tightly regulated according to environmental conditions by two distinct RNA degradation mechanisms. We show that these RNA degradation pathways are critical for cell growth in specific conditions. Our study suggests that environmental modulation of nuclear RNA degradation pathways is a previously unappreciated aspect of gene expression control.
Introduction
DNA in eukaryotes is wrapped around histone octamers to form nucleosomes [1]. The tails of the histone proteins are subject to a diverse set of chemical modifications, including acetylation, phosphorylation, methylation, and ubiquitination, impacting the majority of DNA-based processes, including transcription, heterochromatin formation, DNA replication, and DNA recombination and repair [2], [3]. Non-histone proteins recognize specific tail modifications to mediate the downstream effects [4]. Histone lysine acetylation, one of the best-studied modifications, has important roles in transcription activation, DNA repair and heterochromatin formation [5]. Histone acetylation can increase accessibility of DNA by weakening the interaction between the positively charged histone tail and the nucleosomal DNA [6]. Histone acetylation can also recruit proteins containing bromodomains, which are evolutionarily conserved motifs that recognize acetyl-lysines and play an important role in anchoring chromatin-associated complexes to the nucleosome [7].
S. cerevisiae bromodomain factors 1 and 2 (Bdf1p and Bdf2p) localize throughout the genome at loci enriched for acetylated histones 3 and 4 [5]; [8], where they function in various aspects of transcription initiation and chromatin remodeling [9], as well as protection of euchromatin against heterochromatin spreading [10]. While the bdf1Δbdf2Δ double deletion is lethal, both single deletion mutants are viable, indicating that there is at least partial functional redundancy between the two paralogs [11]. In wild-type cells, Bdf1p is present at nearly 3-fold the levels of Bdf2p [12], and each occupies distinct genomic locations [8]. Cells lacking BDF1 (bdf1Δ) show an upregulation of Bdf2 protein (Bdf2p) levels [13] and a redistribution of Bdf2p to the acetylated histones at genomic loci normally bound by Bdf1p [8]. The deletion of BDF2 affects expression of less than 0.05% of the transcriptome in normal conditions, while the deletion of BDF1 results in a greater than 2-fold change in the levels of ∼15% of all expressed transcripts [10].
In addition to activating TFIID-dependent transcription [11], Bdf1p is part of the SWR1-C chromatin-remodeling complex responsible for replacing histone H2A with the variant H2AZ [14]. The NuA4 histone acetyltransferase acts upstream, depositing an acetyl group on the histone H4, resulting in the recruitment of Bdf1p and SWR1-C [15]. Recent work has revealed that mutations in NuA4 subunits, Bdf1p, SWR1-C, or H2AZ render cells hypersensitive to DNA damage agents, suggesting that histone acetylation-mediated H2AZ deposition plays crucial roles in the DNA damage response [16]; [17]. Furthermore, cells lacking BDF1 display a growth defect in normal conditions and hypersensitivity to a wide range of stress conditions [10], [16], [18]. While cells lacking BDF2 exhibit no growth defect in normal conditions, they display decreased resistance to the DNA damage agents camptothecin and bleomycin [19]. Additionally, recent studies in fission yeast have demonstrated an important role for Bdf2p in the S-phase stress response and the establishment of heterochromatin boundaries [17], [20].
Despite detailed biochemical and functional analysis of the Bdf1p and Bdf2p proteins, little is known about how these factors are regulated in specific environmental conditions. A recent study demonstrated a novel role for the spliceosome in degrading the BDF2 mRNA, through coupling the splicing of an intron encoded within the BDF2 ORF to decay by nuclear exonucleases [13]. Spliceosome-mediated decay (SMD) of BDF2 mRNA was found to be dependent on Bdf1p-mediated recruitment of the spliceosome to the BDF2 locus, and its proposed biological function was to maintain the homeostasis of Bdf2p levels in normal growth conditions. Here we demonstrate that BDF2 expression is subject to extensive control by two distinct nuclear RNA degradation pathways, and that each pathway is regulated in different stress conditions to maintain cellular fitness. These results also establish that tight control of the levels of Bromodomain factors is important for cell survival in specific environmental conditions, underscoring their importance for regulating gene expression during stress.
Results
The S.cerevisiae RNase III Rnt1p cleaves the BDF2 mRNA in vivo and in vitro
Recent studies have revealed the presence of cryptic splicing signals within S.cerevisiae transcripts [13], [21], [22]. In contrast to the canonical role of the spliceosome in promoting gene expression, the usage of these splice sites by the spliceosome promotes degradation of these transcripts [13]. We previously showed that the yeast orthologue of RNase III (Rnt1p) cleaves stem-loop structures within introns of various transcripts to regulate the levels of the spliced transcripts [23]. To test whether these recently identified cryptic introns contain cleavage signals for Rnt1p, we performed a computational RNA secondary structure screen for these intron-containing transcripts using m-Fold [24]. We analyzed the predicted secondary structures for canonical Rnt1p cleavage signals, which consist of double-stranded RNA (dsRNA) hairpin structures capped by NGNN or AAGU tetraloops [25], [26]. This analysis revealed the presence of a canonical Rnt1p stem-loop within the open reading frame (ORF) of the yeast gene encoding bromodomain factor 2 (BDF2) (Fig. 1A). This 40 base-long stem-loop is situated within the ORF-embedded intron of the BDF2 mRNA, 1165 nucleotides downstream of the 5′-SS and 298 nucleotides upstream of the annotated 3′-SS ([13], Fig. 1B). The existence of this stem-loop structure was confirmed in vivo in a transcriptome-wide study of RNA secondary structure that employed dimethyl sulfate (DMS) modification of single-stranded RNA coupled to deep sequencing [27]. Consistent with its potential degradation by Rnt1p, microarray analysis showed that the BDF2 transcript is upregulated 2-fold in cells lacking Rnt1p (rnt1Δ; [28]). Furthermore, recent studies have found that BDF2 mRNA exhibits a high transcription rate in conjunction with a short half-life that is similar to the histone mRNAs, suggesting that RNA degradation may play a major role in its regulation ([29], see Discussion). These initial observations suggested that BDF2 expression is highly regulated at the level of RNA degradation and raised the possibility that Rnt1p may directly control BDF2 expression.

To investigate if the upregulation of BDF2 mRNA in rnt1Δ is specifically due to the loss of Rnt1p catalytic activity, we compared BDF2 transcripts levels in wild-type (WT) and rnt1Δ strains, and in a strain expressing the catalytically inactive rnt1 E320K mutant (Fig. 1C, lanes 1–2, 7–8). This analysis revealed a substantial increase of BDF2 mRNA upon the loss of Rnt1p catalytic function. To formally demonstrate cleavage of BDF2 by Rnt1p in vivo, we analyzed BDF2 species in strains carrying mutations in various exonucleases in order to stabilize the 3′ product of Rnt1p cleavage, which is normally subject to decay by 5′-3′ exonucleases due to the presence of an unprotected 5′-end with an exposed monophosphate. The xrn1Δ background was used to inactivate the major pathway of cytoplasmic 5′ to 3′ degradation. To inactivate the nuclear 5′ to 3′ exonuclease Rat1p, we used the temperature-sensitive rat1-1 mutant. In a parallel approach, we deleted the MET22 gene in the xrn1Δ background. Upon a shift to medium lacking methionine, this strain accumulates 3′-phosphoadenosine-5′-phosphate (pAp), a metabolite that inhibits cellular 5′ to 3′ exonuclease activities [30]. We utilized a probe hybridizing to the 3′UTR of BDF2 mRNA and found that both methods of inactivating the 5′ to 3′ exonucleases revealed a substantial accumulation of a species migrating faster than the full-length BDF2 mRNA (Fig. 1C, lanes 3,4,9,10). This species was no longer detected upon disruption of RNT1 in these strains (Fig. 1C, lanes 5,6,11,12) suggesting that it is the downstream (3′) product of Rnt1p cleavage.
We detected a slower migrating form of BDF2 mRNA in samples from rnt1 mutant backgrounds (Fig1C, asterisk). Previous studies had identified two different transcription start sites for BDF2 corresponding to two distinct nucleosome free regions (NFRs) [31]–[33]. To confirm that two different forms of BDF2 mRNA differing by their 5′-UTR were present, we performed prolonged electrophoresis in high percentage gels (2% agarose). This enabled the detection of two closely migrating forms of the BDF2 mRNA (denoted BDF2-L and BDF2-S), the longer of which was up-regulated in the met22Δ background after a shift to medium lacking methionine (Fig. 1D, upper panel, lanes 4,6). This analysis confirmed that BDF2-S is the predominant form in wild-type cells under normal conditions. We subsequently used probes targeted to the 5′UTR of the transcript arising from the upstream transcription start site (discussed below), to demonstrate that BDF2-L is indeed a 5′-extended form of BDF2-S.
We next tested whether Rnt1p could directly cleave the BDF2 mRNA in vitro by incubating recombinant Rnt1p enzyme with total RNAs extracted from a strain expressing the catalytic mutant rnt1 E320K (Fig. 1D, lanes 7–8). The 3′-UTR probe detected a single band from the in vitro cleavage reaction, and this band co-migrated with the band stabilized in vivo in the xrn1Δ background (Fig. 1D, upper panel, lanes 3,4, and 8). Next, we utilized a 5′UTR probe to detect the upstream product of Rnt1p cleavage. This probe was designed to bind predominantly to BDF2-L, and with only a short stretch of complementarity to the 5′-end of BDF2-S. This resulted in an enhanced signal for BDF2-L relative to BDF2-S, consistent with BDF2-L having a 5′-extension. Furthermore, in vitro cleavage of total RNAs by Rnt1p gave rise to two different 5′-products, the longer of which was also observed as an in vivo cleavage product in the xrn1Δmet22Δ background (Fig. 1D, lower panel, lane 4). This was surprising as the 5′ cleavage product generated by Rnt1p exhibits an exposed 3′-OH, lacks a stabilizing poly(A) tail, and is subject to degradation by 3′ to 5′ exonucleases [23], [34]. The stabilization of this species upon depletion of 5′ to 3′ exonuclease activity may be an indirect consequence of 3′ to 5′ exonucleases being saturated by RNA substrates in these conditions. We tested the ability of Rnt1p to cleave the mRNA encoding the BDF2 paralog (BDF1), and other targets of spliceosome-mediated decay [13] identified in our computational screen, but found no evidence for in vivo or in vitro cleavage of these transcripts (Fig. S1).
To precisely identify the Rnt1p cleavage site on BDF2 mRNA, we performed primer extension analysis using a primer hybridizing 70 nucleotides downstream of the stem-loop structure. We detected two primer extension stops from in vitro cleavage with recombinant Rnt1p (Fig. 1E), corresponding to 14 and 15 bases below the tetraloop for the 5′ and 3′ cleavage sites, respectively (Fig. 1A and 1E, arrows 1 and 2). Primer extension analysis using RNAs extracted from the 5′ to 3′ exonuclease mutants detected the same site in vivo for the 5′ cleavage, and a 3′ cleavage site 17 nucleotides below the tetraloop (Fig. 1E lane 3 and 1A, arrow 3). The distances from the cleavage site to the tetraloop are consistent with the known enzymatic properties of the enzyme in vitro and in vivo ([25] [35]). Importantly, these cleavage sites were no longer detected when Rnt1p was inactivated in the 5′ to 3′ exonuclease mutants (Fig. 1E, lane 4), showing that they are dependent on Rnt1p activity.
To confirm that this stem loop structure is required for Rnt1p cleavage, we constructed a plasmid-borne version of BDF2 fused at the C-terminus to GFP, and disrupted the top six base pairs with synonymous mutations (Fig. 1A). We transformed plasmids expressing either the wild-type BDF2 mRNA or the stem mutant into the xrn1Δ rat1-1 background and tested for the presence of an Rnt1p cleavage product from these constructs. We utilized a GFP probe to avoid hybridization with endogenous BDF2 mRNA, and detected the Rnt1p degradation intermediate from the WT BDF2 construct, which was not observed after disruption of the Rnt1p stem loop (stem mutant, Fig. 1F). These results confirmed that Rnt1p degrades BDF2 mRNA in vivo by recognition of a canonical Rnt1p-target stem loop.
Mutation of the Rnt1p-target stem loop upregulates BDF2-S and stabilizes the intron-exon 2 degradation intermediate of spliceosome-mediated decay (SMD)
A previous study demonstrated that cytoplasmic nonsense-mediated decay (NMD) and nuclear degradation systems can have partially redundant roles in the degradation of various unspliced pre-mRNAs, such that only upon inactivation of both systems does substantial accumulation of these species occur [36]. We found a slight increase in the BDF2-L form upon deletion of the gene coding for the NMD helicase Upf1p (upf1Δ; Fig. 1G), consistent with the presence of multiple upstream ORFs in the extended 5′ UTR that would elicit recognition of a premature-termination codon and NMD in accordance with the faux-3′ UTR model [37]. To test whether NMD cooperates with SMD or Rnt1p in the regulation of BDF2 mRNA, we mutated the BDF2 5′-splice site (5′-SS) or the Rnt1p-target stem loop at the BDF2 chromosomal copy in otherwise wild-type or upf1Δ backgrounds (Fig. 1G). During the course of this study, RT-PCR analysis of full-length BDF2 mRNA and its spliced products generated by SMD revealed that mutating the 5′-side of the top six base pairs of the Rnt1p-target stem loop from UUCAUC to CAGCAG inadvertently created a new 3′ splice site, due to the introduction of two YAG sequences proximal to a polypyrimidine tract, (UUC)3 (Fig. S2). As an alternative approach to inactivating Rnt1p cleavage in cis, we deleted twelve nucleotides at the top of the stem-loop structure, resulting in the in-frame removal of four codons (UCA-UCU-GAU-GAU) encoding an SSDD amino acid sequence (red letters, Fig. 1A). Both mutations of the Rnt1p stem loop resulted in slightly increased BDF2-S mRNA levels (Fig. 1F). These stem-loop mutations did not noticeably affect BDF2-L levels in the upf1Δ background, suggesting that NMD is the primary degradation system regulating BDF2-L. Strikingly, we observed a band migrating faster than BDF2-S in the stem-loop mutants. This band was also detectable at low levels in WT cells, but not in the 5′-SS mutant. This band matches the size expected for the linearized intron–exon2 product of SMD, which would be generated by debranching of the lariat intermediate generated by the first splicing step of the BDF2 mRNA [13], [21]. To definitively demonstrate the identity of this band as the intron-exon2 species, we performed a probe-walking experiment with probes hybridizing to exon1, the intron, and exon2 (Fig. S3). Only the intronic and exon2 probes detected this band, while all three probes detected the full-length BDF2 mRNA (Fig. S3). It is remarkable that the intron-exon2 species is readily detectable in the absence of exonuclease mutations, as it contains an exposed 5′-phosphate and should be subject to rapid 5′ to 3′ decay. The substantial accumulation of this species in stem-loop mutants suggests that a large fraction of BDF2 mRNA is subject to SMD in the absence of Rnt1p cleavage.
Rrp6 and the nuclear retention factors Mlp1/2 regulate BDF2-S
We next tested whether the nuclear exosome co-factor Rrp6p and NMD have functional overlap in regulating the BDF2 mRNA. We found that while both repress BDF2-L, there is no additive increase upon inactivation of both degradation systems (Fig. S4A, right panel). The loss of Rrp6p also resulted in increased BDF2-S levels, as well as in stabilization of the SMD and Rnt1p cleavage products (Fig. S4A, left panel). The Mlp proteins have previously been implicated in the nuclear retention of unspliced mRNAs [38]. To test the hypothesis that these proteins mediate the nuclear retention of the unspliced-like full-length BDF2 mRNA, we inactivated Mlp1p and Mlp2p, both of which function to prevent the export of intron-containing mRNA from the nucleus in a manner that depends on 5′-splice site recognition [38] [36]. The mlp1Δmlp2Δ strain exhibited an upregulation of BDF2-S, suggesting that the leakage of BDF2-S out of the nucleus results in diminished nuclear decay (Fig. S4B). Mlp1/2 had no effect on the NMD-sensitive BDF2-L transcript, consistent with more efficient export and cytoplasmic degradation of this species. In the rrp6Δ background, the inactivation of Mlp1/2 factors did not affect the levels of full-length BDF2 mRNA or its degradation intermediates.
Inactivation of Rnt1p-mediated decay (RMD) of BDF2 rescues the salt sensitivity of the bdf1Δ mutant and its inability to grow on non-fermentable carbon
In non-fermentable carbon sources or hyperosmotic stress conditions, BDF2 levels are limiting for growth in bdf1Δ cells, as overexpression of BDF2 mRNA suppresses bdf1Δ growth defects in a dose-dependent manner [18]. The presence of two distinct nuclear degradation systems acting on the BDF2 mRNA prompted us to investigate the physiological importance of these degradation pathways in these environmental conditions. A previous study found that blocking SMD of BDF2 mRNA by mutating the 5′-SS improved the growth of bdf1Δ in the presence of 0.6 M NaCl [13]. To compare the consequences of SMD inactivation and RMD inactivation on bdf1Δ growth phenotypes, we deleted BDF1 in the BDF2 5′-SS mutant (bdf2 5′SS mut), the stem-loop mutants (bdf2 SLΔ and bdf2 stem), and the double mutant (bdf2 5′SS mut+stem). In synthetic minimal medium (SDC), but not rich medium (YPD), we found that inactivation of RMD by the stem-loop deletion (bdf2 SLΔ) resulted in a modest growth enhancement over wild-type BDF2 (Fig. 2A, top panel). By contrast, the 5′-SS mutant grew at the same rate as the WT in both of these conditions, consistent with previous observations using plasmid-borne BDF2 [13].

At 0.2 M and 0.4 M NaCl, the inactivation of RMD resulted in a slight rescue of the bdf1Δ growth defect, with SMD inactivation showing no effect (Fig. 2A, top panel). The simultaneous inactivation of SMD and RMD showed no additional effect over the stem loop deletion strain in these conditions. Surprisingly, at 0.6 M NaCl, the inactivation of RMD resulted in a substantial growth rescue (Fig. 2A, middle panel). The stem loop mutation (which incidentally introduced an additional 3′-SS) resulted in a reduced rescue compared to the stem loop deletion. Inactivating the 5′-SS in the stem mutant resulted in a similar rescue to that of the stem loop deletion, suggesting that the presence of the new 3′-SS site enhanced the SMD of BDF2 mRNA in these conditions. Importantly, the growth rescue observed upon the deletion of the stem-loop confirmed that the removal of the SSDD amino acid sequence from the Bdf2p protein did not perturb protein function, validating the use of this mutant to assay the phenotypic consequences of increased BDF2 expression due to RMD inactivation.
We next tested different carbon sources, and found that inactivation of RMD, but not SMD, restored the growth of bdf1Δ cells on the non-fermentable carbon sources ethanol and glycerol (Fig. 2B, top panel). Furthermore, inactivation of RMD conferred a substantial growth rescue in galactose, for which optimal growth requires simultaneous respiration and fermentation [39]. By contrast, disabling the 5′-SS failed to rescue growth in galactose, and slightly exacerbated the growth defect of bdf1Δ. It is possible that regulation by SMD occurs at a different phase of the cell cycle than RMD, and that inactivating SMD results in toxic levels of Bdf2p at a particular stage in the presence of galactose. On the other hand, RMD inactivation might increase levels of Bdf2p at a stage where more Bdf2p is beneficial. Overall, these results suggest that the underlying basis for bdf1Δ growth defects in hyper-osmotic stress and respiratory conditions is the degradation of BDF2 mRNA by RNase III.
Simultaneous inactivation of the RMD and SMD pathways of BDF2 suppresses the hypersensitivity of the bdf1Δ mutant to elevated temperatures, high salt and lithium stress
While the inactivation of BDF2 RMD rescued bdf1Δ growth defects in moderate levels of salt stress, we found that only the simultaneous inactivation of RMD and SMD allowed the bdf1Δ strain to grow at elevated temperatures in non-fermentable carbon (34°C and 37°C), or in high salt stress (1.2M NaCl) (Fig. 2B). We also tested lithium chloride stress, as it confers not only osmotic stress but also lithium ion toxicity. Strains lacking Bdf1p are sensitive to 0.1 M lithium chloride and hyper-accumulate lithium ions [40], [41]. The simultaneous inactivation of RMD and SMD of BDF2 mRNA enabled growth of bdf1Δ at 0.3 M lithium chloride (Fig. 2B, bottom right). These results suggest that in these conditions, both SMD and RMD play significant roles in limiting BDF2 expression, as inactivation of either degradation pathway alone is insufficient to enhance the growth of bdf1Δ cells. Furthermore, these results suggested that either bdf1Δ cells require more BDF2 mRNA to survive in stress conditions than is required for growth in normal conditions, or that these stress conditions enhance the activity of the nuclear degradation pathways of BDF2 mRNA, resulting in reduced levels of BDF2 mRNA relative to normal growth conditions. As cells lacking BDF1 are strictly dependent on BDF2 for survival, we hypothesized that stress-induced degradation of BDF2 mRNA might be responsible for the phenotypes detected in the bdf1Δ strain.
RMD limits BDF2 expression in osmotic stress
The previous growth assays suggest that RMD plays an important role in regulating BDF2 expression particularly in salt stress conditions. To understand how BDF2 expression behaves in these conditions, we monitored BDF2 mRNA levels in wild-type cells after a shift from normal to high salt medium. Within 8 minutes of a shift to high osmolarity, we observed a marked drop in the levels of the BDF2 mRNA (Fig. 3A). By contrast, BDF1 and RNT1 mRNA levels increased in the first 8 minutes of osmotic shock. After 1 hour, BDF2 levels were still repressed, while BDF1 and RNT1 levels had returned to pre-treatment levels. To assess BDF2 expression and the levels of its RMD degradation intermediates after prolonged growth in high salt conditions, we monitored the levels of BDF2 mRNA in wild-type and rrp6Δ strains during steady state growth in either normal medium or high salt (Fig. 3B). To test whether the Mlp1/2 factors might mediate increased nuclear retention and RMD of BDF2 mRNA during osmotic stress, we also examined the mlp1Δmlp2Δ and mlp1Δmlp2Δrrp6Δ strains. Under normal growth conditions, we observed a stabilization of the 5′ product of RMD in the rrp6Δ strain, as expected (Fig. 3B). Remarkably, full-length BDF2 was undetectable under steady-state growth in high-salt conditions, while the RMD cleavage products persisted. This suggested that transcription of the BDF2 gene continues after prolonged exposure to osmotic stress, but that the transcripts are continuously degraded by SMD and RMD such that full-length BDF2 mRNA is no longer detected. Inactivation of the Mlp1/2 factors had no effect on the levels of full-length BDF2 mRNA or its degradation intermediates, suggesting that the increased targeting of BDF2 mRNA by Rnt1p is independent of these nuclear retention factors.
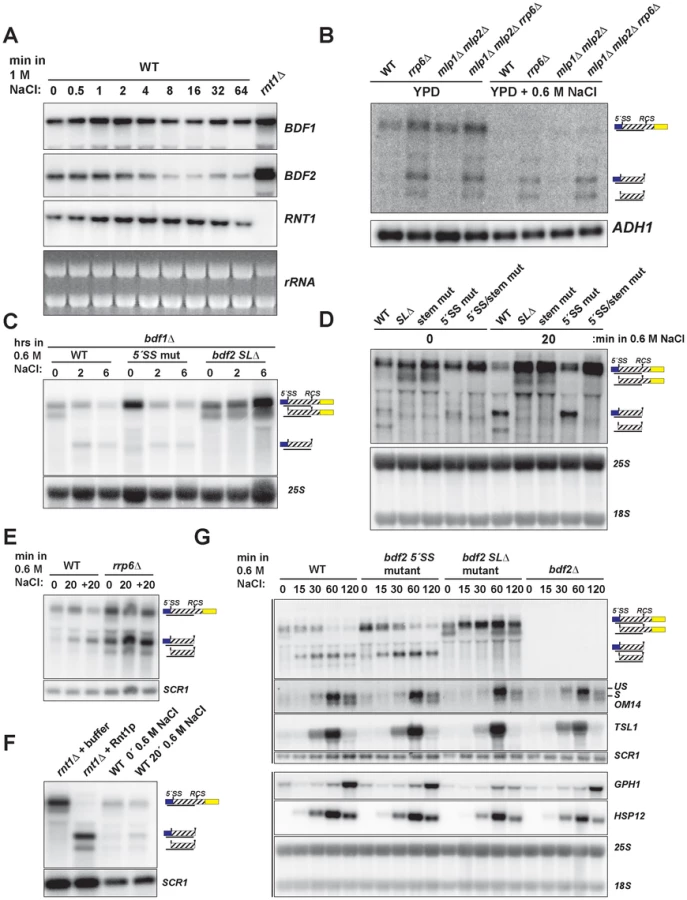
To test whether the salt sensitivity of bdf1Δ is a consequence of increased RMD of BDF2 mRNA, we examined BDF2 mRNA levels in the bdf1Δ background after a prolonged shift to high salt conditions. This revealed a significant reduction of BDF2 mRNA levels for the WT BDF2 and the 5′-SS mutant after six hours (Fig. 3C). Notably, we detected the intron-exon2 product in BDF2 WT and the stem loop deletion strains in normal medium, as observed previously for mutations inactivating RMD in an otherwise wild-type BDF1 background (Fig. 1F). This demonstrates that in normal growth conditions, spliceosome-mediated decay degrades a large fraction of BDF2 mRNA in a manner that does not require Bdf1p, contrary to previous observations [13]. By two hours of shift to high salt, this SMD degradation intermediate disappeared and the RMD degradation intermediate was now detected in both the WT and the 5′-SS mutants. Strikingly, BDF2 mRNA levels in the stem-loop deletion strain actually increased during this time course (Fig. 3C). This demonstrates that BDF2 mRNA repression during osmotic stress is due to increased RMD rather than transcriptional repression. Furthermore, this confirms that bdf1Δ-salt sensitivity is a consequence of increased RMD and loss of BDF2 full-length mRNA in osmotic stress, rather than a specific requirement for Bdf1p in activating osmotic stress response genes.
To examine how RMD affects the regulation of BDF2 mRNA in the initial phase of the salt stress response, we monitored the levels of full-length BDF2 mRNA and the SMD and RMD degradation products in the wild-type strain and strains carrying the 5′-SS and stem loop mutations after 20 minutes of osmotic shock (Fig. 3D). In normal medium, we detected the intron-exon2 product resulting from SMD in the stem-loop mutants. Significantly, we detected the accumulation of the RMD cleavage intermediate in WT and the 5′-SS mutant after 20 minutes in high salt (Fig. 3D). Consistent with the results from the bdf1Δ background, full-length BDF2 mRNA increased in the stem-loop mutants, but not the 5′-SS mutant, suggesting that osmotic stress-induced transcripts are normally rapidly degraded by RMD. A previous study had found that the transcription rate of BDF2 mRNA increases 10 minutes after osmotic shock in 0.4 M NaCl [42]. Our data show that this increase in transcription rate is counter-acted by an even greater increase in BDF2 mRNA degradation through the RMD pathway.
To confirm that the increased abundance of the RMD cleavage intermediate is not due to a decrease in its degradation rate, we monitored levels of the cleavage product in the rrp6Δ strain, as the nuclear exosome is the primary factor involved in the decay of this cleavage intermediate. The RMD cleavage intermediate increased substantially after 20 minutes in osmotic shock in this context, showing that the increased abundance of this cleavage intermediate is due to hyper-activation of RMD (Fig. 3E). We confirmed that the in vivo cleavage bands induced in salt stress match the migration of the bands produced by in vitro Rnt1p cleavage (Fig. 3F). These results indicate that yeast RNase III plays a hitherto unappreciated role in the regulation of gene expression during osmotic stress.
We next tested the impact of an extended shift in 0.6 M NaCl on BDF2 mRNA levels, and found a substantial reduction of full-length BDF2 transcripts by one hour in both the WT and 5′-SS mutant (Fig. 3G). Strikingly, the bdf2 SLΔ mutant exhibited an increase in full-length BDF2 mRNA levels throughout the first hour of high salt exposure, consistent with the increased transcription rate reported previously [42]. Interestingly, the SMD intron-exon2 product, present in bdf2 SLΔ in normal conditions, was absent after 15 minutes of salt stress (Fig. 3G). By one hour, this product re-appeared, suggesting that osmotic stress results in a transient deactivation of SMD (Fig. 3G). The dramatic decrease in BDF2 mRNA levels detected in both the WT and the 5′-SS mutant, but not in the bdf2 SLΔ mutant, demonstrates that RMD is the primary mechanism controlling BDF2 mRNA expression in salt shock conditions. The increase in RMD activity for the BDF2 mRNA is not simply a consequence of decreased competition by SMD, resulting in increased flux of BDF2 mRNA through RMD, because the 5′-SS mutation in normal salt conditions does not phenocopy the RMD hyper-activation that occurs during salt stress (Fig. 3G). Indeed, the BDF2 mRNA harboring the 5′-SS mutation displays a similar profile in salt stress as WT BDF2, where by 60 minutes the RMD degradation product is present at higher levels than full-length BDF2 mRNA (Fig. 3G). Overall, these data confirm that RMD of BDF2 mRNA is hyper-activated in salt stress.
Next we investigated the impact of BDF2 RMD on Bdf2p-regulated genes during osmotic stress. A previous study found that the loss of Bdf2p resulted in the upregulation of 20 transcripts at least 2-fold [10]. These transcripts were enriched in stress-responsive genes, including genes involved in carbohydrate metabolism and the heat shock response. We hypothesized that Bdf2p might normally repress these genes, and that an excess of Bdf2p, due to the inactivation of RMD, might inhibit the activation of these transcripts in response to osmotic stress. To test this hypothesis, we monitored the expression of various genes that were reported to be upregulated in bdf2Δ (Fig. 3G and S5). We found a significant decrease in the induction of the GPH1 transcript in bdf2 SLΔ mutant after two hours, but not in the WT, 5′-SS mutant, or bdf2Δ strains. GPH1 encodes glycogen phosphorylase and is critical for preventing the over-accumulation of glycogen during various stress responses and in stationary phase [43]. Our data suggests that during osmotic stress, Rnt1p must repress BDF2 mRNA levels in order for cells to fully induce GPH1 expression.
Inactivating the SMD of BDF2 sensitizes the bdf1Δ mutant to DNA replication fork stress-inducing agents hydroxyurea and camptothecin
Cells lacking BDF1 are hypersensitive to the DNA damage agents methyl methanesulfonate (MMS) and hydroxyurea (HU) [16]; [44]. The broad range of phenotypes rescued by inactivating RMD and SMD of BDF2 mRNA led us to expect a rescue of bdf1Δ growth defects in DNA damaging conditions as well. Startlingly, inactivation of BDF2 SMD resulted in enhanced toxicity of HU and camptothecin (CPT) for the bdf1Δ strain (Fig. 4A). Both HU and CPT result in the destabilization of replication forks and subsequent induction of double-strand breaks (DSBs) through different mechanisms [45], [46]. Interestingly, the stem loop mutation resulted in a slight rescue of the growth defect conferred by the 5′-SS mutation in 20 µM CPT, and to a lesser extent in 20 mM HU (Fig. 4A). It is possible that SMD and RMD predominate at different stages of the cell cycle, and in the presence of DNA replication stress, additional molecules of Bdf2p gained at one stage by inactivating RMD can compensate for the increased toxicity of excess Bdf2p due to SMD inactivation at another stage. Significantly, these results reveal that SMD plays an important role in the context of DNA replication stress. The lack of an effect from RMD inactivation in 50 mM HU led us to hypothesize that RMD activity on BDF2 mRNA might be decreased in these conditions, and that the flux of BDF2 mRNA degradation through SMD or RMD could be dependent upon specific environmental conditions.

SMD predominates over RMD of BDF2 mRNA in HU-induced DNA damage
The toxic effect of HU observed for the bdf1Δ strain harboring the 5′-SS mutation suggested that SMD protects against HU toxicity by preventing an excess accumulation of Bdf2p protein. Furthermore, the lack of toxicity of the stem-loop mutations indicated that SMD of the BDF2 mRNA might predominate over RMD in DNA damage conditions. Bdf2p protein levels were reported to increase in response to the DNA damage agents MMS and HU [47]. To determine if the increase in protein is due to an increase at the mRNA level, we monitored BDF2 mRNA expression after exposure to these DNA damage agents. Both treatments resulted in a transient increase in BDF2 mRNA in the first hour, with slight differences in the kinetics, followed by a decrease to normal levels after two hours (Fig. 4B).
To further investigate how SMD and RMD affect the levels of BDF2 mRNA in the bdf1Δ background, we exposed the 5′-SS and stem-loop mutants to 50 mM HU for two hours. There was no significant change in the levels of full-length BDF2 mRNA or the intron-exon2 SMD degradation intermediate throughout this time course (Fig. 4C). Nonetheless, the time course demonstrated that SMD of BDF2 mRNA remains active in the presence of HU. Because of the lack of an effect of the stem mutations on bdf1Δ growth in HU, we predicted that Rnt1p cleavage activity might decrease in HU. To test how RMD on BDF2 mRNA behaves during HU exposure in the wild-type BDF1 background, we treated wild-type, the 5′-SS mutant, the stem-loop deletion mutant, and rrp6Δ strains with 200 mM HU and monitored the expression of BDF2 mRNA and its degradation products. Consistent with reduced RMD, there was a substantial drop in the levels of the RMD intermediate within 30 minutes of HU treatment in the rrp6Δ background (Fig. 4D). Furthermore, the intron-exon2 SMD product, which is normally degraded by Rnt1p due to the presence of the Rnt1p target stem loop in the intron, actually increased in the WT strain throughout the HU treatment. Together these results indicate that RMD of BDF2 mRNA is inhibited during DNA replication stress, and that the increased flux of BDF2 mRNA through SMD is important to protect bdf1Δ cells from the toxic effects of excess Bdf2p accumulation.
Discussion
BDF2 expression is predominantly regulated at the level of RNA decay
In this study we demonstrate that two major nuclear RNA degradation pathways, Rnt1p - and spliceosome-mediated decay, limit the expression of BDF2 mRNA, and that BDF2 expression is tightly regulated at the post-transcriptional level in specific environmental conditions. Strikingly, in normal growth conditions, the BDF2 gene exhibits a high transcription rate (0.51 RNA molecules per minute per cell) in the top 4.6% of all RNA polymerase II (Pol II)-transcribed genes [29], with an mRNA stability that is among the lowest of all S.cerevisiae Pol II transcripts (∼5 minute half life, lowest 1.1% [48]). During osmotic stress, a condition where BDF2 mRNA levels rapidly drop, the Pol II transcription rate across the BDF2 gene paradoxically increases. However, RNase III-mediated degradation (RMD) over-compensates for the increased transcription, and BDF2 mRNA levels drop rapidly. Simultaneously, there is a transient decrease in spliceosome-mediated decay (SMD) of BDF2 mRNA during salt stress. A decrease in spliceosome activity during osmotic stress is consistent with a recent report that intron-containing ribosomal protein pre-mRNAs, the predominant substrates for the spliceosome, accumulate as unspliced transcripts in osmotic stress conditions when their degradation by nonsense-mediated decay is inactivated [49]. Therefore, both nuclear and cytoplasmic RNA degradation systems have evolved to degrade intron-containing transcripts that accumulate during osmotic stress. By contrast, SMD of BDF2 mRNA plays a critical role during DNA replication stress, as evidenced by the toxicity of the 5′-SS mutation to cells lacking BDF1 when grown in the presence of DNA double-strand break agents (Fig. 4A). Conversely, we found that RMD of BDF2 mRNA is diminished during hydroxyurea exposure and its inactivation confers no phenotype towards bdf1Δ cells in HU or CPT (Fig. 4A,D). Interestingly, there was not a significant effect of the 5′-SS mutation on BDF2 mRNA levels in HU during the time window we examined (Fig. 4C,D). It is possible that an effect of HU on BDF2 mRNA levels in the 5′SS mutant requires longer exposure times to HU. It is also possible that the 5′SS mutation results in enhanced leakage of the “unspliced-like” BDF2 mRNA to the cytoplasm and thus increased levels of Bdf2p translation, as the nuclear retention of intron-containing transcripts by Mlp1p is known to require an intact 5′-splice site [38]. This would be consistent with our finding that Mlp1/2p proteins regulate BDF2 mRNA levels (Fig. S4B).
Our results reveal that the relative flux of BDF2 mRNA degradation through RMD or SMD is dependent on the particular environmental condition, such that when one degradation system is inactivated, the other system predominates (Fig. 5). In particular, we found that levels of the SMD degradation intermediate decrease in salt stress, whereas the RMD intermediate increases in abundance. During DNA replication stress, the RMD intermediate decreases while the SMD degradation intermediate increases. These molecular phenotypes are consistent with the growth phenotypes of the BDF2 5′-SS and BDF2 stem loop mutations in the bdf1Δ background in these stress conditions. It appears that the BDF2 gene has evolved to retain the degradation signals for both systems, such that its tight post-transcriptional control is maintained in conditions that block either RMD or SMD. Therefore, the presence of two independent nuclear degradation pathways acting on the BDF2 mRNA guards against excess accumulation of BDF2 mRNA across a range of stress conditions. The observation that BDF2 mRNA is detectable in WT cells in normal growth conditions suggests that a substantial fraction of BDF2 mRNA normally escapes nuclear degradation by SMD and RMD. In salt stress, an increase in the absolute activity of RMD leads to a substantial increase in the overall fraction of full-length BDF2 mRNA subject to nuclear degradation. Furthermore, inhibiting both RMD and SMD has an additive effect compared to inhibiting either pathway alone in specific conditions (Fig. 1D and Fig. 2B), demonstrating that the pathways are non-redundant and independently regulate BDF2 mRNA in a condition-specific manner (Fig. 5).

RMD and SMD modulate Bromodomain factor 2 expression to maintain cellular fitness in response to adverse environmental conditions
Despite detailed biochemical work on the binding affinities of Bdf1p and Bdf2p for different acetylated histone tail peptides [50], and extensive chromatin immunoprecipitation studies mapping genomic binding sites [8], it is largely unknown how Bdf1p and Bdf2p achieve their specificity in vivo and how these factors are regulated in different conditions. We found that during osmotic stress, degradation of BDF2 mRNA is required for the full induction of glycogen phosphorylase (GPH1, Fig. 3F), suggesting that Bdf2p may normally repress a subset of salt-stress induced genes. We also found that inactivating SMD or RMD in a wild-type background resulted in sensitivity to lithium chloride (Fig. S6), suggesting that decreasing BDF2 mRNA levels via nuclear RNA decay constitutes an important mechanism for adapting gene expression to lithium ion stress.
The regulation of BDF2 expression by these two RNA degradation mechanisms controls highly specific phenotypes. Strikingly, the inactivation of SMD, but not RMD, sensitized bdf1Δ cells to hydroxyurea (HU) and camptothecin (CPT), but not methyl methanesulfonate (MMS) (Fig. 4A). While all three agents stall replication fork progression, only HU and CPT induce in vivo double-strand DNA breaks (DSBs) [51]. This suggests that an excess of Bdf2p in the absence of Bdf1p is toxic specifically in the presence of DSBs. This finding is consistent with a report that the fission yeast homolog of BDF2 is toxic to cells with unstable replication forks, as the deletion of BDF2 suppressed the HU sensitivity of numerous S-phase checkpoint mutants [17]. It remains to be determined whether the toxicity of Bdf2p is due to a direct effect on chromatin structure, or rather due to its effects on gene expression in DNA damage conditions. Notably, the human homolog of Bdf2p, BRD4, has been shown to activate cell death in response to DNA damage by promoting chromatin condensation [52]. It is therefore possible that at least some of the functions of Bdf2p in the DNA damage response have been conserved from yeast to humans. Significantly, we found that BDF2 is required for resistance of wild-type cells to DMSO (Fig. S6), which has been implicated in causing DNA damage by an unknown mechanism [53]. Therefore, Bdf2p is beneficial in specific conditions, and can play antagonizing roles in the DNA damage response depending on the specific type of DNA damage.
Overall, our study highlights how environmental stress conditions can differentially control RNA degradation systems to regulate gene expression (Fig. 5), and emphasizes the importance of modulating bromodomain factors expression in the cellular response to environmental changes. The ability of the cell to degrade or stabilize specific transcripts by environmentally-controlled RNA degradation pathways mirrors the control that transcriptional activators and repressors can provide in response to stress conditions. We speculate that the presence of SMD and/or RMD degradation signals in other mRNAs may allow for broad post-transcriptional fine-tuning of gene expression in conditions where the activity of these degradation systems is modulated. Future work will unravel precisely what mechanisms regulate RNase III and spliceosome-mediated decay in different environmental conditions, as well as uncover the repertoire of transcripts targeted by these pathways.
Materials and Methods
RNA secondary structure screen
Transcripts targeted by spliceosome-mediated decay were obtained from Supplementary Table 2 from [13]. The 5′ and 3′ UTRs for each transcript were obtained from [31] from Saccharomyces Genome Database. If the transcript did not have an annotated UTR, the open reading frame start or stop coordinates were used. Each sequence was first split into 200 base fragments in 100 base overlapping steps. mFold [24] was used to predict the fold of each 200 base sequence, and sequences capable of adopting stem-loop structures with an NGNN or AAGU tetraloop were identified for further study.
Strains
The BMA64 (mating type a) background was used to generate the mutant strains used in this study (Table S1). The indicated deletion mutants were constructed by the lithium acetate/PEG method [54] by replacing the open reading frames with PCR products encoding either S. cerevisiae TRP1, S. kluyveri HIS5, or the KANMX6 gene [55], [56]. Strains containing the rat1-1 mutation were previously described [28]. Mutations were introduced into the endogenous BDF2 gene by the delitto perfetto method [57]. First, the CORE cassette encoding the URA3 and KANMX6 genes was inserted at Rnt1p-target stem loop (chrIV:332367-80). G418 resistant colonies were screened for successful integration of the CORE cassette by PCR. For the second step, we used the pUG23-BDF2 plasmid (described below), containing either the WT or various BDF2 mutations, as a template to generate high-fidelity PCR products (NEB Phusion). Primers were designed so that the resulting PCR products contained >100 bases of homology flanking the mutation sites (Table S1). 200 µl of each PCR product were precipitated in 1 ml ethanol and 40 µl 3 M sodium acetate pH 5.2. Pellets were washed in 70% ethanol and resuspended in 34 µl water and used for excision of the CORE cassette by standard yeast transformation techniques [54]. Transformations were plated overnight on YPD, and then replica-plated to 5-FOA to select for loss of URA3. Colonies were screened for sensitivity to G418 to confirm loss of the CORE cassette, and mutations were confirmed by PCR amplification of the BDF2 locus followed by sequencing (Laragen, Inc).
Plasmids
The BDF2 open reading frame without the stop codon was amplified by high-fidelity PCR (NEB Phusion) from WT genomic DNA with the forward primer containing the SpeI restriction site and the reverse primer containing the ClaI restriction site (Table S1). The insert was cloned into the pUG23 vector and 5′-splice site and stem-loop mutations were constructed with the Quick-Change site-directed mutagenesis kit (Agilent Technologies, Inc). All plasmids and mutations were confirmed by sequencing (Laragen, Inc.).
Media and culturing
Unless otherwise indicated, cultures were grown in YPD (1% yeast extract, 2% bacto-peptone, 2% dextrose) at 30°C at 200 rpm. SDC (.67% yeast nitrogen base with ammonium sulfate, 2% dextrose, and 0.2% complete amino acid mixture) was used to culture strains containing the met22Δ deletion, and where indicated these cultures were pelleted, washed once with SD medium lacking methionine (SD-MET), and resuspended in SD-MET for 12 hours. Strains containing the pUG23 vector were grown in SD-URA-MET. Strains containing the rat1-1 mutation were grown at 25°C and shifted to pre-warmed flasks containing the same medium at 37°C. All cultures were harvested by centrifugation at 4000 rpm for 2 minutes, washed in deionized water, and spun down in microcentrifuge tubes. The supernatant was removed and pellets were flash-frozen in liquid nitrogen and stored at −80°C.
RNA extraction and northern blot analysis
RNA extractions were performed by the addition of 400 µl acid-washed glass beads, 500 µl phenol∶chloroform∶isoamyl alcohol (25∶24∶1, pH 6.7, Fisher Scientific), and 500 µl of RNA-SDS buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM EDTA, 2% SDS) to the frozen pellets. Samples were vortexed for 1 minute, heated in a 65°C water bath for 5 minutes, and vortexed for an additional minute. Samples were spun at 13,200 rpm for 5 minutes, and 450 µl of the supernatant was transferred to a new tube containing 450 µl of phenol∶chloroform∶isoamyl alcohol. Samples were vortexed for 1 minute, and spun at 15,000 rpm for 5 minutes. 400 µl of supernatant were precipitated in 1 ml EtOH and 40 µl of 3M NaOAc pH 5.2, and incubated at −80°C for 30 minutes. Samples were spun at 15,000 rpm for 5 minutes, and pellets were washed with 70% EtOH and resuspended in nuclease-free water (Ambion). In vitro cleavage of 50 µg total RNA with 4 pmol recombinant Rnt1p was performed at 30°C for 20 minutes as described [58], [59]. 1 volume of RNA was denatured in 5 volumes of glyoxal buffer [60% DMSO (Sigma-Aldrich), 8% glyoxal w/v (Sigma-Aldrich), 5% glycerol, 40 µg/ml ethidium bromide, 1× BPTE pH 6.5 (10 mM PIPES (Sigma-Aldrich), 30 mM Bis-Tris (Sigma-Aldrich), 10 mM EDTA pH 8.0)]. Samples were vortexed and denatured at 55°C for one hour. Samples were chilled on ice for 10 minutes, and 10× BPTE-RNA loading dye (0.25% xylene cyanol, 0.25% bromophenol blue, 5% glycerol, 1× BPTE) was added to each sample before loading on 2% agarose gels with 1× BPTE buffer. Electrophoresis was performed at 4 V/cm while mixing the buffer with magnetic stir bars. Gels were visualized and washed for 10 minutes in deionized water, 20 minutes in 75 mM NaOH, an additional 10 minutes in deionized water, and 10 minutes in 10× SSPE (1.5 M NaCl, 100 mM sodium phosphate, 100 mM EDTA, pH 7.4). Gels were passively transferred in 10× SSPE to positively charged nylon membranes (Hybond-N+, GE Healthcare Life Sciences). Blots were cross-linked with the auto cross-link function (120,000 joules/cm2) on the Stratalinker UV Crosslinker 2400 (Stratagene), and stored in 2× SSPE at 4°C. RNA probes were constructed by in vitro transcription with the T3 RNA polymerase (Promega) and α-32P - UTP (Perkin-Elmer) following the manufacturer's protocol, with the exception that radioactive UTP was used instead of radioactive CTP. Templates were constructed by PCR using genomic DNA and primers hybridizing to the target gene (Table S1). The reverse primer contained at its 5′-end the T3 promoter sequence (5′-AATTAACCCTCACTAAAGGGA-3). Blots were pre-hybridized in Church's buffer (1% BSA, 1 mM EDTA, 0.5 M NaPO4 pH 7.2, 7% SDS) at 65°C for 1 hour, and transcription reactions were diluted in 100 µl of deionized water and added directly to the hybridization bottles. Hybridizations were conducted overnight at 65°C, and blots were washed for 20 minutes with 2× SSPE+0.1% SDS at 65°C, followed by two washes for 20 minutes with 0.1× SSPE+0.1% SDS at 65°C. Blots were exposed to K-screens (Kodak) and scanned with the BioRad FX Phosphorimager.
Primer extension mapping of the Rnt1p cleavage site
20 pmol of BDF2 downstream RCS reverse primer (5′-CAATTGTTGCAATTCAACCTCC-3′) was labeled with γ-32P-ATP with T4 polynucleotide kinase (NEB) according to the manufacturer's protocol. Primer extension was performed using the SuperScript III reverse transcriptase (Invitrogen/Life Technologies) according to manufacturer's protocol, except that denaturation was performed at 75°C for 4 minutes in a thermocycler, followed by primer annealing at 45°C for 5 minutes. Primer extension was performed at 55°C for 60 minutes, followed by 70°C for 15 minutes to inactivate the enzyme. An equal volume of 2× formamide loading dye (Ambion) was added to the primer extension reactions, heated at 75°C for 3 minutes, and 10 µl was loaded onto 8% poly-acrylamide sequencing gels. To obtain the sequencing ladder, a PCR product was first generated using the reverse primer and the BDF2 exon1 forward primer (5′-GCACATTCTGCTTTACTGGCAGC-3′) and pUG23-BDF2 as template. The PCR product was used as a template with the Thermo Sequenase Cycle Sequencing Kit using 0.5 picomoles of labeled primer for each of the 4 dideoxy-nucleotide sequencing reactions (according to manufacturer's protocol). Gels were dried at 80°C for one hour on a vacuum gel drier (Amersham Pharmacia), and exposed to K-screens (Kodak) and scanned with the BioRad FX Phosphorimager.
RT-PCR analysis
40 µg total RNA of total RNA was treated with DNase I (Ambion) according to manufacturer's protocol. 5 µg of DNase I-treated RNA was reverse-transcribed with random hexamers and M-MLV reverse transcriptase (Invitrogen) according to manufacturer's protocol. 2 µl of cDNA was used as a template in standard 50 µl PCR with the forward primer 5′-AGCAGCGAGAGTAGTAGTAACAAAAAC-3′ and either reverse primer R1 5′-ATCTCTCATAAATTCTTGCAATAGTCG-3′ or R2 5′-TGCTTCATTGCCTTCCTACG-3′. The following thermo-cycler parameters were used: 94°C 2 min, 35 cycles of 94°C 30 s, 60°C 30 s, 72°C 60 s. RT-PCR products were analyzed on 1× TAE 2% agarose gels and stained with ethidium bromide. The identity of each band was verified by gel extraction of the excised band followed by Sanger sequencing (Laragen, Inc.).
Plate growth assays
Cultures were grown to mid-log phase (OD600 nm 0.4–0.6) and 1 ml of each culture was spun at 13,200 rpm for 30 seconds. Pellets were resuspended in 1 ml of sterile water, and diluted to OD600 nm 0.1 followed by four 5-fold serial dilutions in sterile water. 4 µl of each dilution was spotted onto the indicated media. Hydroxyurea was purchased from Alfa-Aesar, and camptothecin and methyl methanesulfonate were purchased from Sigma-Aldrich.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KornbergRD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184 : 868–871.
2. KornbergRD, LorchY (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98 : 285–294.
3. WuJ, GrunsteinM (2000) 25 years after the nucleosome model: chromatin modifications. Trends Biochem Sci 25 : 619–623.
4. StrahlBD, AllisCD (2000) The language of covalent histone modifications. Nature 403 : 41–45 doi:10.1038/47412
5. KurdistaniSK, GrunsteinM (2003) Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol 4 : 276–284 doi:10.1038/nrm1075
6. ZentnerGE, HenikoffS (2013) Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 20 : 259–266 doi:10.1038/nsmb.2470
7. ZengL, ZhouMM (2002) Bromodomain: an acetyl-lysine binding domain. FEBS Lett 513 : 124–128.
8. DurantM, PughBF (2007) NuA4-directed chromatin transactions throughout the Saccharomyces cerevisiae genome. Mol Cell Biol 27 : 5327–5335 doi:10.1128/MCB.00468-07
9. KroganNJ, KeoghM-C, DattaN, SawaC, RyanOW, et al. (2003) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12 : 1565–1576.
10. LadurnerAG, InouyeC, JainR, TjianR (2003) Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol Cell 11 : 365–376.
11. MatangkasombutO, BuratowskiRM, SwillingNW, BuratowskiS (2000) Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev 14 : 951–962.
12. GhaemmaghamiS, HuhW-K, BowerK, HowsonRW, BelleA, et al. (2003) Global analysis of protein expression in yeast. Nature 425 : 737–741 doi:10.1038/nature02046
13. VolanakisA, PassoniM, HectorRD, ShahS, KilchertC, et al. (2013) Spliceosome-mediated decay (SMD) regulates expression of nonintronic genes in budding yeast. Genes Dev 27 : 2025–2038 doi:10.1101/gad.221960.113
14. KoborMS, VenkatasubrahmanyamS, MeneghiniMD, GinJW, JenningsJL, et al. (2004) A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol 2: E131 doi:10.1371/journal.pbio.0020131
15. LuPYT, LévesqueN, KoborMS (2009) NuA4 and SWR1-C: two chromatin-modifying complexes with overlapping functions and components. Biochem Cell Biol 87 : 799–815 doi:10.1139/O09-062
16. ChuaP, RoederGS (1995) Bdf1, a yeast chromosomal protein required for sporulation. Mol Cell Biol 15 : 3685–3696.
17. GarabedianMV, NoguchiC, ZieglerMA, DasMM, SinghT, et al. (2012) The double-bromodomain proteins Bdf1 and Bdf2 modulate chromatin structure to regulate S-phase stress response in Schizosaccharomyces pombe. Genetics 190 : 487–500 doi:10.1534/genetics.111.135459
18. FuJ, HouJ, LiuL, ChenL, WangM, et al. (2013) Interplay between BDF1 and BDF2 and their roles in regulating the yeast salt stress response. FEBS J 280 : 1991–2001 doi:10.1111/febs.12219
19. KapitzkyL, BeltraoP, BerensTJ, GassnerN, ZhouC, et al. (2010) Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol Syst Biol 6 : 451 doi:10.1038/msb.2010.107
20. WangJ, TadeoX, HouH, TuPG, ThompsonJ, et al. (2013) Epe1 recruits BET family bromodomain protein Bdf2 to establish heterochromatin boundaries. Genes Dev 27 : 1886–1902 doi:10.1101/gad.221010.113
21. HarigayaY, ParkerR (2012) Global analysis of mRNA decay intermediates in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 109 : 11764–11769 doi:10.1073/pnas.1119741109
22. KawashimaT, DouglassS, GabunilasJ, PellegriniM, ChanfreauGF (2014) Widespread Use of Non-productive Alternative Splice Sites in Saccharomyces cerevisiae. PLoS Genet 10: e1004249 doi:10.1371/journal.pgen.1004249
23. Danin-KreiselmanM, LeeCY, ChanfreauG (2003) RNAse III-mediated degradation of unspliced pre-mRNAs and lariat introns. Mol Cell 11 : 1279–1289.
24. ZukerM (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31 : 3406–3415.
25. ChanfreauG, BuckleM, JacquierA (2000) Recognition of a conserved class of RNA tetraloops by Saccharomyces cerevisiae RNase III. Proc Natl Acad Sci U S A 97 : 3142–3147 doi:10.1073/pnas.070043997
26. WangZ, HartmanE, RoyK, ChanfreauG, FeigonJ (2011) Structure of a yeast RNase III dsRBD complex with a noncanonical RNA substrate provides new insights into binding specificity of dsRBDs. Structure 19 : 999–1010 doi:10.1016/j.str.2011.03.022
27. RouskinS, ZubradtM, WashietlS, KellisM, WeissmanJS (2014) Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505 : 701–705 doi:10.1038/nature12894
28. LeeA, HenrasAK, ChanfreauG (2005) Multiple RNA surveillance pathways limit aberrant expression of iron uptake mRNAs and prevent iron toxicity in S. cerevisiae. Mol Cell 19 : 39–51 doi:10.1016/j.molcel.2005.05.021
29. PelechanoV, ChávezS, Pérez-OrtínJE (2010) A complete set of nascent transcription rates for yeast genes. PLoS ONE 5: e15442 doi:10.1371/journal.pone.0015442
30. DichtlB, StevensA, TollerveyD (1997) Lithium toxicity in yeast is due to the inhibition of RNA processing enzymes. EMBO J 16 : 7184–7195 doi:10.1093/emboj/16.23.7184
31. YassourM, KaplanT, FraserHB, LevinJZ, PfiffnerJ, et al. (2009) Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci U S A 106 : 3264–3269 doi:10.1073/pnas.0812841106
32. ZhangZ, DietrichFS (2005) Mapping of transcription start sites in Saccharomyces cerevisiae using 5′ SAGE. Nucleic Acids Res 33 : 2838–2851 doi:10.1093/nar/gki583
33. LeeW, TilloD, BrayN, MorseRH, DavisRW, et al. (2007) A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 39 : 1235–1244 doi:10.1038/ng2117
34. EgeciogluDE, HenrasAK, ChanfreauGF (2006) Contributions of Trf4p - and Trf5p-dependent polyadenylation to the processing and degradative functions of the yeast nuclear exosome. RNA 12 : 26–32 doi:10.1261/rna.2207206
35. LiangY-H, LavoieM, ComeauM-A, Abou ElelaS, JiX (2014) Structure of a eukaryotic RNase III postcleavage complex reveals a double-ruler mechanism for substrate selection. Mol Cell 54 : 431–444 doi:10.1016/j.molcel.2014.03.006
36. SayaniS, ChanfreauGF (2012) Sequential RNA degradation pathways provide a fail-safe mechanism to limit the accumulation of unspliced transcripts in Saccharomyces cerevisiae. RNA 18 : 1563–1572 doi:10.1261/rna.033779.112
37. AmraniN, GanesanR, KervestinS, MangusDA, GhoshS, et al. (2004) A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432 : 112–118 doi:10.1038/nature03060
38. GalyV, GadalO, Fromont-RacineM, RomanoA, JacquierA, et al. (2004) Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116 : 63–73.
39. FendtS-M, SauerU (2010) Transcriptional regulation of respiration in yeast metabolizing differently repressive carbon substrates. BMC Syst Biol 4 : 12 doi:10.1186/1752-0509-4-12
40. ChenL, LiuL, WangM, FuJ, ZhangZ, et al. (2013) Hal2p functions in Bdf1p-involved salt stress response in Saccharomyces cerevisiae. PLoS ONE 8: e62110 doi:10.1371/journal.pone.0062110
41. ZhaoJ, LinW, MaX, LuQ, MaX, et al. (2010) The protein kinase Hal5p is the high-copy suppressor of lithium-sensitive mutations of genes involved in the sporulation and meiosis as well as the ergosterol biosynthesis in Saccharomyces cerevisiae. Genomics 95 : 290–298 doi:10.1016/j.ygeno.2010.02.010
42. Romero-SantacreuL, MorenoJ, Pérez-OrtínJE, AlepuzP (2009) Specific and global regulation of mRNA stability during osmotic stress in Saccharomyces cerevisiae. RNA 15 : 1110–1120 doi:10.1261/rna.1435709
43. FavreC, AguilarPS, CarrilloMC (2008) Oxidative stress and chronological aging in glycogen-phosphorylase-deleted yeast. Free Radic Biol Med 45 : 1446–1456 doi:10.1016/j.freeradbiomed.2008.08.021
44. ChangM, BellaouiM, BooneC, BrownGW (2002) A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA 99 : 16934–16939 doi:10.1073/pnas.262669299
45. KoçA, WheelerLJ, MathewsCK, MerrillGF (2004) Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem 279 : 223–230 doi:10.1074/jbc.M303952200
46. TesauroC, Morozzo della RoccaB, OttavianiA, ColettaA, ZuccaroL, et al. (2013) Molecular mechanism of the camptothecin resistance of Glu710Gly topoisomerase IB mutant analyzed in vitro and in silico. Mol Cancer 12 : 100 doi:10.1186/1476-4598-12-100
47. TkachJM, YimitA, LeeAY, RiffleM, CostanzoM, et al. (2012) Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14 : 966–976 doi:10.1038/ncb2549
48. WangY, LiuCL, StoreyJD, TibshiraniRJ, HerschlagD, et al. (2002) Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA 99 : 5860–5865 doi:10.1073/pnas.092538799
49. GarreE, Romero-SantacreuL, Barneo-MuñozM, MiguelA, Pérez-OrtínJE, et al. (2013) Nonsense-mediated mRNA decay controls the changes in yeast ribosomal protein pre-mRNAs levels upon osmotic stress. PLoS ONE 8: e61240 doi:10.1371/journal.pone.0061240
50. MatangkasombutO, BuratowskiS (2003) Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell 11 : 353–363.
51. LundinC, NorthM, ErixonK, WaltersK, JenssenD, et al. (2005) Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res 33 : 3799–3811 doi:10.1093/nar/gki681
52. FloydSR, PacoldME, HuangQ, ClarkeSM, LamFC, et al. (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498 : 246–250 doi:10.1038/nature12147
53. GaytánBD, LoguinovAV, La Rosa DeVY, LerotJ-M, VulpeCD (2013) Functional genomics indicates yeast requires Golgi/ER transport, chromatin remodeling, and DNA repair for low dose DMSO tolerance. Front Genet 4 : 154 doi:10.3389/fgene.2013.00154
54. GietzRD, SchiestlRH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2 : 31–34 doi:10.1038/nprot.2007.13
55. LongtineMS, McKenzieA, DemariniDJ, ShahNG, WachA, et al. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 : 953–961 doi:;10.1002/(SICI)1097-0061(199807)14 : 10<953::AID-YEA293>3.0.CO;2-U
56. WachA, BrachatA, PöhlmannR, PhilippsenP (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10 : 1793–1808.
57. StuckeyS, StoriciF (2013) Gene knockouts, in vivo site-directed mutagenesis and other modifications using the delitto perfetto system in Saccharomyces cerevisiae. Meth Enzymol 533 : 103–131 doi:10.1016/B978-0-12-420067-8.00008-8
58. ChanfreauG, RotondoG, LegrainP, JacquierA (1998) Processing of a dicistronic small nucleolar RNA precursor by the RNA endonuclease Rnt1. EMBO J 17 : 3726–3737 doi:10.1093/emboj/17.13.3726
59. LamontagneB, ElelaSA (2001) Purification and characterization of Saccharomyces cerevisiae Rnt1p nuclease. Meth Enzymol 342 : 159–167.
60. RondónAG, MischoHE, KawauchiJ, ProudfootNJ (2009) Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol Cell 36 : 88–98 doi:10.1016/j.molcel.2009.07.028
61. GhazalG, GagnonJ, JacquesP-E, LandryJ-R, RobertF, et al. (2009) Yeast RNase III triggers polyadenylation-independent transcription termination. Mol Cell 36 : 99–109 doi:10.1016/j.molcel.2009.07.029
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 9
Nejčtenější v tomto čísle
- Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals
- Nipbl and Mediator Cooperatively Regulate Gene Expression to Control Limb Development
- Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel
- Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation