
The MAPK p38c Regulates Oxidative Stress and Lipid Homeostasis in the Intestine
The p38 mitogen-activated protein (MAP) kinase is a signaling pathway that is involved in both stress and immunity in various species from yeast to human. p38 kinases regulate transcription factors of the ATF family and other protein kinases that then induce cellular adaptation to stress to a wide variety of physical, chemical and biological stresses. The Drosophila genome encodes three p38 kinases named p38a, p38b and p38c. In this study, we have analyzed the role of p38c in the Drosophila intestine. The p38c gene is expressed in the digestive tract and up-regulated upon intestinal infection. We observed a lower production of Reactive Oxygen Species (ROS) in the gut of p38c mutants upon bacterial infection. Consistent with this observation, the transcription of the Duox, a gene encoding an enzyme that produces ROS, is reduced in p38c mutant flies. Our analysis shows that p38c induces the phosphorylation of Atf-2, a transcription factor that controls Duox expression. Interestingly, our study also shows that p38c and Atf3 function in a common pathway in the intestine to regulate lipid metabolism and immune homeostasis. Collectively, our study demonstrates that p38c plays a central role in the intestine of Drosophila.
Published in the journal:
. PLoS Genet 10(9): e32767. doi:10.1371/journal.pgen.1004659
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004659
Summary
The p38 mitogen-activated protein (MAP) kinase is a signaling pathway that is involved in both stress and immunity in various species from yeast to human. p38 kinases regulate transcription factors of the ATF family and other protein kinases that then induce cellular adaptation to stress to a wide variety of physical, chemical and biological stresses. The Drosophila genome encodes three p38 kinases named p38a, p38b and p38c. In this study, we have analyzed the role of p38c in the Drosophila intestine. The p38c gene is expressed in the digestive tract and up-regulated upon intestinal infection. We observed a lower production of Reactive Oxygen Species (ROS) in the gut of p38c mutants upon bacterial infection. Consistent with this observation, the transcription of the Duox, a gene encoding an enzyme that produces ROS, is reduced in p38c mutant flies. Our analysis shows that p38c induces the phosphorylation of Atf-2, a transcription factor that controls Duox expression. Interestingly, our study also shows that p38c and Atf3 function in a common pathway in the intestine to regulate lipid metabolism and immune homeostasis. Collectively, our study demonstrates that p38c plays a central role in the intestine of Drosophila.
Introduction
In addition to its central role in digestion and absorption, the intestine serves as an interactive barrier against a wide variety of pathogens and commensals. This is especially true for insects such as Drosophila, which feed on rotting fruits and continuously ingest microbes. In recent years, D. melanogaster has emerged as a powerful model to investigate intestinal homeostasis and immunity [1]–[4].
Recent studies have shown that the Drosophila gut defense against bacterial infection involves (i) the production of ROS through the NADPH oxidase Duox, (ii) the production of antibacterial peptides through the Imd pathway, and (iii) the maintenance of gut homeostasis through regulation of stem cell activity [2]. Oral ingestion of bacteria induces the rapid synthesis of microbicidal ROS in the Drosophila gut by Duox [3]. The activity of Duox is triggered by the Gαq-phospholipase C-ß-Ca2+ pathway, which is itself initiated upon binding of an uncharacterized G-protein coupled receptor to uracil, a microbial ligand released from pathogenic bacteria [5], [6]. Duox is also regulated at the transcriptional level by the transcription factor Atf-2, downstream of a p38a-Mkk3-Mekk1-PGRP-LC (peptidoglycan recognition protein LC) pathway [7]. In addition to this ROS response, several antimicrobial peptides (e.g., Diptericin, Attacin) are produced in the gut under the control of the NF-kB protein Relish downstream of the Imd pathway [8]–[10]. This local immune response is triggered by the recognition of peptidoglycan from Gram-negative bacteria by the pattern recognition receptors PGRP-LC and PGRP-LE [11], [12]. Infection can also lead to intestinal damage, induced either by bacterial toxins or by the excessive production of ROS [13]–[17]. Stress response programs and increased epithelial renewal can then be deployed to repair the intestinal epithelium and maintain the integrity of the gut barrier. Epithelium renewal of the Drosophila gut is stimulated by the release of secreted ligands of the Unpaired and EGF families, which activate respectively the JAK/STAT and EGFR pathways in stem-cell like progenitor cells to promote their division and differentiation, thereby establishing compensatory homeostatic regulatory loops [2], [18].
We have recently described how an entomopathogenic bacterium, Pseudomonas entomophila, disrupts gut homeostasis in Drosophila. Although P. entomophila ingestion by D. melanogaster stimulates the transcription of genes encoding antimicrobial peptides (e.g. Diptericin) and epithelium renewal inducers (e.g. upd3), neither immune response nor epithelium renewal is observed. This is due to a general inhibition of translation in the intestine that affects all newly synthesized transcripts [16]. As a consequence, D. melanogaster succumb to P. entomophila infection because they are unable to repair the gut damage ensuing infection. This reduction of translation is a consequence of cellular damage to the intestine, caused by both host-derived ROS and the direct action of a pore-forming toxin produced by the pathogen. Thus the P. entomophila infection model provides a good system to probe the complex cross-talks between stress, repair and immune pathways during microbial infection [19].
To further study the interaction between stress response and host defense, we have now analyzed the role of the conserved p38 MAPK pathway in the Drosophila gut response. The p38 MAPK family has been involved in stress and immunity in both mammals and Drosophila [20]. In response to a wide range of physical, chemical and biological stresses, p38 kinases phosphorylate various substrates, such as transcription factors of the Activating Transcription Factor (ATF) family and other protein kinases, so as to regulate cellular adaptation to stress [21]. The Drosophila genome encodes for three p38 kinases named p38a, p38b and p38c. Mutations in p38a or p38b lead to an increased susceptibility to environmental stresses such as osmotic stress, heat stress or intestinal infection [22]–[24]. In contrast, p38c has been shown to regulate dopadecarboxylase, which encodes an enzyme involved in the formation of melanin in the epidermis following septic injury. This points to a possible role of p38c in wound healing [25].
In this study, we have analyzed the contribution of Drosophila p38c to the intestinal response to bacteria. We showed that p38c mutant flies are more resistant to P. entomophila, but are more susceptible to the non-pathogenic bacterium Erwinia carotovora carotovora 15 (Ecc15). This phenotype is linked to a lower production of ROS in p38c mutants. We observed that the transcription of the ROS producing enzyme Duox is reduced in p38c mutant flies. In addition, we showed that Mekk1, Mkk3, p38c and Atf-2 function in a common pathway to control Duox transcription following infection. Finally, we observed that this pathway also regulates lipid homeostasis and basal antimicrobial peptide gene expression in an Atf3 dependent manner. Thus, our study delineates a pathway comprising Mekk1, Mkk3, p38c and Atf-2 that regulate the oxidative stress, immune response and lipid homeostasis in the intestine of Drosophila.
Results
p38c expression is up-regulated in the gut upon oral bacterial infection
In Drosophila, three p38-MAPK-encoding genes, p38a (initially described as mpk2), p38b and p38c, have been identified (Figure S1A; [25]–[27]). Recent studies have revealed that p38a and p38b contribute to stress and immune responses in the Drosophila digestive tract [28], [29]. To date, the function of p38c in the intestine has not been described. Microarray data from FlyAtlas [30] showed that p38c transcripts are enriched in the midgut, Malpighian tubules and fat body of both larvae and adults when compared to p38a and p38b (Figure 1A). To characterize further the immune role of these MAPK genes, we monitored by RT-qPCR their expression in the intestine of flies orally infected with two Gram-negative bacteria, Ecc15 and P. entomophila. This analysis revealed that p38c, and to a lesser extent p38a, is induced following infection with Ecc15 and P. entomophila (Figure 1B). These results are consistent with previous microarray datasets analyzing gene expression profile in the gut of flies orally infected with Ecc15 or P. entomophila (Figure S1B; [15], [16]). The enrichment of p38c in the gut and its high induction upon infection prompted us to investigate the role of this MAPK in the intestine. We first analyzed p38c localization in unchallenged and P. entomophila-infected intestine using a newly generated anti-p38c antibody, which was validated by an absence of signal in the p38c null mutant (p38c7B1 [25]) (Figure S1C and S1D). p38c protein was localized to the cytoplasm of enterocytes (identified by their large nuclei) under unchallenged condition. Following P. entomophila oral infection the intensity of the p38c signals modestly increased (Figure 1C).

It has been suggested that the p38c kinase cannot be activated by phosphorylation due to a mutation that converts the TGY dual-phosphorylation site to TDH (Figure S2A). To test whether p38c is capable of kinase activity, we expressed and purified both GST - and Histidine - fusion derivatives of p38c in bacteria and carried out an in vitro kinase assay using the non-radioactive Kinase-Glo (Promega) kit with a mammalian GST-ATF2 fusion protein as an exogenous substrate (Figure S2B). We observed that GST-ATF2 was phosphorylated by both GST - and His-p38c fusion proteins. In addition, this kinase activity decreased in the presence of SB203580, a p38 inhibitor, which reduces its catalytic activity by binding to the ATP-binding pocket (Figure S2C). Altogether, our study shows that p38c is expressed in the gut, up-regulated upon infection and that the protein can function as a kinase at least in vitro, which suggests an important function of this MAPK in this organ.
p38c mutants show a higher susceptibility to oral bacterial infection and to H2O2
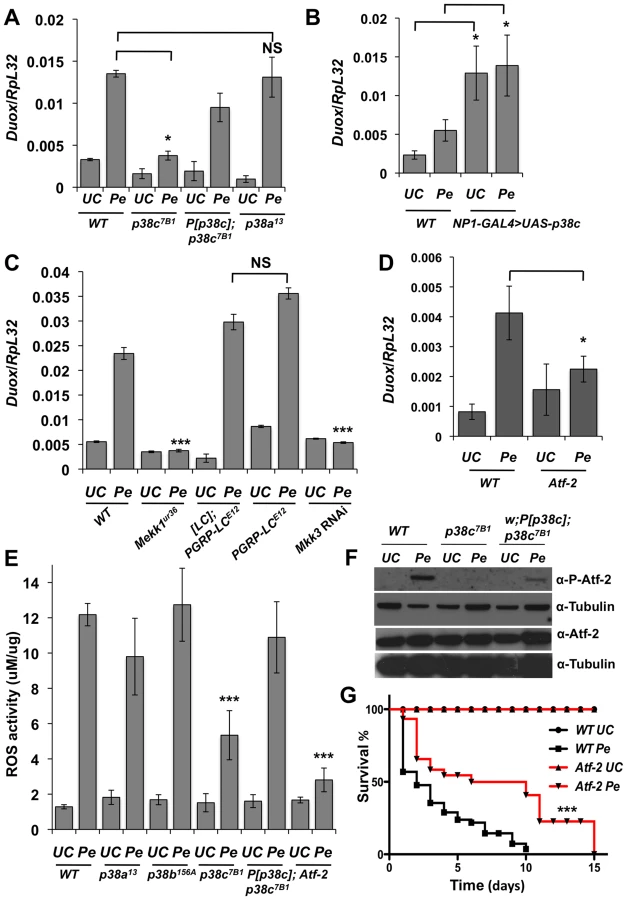
Previous studies have shown that the synthesis of antibacterial peptides under the control of the Imd pathway and production of ROS by Duox provide two complementary inducible defense mechanisms in the gut [5], [7], [10], [30]. The enrichment of p38c transcripts in this tissue and its induction post-infection pointed to a specific role of this MAPK in intestinal immune responses. We therefore analyzed the role of p38c in the resistance to oral infection with the non-lethal bacterium Ecc15. Figure 2A shows that p38c7B1 mutant flies are more susceptible to Ecc15 infection than wild-type flies. The level of susceptibility of p38c7B1 flies is similar to that observed for p38a or p38b mutant flies (Figure 2A). To confirm that the higher susceptibility of p38c7B1 flies is not due to the genetic background, we generated a fly line carrying both the p38c7B1 mutation and a rescue transgene containing the p38c locus including 300 bp of upstream sequences (referred to as P[p38c]). P[p38c];p38c7B1 flies showed a better survival to Ecc15 infection compared to p38c7B1 flies (Figure 2A). RT-qPCR analysis showed that p38c susceptibility is not due to an effect of p38c on the expression of p38a or p38b (Figure S3A).

We then investigated whether p38c affects the Imd pathway by measuring the expression of two antibacterial peptide genes, Diptericin and Attacin-A, in the gut of p38c deficient flies upon infection with Ecc15. We did not detect any effect of the p38c mutation on the expression levels of Diptericin after oral infection with this bacterium (Figure S3A). On the other hand, the basal levels of Diptericin and Attacin-A, as well as the induced levels of Attacin-A were higher in the intestines of p38c mutant flies (Figure S3A and B), but not in the rescued P[p38c];p38c flies. Thus the high susceptibility of p38c7B1 flies to oral infection with Ecc15 cannot be attributed to a lower activation of the Imd pathway.
As infection induces a ROS burst, we next investigated a possible link between p38c and resistance to oxidative stress. Previous studies have reported that p38a is required to resist oxidative stress [22]. However, the p38a strain used in these experiments (p38a1 also called mpk2) was later shown to carry a deletion affecting p38a and its neighboring gene p38c (Figure S1A) [28]. To re-evaluate the contribution of each p38 member to oxidative stress resistance, flies carrying null mutations for either p38a, p38b or p38c were fed on a diet containing 1% H2O2. Figure 2B shows that p38c7B1 but not p38a13, p38b156A or P[p38c];p38c flies, are more susceptible than wild-type to 1% H2O2. This indicates that p38c (and not p38a as initially suggested) contributes to resistance to H2O2. Of note, survival of p38c over-expressing flies to 1% H2O2 was not significantly different compared to wild-type (Figure S3C). Collectively, our data show that all three p38 genes contribute to survival to oral bacterial infection. It also highlights an important role of p38c in oxidative stress resistance.
p38c is required for P. entomophila-induced translation inhibition
As opposed to Ecc15, P. entomophila is highly pathogenic to flies when fed at high doses. P. entomophila pathogenicity has been linked to its capacity to induce severe intestinal damage [15], [16]. We investigated the role of p38c in the defense against P. entomophila oral infection. Surprisingly, we observed that p38c7B1 mutant flies are more resistant to infection with P. entomophila than either wild-type, p38a13, p38b156A mutants or P[p38c]; p38c7B1 flies (Figure 2C and S4B). In this experiment, the survival of P[p38c]; p38c7B1 was not statistically different from the wild-type. Nevertheless, the rescue effect was not observed at early time points possibly due to a lower level of p38c expression in P[p38c]; p38c7B1 flies compared to wild-type (Figure S1C). We also analyzed the survival of the p38c over-expressing flies to P. entomophila infection and observed that the p38c over-expressing flies died slightly faster than the wild-type (Figure 2D).
Infection with high doses of P. entomophila leads to a rupture of gut integrity caused by the loss of stem cell activity and hence an absence of epithelium renewal [15], [16]. To decipher how p38c influences susceptibility to P. entomophila infection, we monitored the stem cell division rate in wild-type, p38c7B1 and P[p38c]; p38c7B1 flies upon P. entomophila infection. Epithelium renewal can easily be monitored by counting the number of mitotic stem cells along the midgut using an anti-phospho-histone 3 antibody. Figures 3A and 3B show that there was a higher mitotic index in the infected guts of p38c7B1 mutants compared to wild-type or P[p38c]; p38c7B1 flies. The p38c7B1 mutant show a low level of stem cell activity under basal conditions and an overall gut structure similar to wild-type flies (Figure S4A), hence the increased intestinal stem cell activity upon infection is unlikely due to a defective gut organization but rather reflect a better capacity to repair the gut.

The lower epithelium renewal rate in P. entomophila infected guts is caused by a general inhibition of translation that impairs both immune and repair gene programs [16]. Translation inhibition can be monitored in the gut with a Dpt-lacZ transgene by analyzing the ratio between Dpt-lacZ ß-galactosidase activity and Dpt-lacZ transcripts, which reflect the extent of Dpt-lacZ translation and transcription respectively. A decrease in the ratio between ß-galactosidase activity and Dpt-lacZ transcript levels is indicative of a translation inhibition. As expected, the ratio of Lac-Z activity/Lac-Z mRNA was low in the gut of P. entomophila infected flies as compared to Ecc15 infected flies (Figure 3C). This assay revealed an increased level of translation in P. entomophila infected gut of p38c flies (Figure 3C). In contrast, both p38a13 and p38b156A flies exhibited a severe reduction in Dpt-lacZ translation, similar to the wild-type (Figure 3C). After infection or damage, epithelium renewal is stimulated by the release of a secreted ligand, Upd3, from stressed enterocytes, which activates the JAK/STAT pathway in progenitor cells to stimulate their division and their differentiation, thus establishing a homeostatic regulatory loop [14], [15]. Previous studies have shown that as a consequence of inhibition of translation, Upd3 was not produced in P. entomophila infected guts despite the strong induction of the upd3 gene. Figure 3D shows that, following P. entomophila infection, the level of Upd3 is higher in the intestines of the p38c mutants than in wild-type or P[p38c];p38 flies. These results indicate that p38c participates in P. entomophila-induced translation inhibition in the gut. Thus, increased level of translation in p38c7B1 flies could explain why p38c7B1 flies survive better than wild-type.
p38c regulates Duox transcription after infection
P. entomophila-mediated translation inhibition is largely a consequence of the ROS produced by the Duox enzyme. Indeed, knocking-down Duox alleviates the inhibition of translation induced by P. entomophila [16] and increase short-term survival to P. entomophila (Figure S4B). A study has shown that Duox gene expression is regulated by the transcription factor Atf-2 downstream of p38a-Mekk3-Mekk1-PGRP-LC pathway [7]. However, the p38a1 mutant used to analyze Duox regulation was the one that also contains a deletion affecting its neighboring gene p38c [28]. This raises the possibility that Duox is regulated by p38c and not by p38a as initially proposed. To test this hypothesis, we monitored Duox transcriptional activation in p38a13 and p38c7B1 single mutants. Figure 4A shows that Duox expression upon P. entomophila infection is lower in p38c compared to wild-type or to p38a13 mutant flies. The genomic P[p38c] element rescues the loss of Duox induction in p38c mutants. Moreover, Figure 4B shows that overexpression of p38c is sufficient to induce Duox in the absence of infection. An increase in Duox gene expression should lead to higher levels of ROS, which are known to cause damage and to stimulate an epithelium renewal [15]. Consistent with this notion, a higher number of mitotic stem cells and a higher amount of Upd3 was found in flies that over-express p38c in absence of infection (Figures S4C–E). Using null mutations (Mekk1Ur36, PGRP-LCE12) and an RNAi construct (Mkk3), we then tested the effect of PGRP-LC, Mkk3 and Mekk1 mutations on the transcriptional induction of Duox by P. entomophila. Contrary to Ha et al (2009), we did not observe any effect of PGRP-LC on Duox expression but confirmed the requirement of both Mkk3 and Mekk1 (Figure 4C). Hence, p38c, Mkk3 and Mekk1, but not PGRP-LC or p38a, control the up-regulation of Duox in response to intestinal infection. This observation raises the hypothesis that the increased resistance of the p38c7B1 mutants to P. entomophila infection is due to a reduced production of ROS by Duox. To test this hypothesis, we compared ROS levels in p38c7B1 and wild-type fly guts upon P. entomophila infection using the Amplex Red reagent (Invitrogen). The data showed that ROS levels are indeed lower in the intestines of p38c7B1 mutant flies than in wild-type (Figure 4E). Collectively, our study shows that Duox induction upon P. entomophila infection depends on p38c rather than p38a. The lower level of Duox-mediated ROS activity in p38c flies provides an explanation why translation is not inhibited in these flies and consequently why they survive better to P. entomophila infection.
p38c and Atf-2 function in a same pathway to control Duox transcription following infection
A previous study combining an in vivo RNAi approach and promoter analysis has provided compelling evidence that Duox is directly regulated by the transcription factor Atf-2 [7]. We used a recently described fly line deleted for atf-2 [31] to confirm that Atf-2 is indeed required for Duox transcription upon P. entomophila infection (Figure 4D). We observed that atf-2 mutant flies exhibit lower ROS levels in the intestine following P. entomophila ingestion (Figure 4E) and are more resistant to this pathogen than wild-type (Figure 4G). Thus, the atf-2 mutant phenocopies the p38c7B1 mutants suggesting that p38c and Atf-2 may function in a common pathway to regulate Duox expression. In mammals, ATF2 is activated upon phosphorylation by p38 in response to various stresses [32]. Atf-2 is also phosphorylated following heat and osmotic stress in Drosophila S2 cells [31]. Figure 4F showed that Atf-2 is phosphorylated in the intestine in response to P. entomophila infection and that this phosphorylation is lost in p38c null flies. The genomic P[p38c] element partially rescues Atf-2 phosphorylation in p38c flies. As the total amount of Atf2 protein is not affected in p38c mutants, we conclude that p38c regulates Atf2 at the post-transcriptional level. Furthermore, the over-expression of p38c in the intestine is sufficient to phosphorylate Atf2 in the absence of infection (Figure S5A). This together with our in vitro analysis showing that p38c phosphorylates mammalian ATF2 (Figure S2B) strongly suggests that p38c directly phosphorylates Atf-2. Our results led us to conclude that a Mekk1/Mkk3/p38c/Atf-2 signaling pathway controls the expression of Duox expression upon intestinal infection.
p38c and Atf3 function in a pathway regulating intestinal lipid homeostasis
We next explored whether p38c has additional roles in the intestine beyond regulating Duox expression. To gain insight into p38c function, we profiled genome expression in dissected intestines from unchallenged w1118 and w1118, p38c7B1 adult female flies using Affymetrix GeneChip Drosophila Genome 2.0 Arrays. Loss of p38c affected the expression of 408 transcripts, with 264 up-regulated and 144 down-regulated by at least 2-fold relative to the control. We used the GO clustering analysis tools and manual annotation to find functional categories within the 408 transcripts (Figure 5A–5B, see Table S1 for complete data set). As expected, one of the most represented GO category was the stress response with 48 genes modulated in this category. Consistent with the results described above, genes involved in oxido-reduction (e.g. oxidoreductases, GSTs) were differentially regulated in p38c intestine as compared to the wild-type. Our microarray analysis indicates that 26 immunity genes were up-regulated or down-regulated in p38c mutant flies. The Drosophila midgut is lined by a chitinous matrix, the peritrophic matrix (PM), which protects the midgut epithelium from abrasive food particles, digestive enzymes, and pathogen toxins [2]. 18 genes encoding for chitin-binding proteins were found to be modulated in the p38c7B mutant pointing to a role of this MAPK in the remodeling of the PM barrier (Figure 5B). The GO category with the largest number of genes was metabolism, notably sugar and lipid metabolism. Interestingly, many genes shown to be regulated in our analysis, notably genes involved in immunity, chitin and lipid metabolism (indicated with a # in Figure 5B) have previously been identified to be regulated by Atf3, a bZIP - transcription factor related to Atf-2 [33]. This finding and the observation that p38c expression is increased in atf376 mutant larvae [33] led us to explore a link between p38c and Atf3. One of the most striking phenotype of atf3 mutant is an overload of lipids in the intestine of larvae [34]. Using an RNAi approach to knockdown Atf3 in the midgut of adult flies we also found a similar phenotype (Figure S6A). We also observed that p38c mutant flies accumulate lipids especially in two domains (R2b and R5) in the gut (Figure 6A for Nile-red in R2b and S6B for Oil-redO in whole intestine). To determine if the effect of the p38c knockdown on lipid metabolism is cell autonomous, we made positively marked clones of p38c RNAi knockdown using the esgtsF/O system [14], and examined lipid accumulation by Nile-red staining on whole guts. Whereas lipid accumulation was normal in cells outside the clones, there was accumulation of lipid in p38c knockdown clones (Figure 6B). Thus, the increased lipid accumulation in p38c mutant guts was not caused by altered feeding but to a specific requirement of p38c in the enterocytes. Of note, no lipid accumulation was observed in the intestine of p38a and p38b mutant flies (Figure S6C), whereas Mekk1 flies showed a similar lipid accumulation in the intestine as p38c mutants (Figure 6A).


In mammals, the p38 pathway controls the transcription of the ATF3 gene in response to oxidative stress [34]. We thus investigated whether p38c regulates Atf3 at the transcriptional level. Figure 6C shows that the atf3 gene is expressed at a significantly lower level in unchallenged p38c mutant flies compared to wild-type. Conversely, over-expression of p38c in wild-type flies leads to an increased atf3 expression (Figure S6D). Finally, we confirmed that atf3 is epistatic to p38c as the over-expression of Atf3 in the p38c mutant restores a wild-type level of lipids in the gut (Figure 6D). These data indicates that Mekk1, p38c and Atf3 function in a pathway required in the gut for lipid metabolism.
The results in the first part of this manuscript have shown that Mekk1 and p38c regulate the activity the transcription factor Atf-2 through its phosphorylation. Since p38c also regulates Atf3 gene expression, we investigated whether the effect of p38c on Atf3 transcription is mediated by Atf-2. Figure 6C shows that the amount of Atf3 transcripts in the intestine is lower in Atf-2 mutants than in wild-type, however the reduction is less marked than that observed in the p38c mutant background. Moreover, Atf-2 deficient flies also accumulate lipids in the two gut regions but at a lower level than Atf3 and p38c mutants (Figure 6A). Altogether, these results show that the effect of p38c on Atf3 expression is partially mediated by Atf-2.
Discussion
The p38 MAPKs have been implicated in the regulation of stress and immune responses in eukaryotes [21]. Despite several studies, the p38 MAPK pathway remains poorly characterized in Drosophila. In this study, we have analyzed the function of p38 MAPKs in the context of intestinal host defense and metabolic homeostasis, with an emphasis on p38c. Our study confirms a previous study indicating that both p38a and p38b are required to resist oral bacterial infection [28]. This function is not mediated through the Imd pathway, which regulates the antimicrobial response ([28], our data). Chen et al (2010) have proposed that p38a and p38b contribute to host defense by regulating Hsf1, a transcription factor which activates the expression of a large number of stress response genes, including many molecular chaperones (e.g. Heat Shock Proteins).
In this study, we have focused our attention on p38c, which has been somewhat neglected so far. The only function that had been previously attributed to p38c was the regulation of the dopadecarboxylase gene in the wounded epithelium [25]. Here, we show that the p38c gene is strongly expressed in the gut compared to p38a and p38b and is strongly up-regulated upon bacterial infection (Buchon 2009, this study). Like p38a and p38b, p38c is also required to survive an infection with the non-lethal bacterium Ecc15. Moreover, we show in vitro assay reveals that p38c has indeed a kinase activity and can phosphorylate mammalian ATF2. Although it remains to be shown how p38c gets activated following infection, our results suggest that it functions as a kinase rather than a scaffold protein. A role for p38c as a kinase is also supported by the observation that Atf-2 phosphorylation is impaired in p38c deficient flies.
A first surprising result was the observation that p38c flies survive better to oral infection with P. entomophila. As P. entomophila pathogenesis is caused by an excessive activation of stress pathways caused by ROS-induced damage, we investigated the link between Duox activation and p38c. Our study shows that Duox expression is regulated by p38c and not by p38a as previously reported [7]. Consistent with this, a lower level of ROS was observed in p38c flies infected with P. entomophila. The lower expression of Duox in p38c flies provides an explanation why these flies are susceptible to Ecc15, an infection model in which Duox contributes to survival [30], while being more resistant to P. entomophila, an infection model in which ROS production by Duox contributes to pathogenesis [16].
Consistent with Ha et al. 2009 we observed that Duox expression is regulated by the Atf-2 transcription factor upon phosphorylation by a MAPK pathway involving p38c, Mkk3 and Mekk1. However, we did not find PGRP-LC to be involved in Duox regulationin response to P. entomophila infection, indicating that this cascade is not triggered by peptidoglycan recognition as previously reported [7]. To date, the factors that trigger the p38c MAPK cascade following infection remains to be identified. One possibility is that uracil, a bacterial product shown to modulate Duox activity by the Gαq-Phospholipase Cß-Ca2+ pathway [6], is involved in the activation of the p38c MAPK pathway. Our study further shows that p38c (and not p38a as initially proposed) contributes to resistance to H2O2. This indicates that the p38c is involved both in the production of extracellular ROS by regulating Duox transcription and in the protection against the cytotoxic effect of ROS. Our identification of a role of p38c in the antioxidant response is an important step toward a better understanding of ROS metabolism in the intestine. This response may not involve Atf-2 because we did not observed an increased susceptibility of atf-2 mutant flies to H2O2 or a phosphorylation of Atf-2 in wild-type flies in response to H2O2 (Figure S5C and D). It would be interesting to characterize how p38c mediates this antioxidant response. A recent paper has suggested that p38 could affect the activity of glutathione-S-transferases (GSTs) by modulating its substrate specificities in Drosophila [35]. In this line, our microarray shows that the expression of many genes involved in oxido-reduction (GSTs) and detoxification are modulated by p38c.
Previous study using an in vivo RNAi study has shown that reduction of Atf-2 expression in the fat body results in reduced triglyceride storage and a decreased survival under starvation conditions [36]. Multiple genes that control triglyceride metabolism, including the PEPCK gene, which encodes a key enzyme required for triglyceride synthesis via glycerol-3-phosphate, are expressed at lower levels in Atf-2 knockdown flies. During the course of our study, we noticed that p38c mutant flies are leaner than wild-type flies and have also reduced TAG levels (Figure S7A and S7B). Consistent with the decrease in TAG reserves, p38c7B1 flies (but not p38a13 and p38b156A) succumb more rapidly to acute starvation, in which flies are only given a source of water but no source of nutrition (Figure S7C). These observations suggest that p38c and Atf-2 also function in a common pathway to regulate lipid homeostasis in the fat body. Hence, it is likely that the regulation Atf2 transcription factor by p38c is not restricted to the intestine.
Another unexpected observation was that p38c deficient flies accumulate lipids in the intestine. Such accumulation is similar to the phenotype described for Atf3 mutant larvae [33]. In addition, in both atf3 and p38c deficient flies, genes involved in lipid metabolism and immunity are up-regulated in the intestine. This observation suggested that p38c and Atf3 function in a common pathway in the gut to regulate lipid metabolism and immune homeostasis. In agreement with this, we could show that Atf3 is regulated in the intestine at the transcriptional level by p38c. This effect is partially mediated by Atf-2 as supported by a RT-qPCR analysis and the intermediate lipid accumulation phenotype of Atf-2 mutants. Thus, Atf3 gene expression would be regulated by p38c in the intestine by both Atf2-dependent and Atf2-independent pathways. Many immune genes are expressed at higher than wild-type levels in the intestine of p38c deficient flies in the absence of infection. The mechanism underlying the higher activation of these immune genes in p38c flies was not identified, however Rynes and colleagues have shown that immune gene activation in atf3 larvae requires Relish, the Imd pathway transcription factor [33]. Nevertheless, the effect of p38c on antimicrobial peptide gene expression could be direct if Atf-2 or Atf3 directly regulated the transcription of these genes under basal conditions. Alternatively, a higher Imd pathway activity could be an indirect consequence of the rupture of gut homeostasis.
The present study allowed us to better delineate the p38 MAPK responsive pathway in Drosophila, revealing a central role for p38c in the gut. It also revealed that many roles initially attributed to p38a [22], notably in the resistance to H2O2 and to starvation, are in fact mediated by p38c. Seisenbacher et al. [29], have previously shown that p38a also contribute to the resistance to osmotic shock using a fly line deleted for both p38a and p38c. To clarify this point, we monitored the resistance to osmotic stress of single p38 mutants. Figure S8B shows that only the double mutant p38a1 (Mpk2) affecting both p38a, p38c but neither p38a nor p38c single mutant exhibit an increase susceptibility to high salt (Figure S8). Our study also confirms a previous study showing that p38b contributes to the resistance to osmotic shock ([29], Figure S8). To avoid any confusion in the future, we propose that the p38a1 (also called mpk2) strain should be renamed p38a-cdel.
There are substantial parallels between the regulation of ATF transcription factors and p38 in Drosophila, C. elegans, and mammals. This is probably a legacy of the ancestral role of this pathway in animal stress responses. In C. elegans, PMK1 (the p38 homolog) regulates the phosphorylation of ATF7 (the Atf-2 homologue) to control the transcription of immune genes in the intestine upon ingestion of pathogenic bacteria [37]. The regulation of ATF2 by p38-mediated phosphorylation [38], as well as interactions between ATF2, ATF3 and p38 have also been observed in mammals. ATF3 is up-regulated through the p38 pathway in response to oxidative or anisomycin stresses [39], [40]. Another study reports that ATF2 can activate the expression of ATF3 in response to stress in colonic cancer cells [41]. Moreover, ATF3 has also been shown to regulate lipid metabolism in pancreatic ß-cells of mice [42]. Our study suggests that ATF2 and ATF3 could play a fundamental role in the intestine of mammals. We believe that inter-specific comparisons of p38 MAPK pathway functions should allow us to better understand the mechanism underlying p38 activation and to decipher the specific roles that this pathway has acquired in different organs.
Materials and Methods
Drosophila stocks and rearing
CantonS (CanS) and w1118 flies were used as wild-type controls. The following fly lines were used in this study: y1 w67c23; p38a13 [28], y1 w67c23; p38b156A [28], yw; p38bd27/CyO,y+ [23], PGRP-LCE12 [43], p38a1 [22], MEKK1Ur36 [44], p38c7B1 [25], [PGRP-LC];PGRP-LCE12 [12], Atf-2 (PBac[32]Atf-2c06467) [31], Gαq1 [5], UAS-Atf3 [45], w*; Df(3R)w6/TM6C (Bloomington stock 7251), NP1-GAL4 (II) (or Myo1A-Gal4 [10], UAS-Duox-IR (GD2593, VDRC), UAS-mkk3-IR (KK108550, VDRC) and UAS-Atf3-IR (26741, Bloomington TRiP line). esg-Flip-Out system (esgF/O, [14]: esg-Gal4, tub-Gal80TS; UAS-FLP actFRTCD2FRTGal4, UAS-GFP flies were crossed with UAS-p38c-IR (KK103439, VDRC).
For RNAi (IR) studies, F1 progeny carrying one copy of the driver as well as one copy of the UAS-IR were raised at 18°C during their larval and pupal development, and then moved to 29°C for 8 days to activate the UAS-IR. Drosophila stocks were maintained using standard fly medium comprising of 6% cornmeal, 6% yeast, 0.62% agar, 0.1% fruit juice, supplemented with 10.6 g/L moldex and 4.9 ml/L propionic acid. All stocks were maintained at 25°C on a 12 h light/12 h dark cycle unless otherwise stated.
Plasmids and transgenic lines
UAS-p38c construct: A full-length cDNA of p38c was amplified from total cDNA of OregonR flies and cloned into the pDONR207 Gateway vector (Invitrogen) and subsequently sub-cloned in the pTW (Drosophila Genomics Resource Center plasmid) transgenesis vector and used to generate transgenic flies. A fly line carrying the transgene on the third chromosome was established and used as UAS-p38c.
P[p38c] rescue construct: To generate a rescue transgene of p38c, we amplified by PCR a fragment comprising the coding region of p38c with the 3′UTR and 300 bp of the upstream sequence (corresponding to the sequence between p38c and p38a,). The amplicon was cloned into pCasper4 plasmid using the restriction sites Not1 and Xho1, and used for generating transgenic flies according to standard procedures. Fly line carrying the transgene on the second chromosome, and was introgressed into the p38c7B1 mutant.
Bacterial strains and infection experiments
Erwinia carotovora carotovora 15 (Ecc15) and Pseudomonas entomophila (Pe) are Gram-negative bacteria described in [9], [46]. Both bacteria were cultured overnight in LB medium at 29°C. For oral infection, batches of 20 adult female flies of 3 to 5 day old age were starved for 2 h at 29°C in an empty vial before being transferred to a fly vial with infection solution and maintained at 29°C. The infection solution consisted of an equal volume of 100× concentrated pellet from an overnight culture of Ecc15 or Pe (OD600 = 200) with a solution of 5% sucrose (1∶1) was deposited on a filter disk that completely covered the surface of standard fly medium. Flies were incubated for one day at 29°C on the contaminated filter, after which they were transferred to fresh vials containing standard medium without living yeast.
Oxidative stress, osmotic stress and starvation stress assay
For survivals to oxidative stress, flies were fed on a standard medium containing 1% H2O2 (Sigma). To assess sensitivity to osmotic stress, eggs were collected on apple agar plates and 20–30 eggs per genotype were transferred on to standard medium containing 0.2 M NaCl and survival to adulthood was recorded. Starvation experiments were performed with 2–3 day old female flies in batched of 20 flies. Flies were transferred to vial containing 0.62% agarose to prevent dessication of flies (supplemented with moldex and propionic acid). The survival analysis was done by counting dead flies every 10 h.
RT-qPCR
Twenty dissected guts (crop, midgut and hindgut without Malpighian tubules) were collected in Trizol (Invitrogen) and total gut RNA was extracted according to manufacturer's instructions. Quality and quantity of RNA was determined using NanoDrop ND-1000 spectrophotometer. 1 µg of RNA was used to generate cDNA using SuperScript II (Invitrogen, Carlsbad, California, United States). RT-qPCR was performed using dsDNA dye SYBR Green I (Roche Diagnostics, Basel, Switzerland). Expression values were normalized to RpL32. Primers sequences used is provided in Table S2.
Quantitative measurements of β-galactosidase activity
Ten to twenty female adult guts were dissected and homogenized in Z buffer [47] and centrifuged for 10 minutes at 10 000 r.p.m. (4°C). β-Galactosidase activity was measured as described in [48] and normalized to the protein concentration determined by Bradford assay (Sigma). Results are represented as nmol product formed/min/mg protein.
Monitoring the level of translation
To determine the level of translation we monitored the ratio between Dpt-lacZ (ß-galactosidase) activity (as described above) and Dpt-lacZ transcript level (normalized on the amount of RpL32) in gut extracts of Dpt-lacZ flies. The ratio obtained from flies collected at 16 h post Ecc15 infection was set up as 1. Reduction of this ratio indicates an inhibition of translation.
Production of the antibody anti-p38c
The anti-p38c antibody was produced by immunizing rabbits with GST-p38c fusion protein following the 28-day protocol (Eurogentec, Belgium).
Imaging and immunohistochemistry
For immunofluorescence, guts were dissected in 1X PBS, fixed for 20 minutes in PBS and 0.1% Tween 20 (PBT), and 4% paraformaldehyde; then stained with primary antibody [1/100 anti-p38c (this study); 1/500 anti-PH3 (Upstate/Millipore)] in PBT+2% BSA. Secondary staining was performed with Alexa594 anti-rabbit antibodies (Invitrogen). DNA was stained with 4′,6 - diamidino-2-phenylindole DAPI (Sigma). The stained gut tissue was mounted in the antifading agent Citifluor AF1 (Citifluor Ltd.). PH3 positive cells were counted along the gut with Axioplot imager (Zeiss).
Reactive oxygen species measurement
Ten adult female guts (including the crop) per genotype were dissected and collected in 300 µl Ringer's solution (pH 7.2) 45 minutes after oral infection with P. entomophila. Dissected guts were homogenized and centrifuged to remove debris. ROS level in the adult guts was monitored by the addition of 100 µl reaction buffer (50 µM Amplex Red, Invitrogen # A12222; 0.2 µM horseradish peroxidase from Sigma prepared in Drosophila Ringer's solution) to 20 µl homogenized gut extract. Fluorescence intensity was measured with a fluorescence microplate reader using excitation wavelength of 530 nm and emission wavelength of 590 nm. A standard curve was plotted with a range of H2O2 dilutions and used to determine the amount of H2O2 and normalized to the amount of protein.
Western blot analysis
At least 30 female adult guts were dissected and homogenized in lysis buffer (0.5%NP40, 500 mM NaCl, 500 mM Tris-HCl pH 7.4, 20 mM EDTA, 10 mM NaF, 2 mM benzamidine, and a cocktail of protease and phosphatase inhibitors from Roche). Protein quantification was done with Bradford reagent (Sigma), before equal amount of protein was loaded and separated on a NUPAGE 4–12% Bis-Tris precast gel (Invitrogen) under reducing conditions. Following transfer onto a nitrocellulose membrane and blocking in 2% bovine serum albumin in PBT for 1 h, the membrane was incubated with the appropriate primary antibodies at 4°C overnight. Secondary antibodies were incubated for 2 h at room temperature and chemiluminescence signal was detected using ECL (GE Healthcare) according to the manufacturer's instructions. Primary antibodies used were at the following dilutions: α-tubulin antibody 1∶10000 (Sigma), α-Atf-2 1∶1000 and α-P-Atf-2 1∶1000 [31], α-Upd3 1∶1000 ([16]; gift from Yu-Chen Tsai) and α-p38c 1∶1000. Secondary antibodies used were: anti-rabbitHRP 1∶1000 (Dako); anti-mouseHRP 1∶1000 (GE Healthcare).
Nile Red and Oil Red O staining
Three to four day old female fly guts were dissected and fixed in 500 µl of 4% paraformaldehyde for 20 minutes at room temperature. For nile Red staining (to stain intracellular lipid droplets), fixed guts were incubated in Nile Red solution (diluted 1∶100 in 1X PBS from a 100 µg/ml stock solution in acetone) [48] for 15 minutes at room temperature and then washed with 1X PBS three times before mounting on a slide in 70% Glycerol. The tissue was imaged with a Zeiss LSM700 upright confocal microscope under the 20X/0.8 NA objective. At least 10 guts per genotype were imaged per experiment.
For Oil Red O staining, fixed guts washed twice with MilliQ water, 100% propylene glycol and the incubated in propylene glycol for 10 minutes. The tissue was then incubated in 0.5% Oil Red O in propylene glycol at 60°c for 30 minutes. 10 ml of Oil Red O staining solution was prepared by mixing 6 ml (0.1% Oil Red O) in isopropanol and 4 mL MilliQ H2O, which was filtered through a 0.45 um syringe filter. After 30 minutes incubation in Oil Red O solution the gut tissue was washed twice in 85% propylene glycol at RT, thrice in MilliQ water before mounting onto a slide in 70% Glycerol. Samples were imaged with a Leica MZ16F dissecting microscope.
Triacylgylceride assay
For colorimetric triacylglyceride assays, five 3–5 day old female flies were washed in phosphate-buffered saline (1X PBS) and then homogenized in 200 µl of PBST. The homogenate was heat inactivated at 70°C and then centrifuged (3,000 rpm; 5 minutes; 4°C). 20 µl aliquots of the supernatant were assessed in 96-well plates with the Triglyceride reagent and Free Glycerol Reagent (Sigma). Lipid levels were normalized to the protein contents.
Promega's Kinase-Glo luminescent kinase assay
Kinase-Glo is a homogeneous non-radioactive method for determining the activity of purified kinases by quantifying the amount of ATP remaining in a solution following a kinase reaction. Luminescence signal correlated with the amount of ATP present and inversely proportional to the amount of kinase activity. To determine EC50 of P38c-his, a serial dilution of the kinase was added to the reaction buffer (40 mM Tris-HCl, 0.1 mg/ml BSA, 20 mM MgCl2 and 10 µM ATP) with 1 µg ATF2-GST in a final volume of 50 µl. After 30 minutes of reaction, 50 µl Kinase-Glo Reagent was added to each well and incubated for another 10 minutes at room temperature. The p38 kinase inhibitor SB203580 compound (Invitrogen) was dissolved in dimethyl sulfoxide (DMSO) and then added to the reaction buffer with the kinase prior to the addition of the substrate. For the 0 µM SB203580 condition, DMSO was added alone. Luminescence of each well was measured by TecanF200 microplate spectrofluorometer.
Analysis of whole genome mRNA expression by Affymetrix Droso2.0 chips
The microarray analysis was performed on 3 independent biological repeats. RNA from 30 guts (crop, midgut and hindgut without Malpighian tubules) per genotype (w1118 and p38c7B1) of 3 to 5 days old females from was isolated by TRIzol extraction, and purified with RNA clean up purification kits (Macherey Nagel). The quality of RNA was assessed on Agilent 2100 bioanalyzer chips. For each sample, 100 ng of total RNA was amplified and labeled using the GeneChip IVT Labeling Kit according to the manufacturer's protocol. Affymetrix Drosophila Genome 2.0 arrays were hybridized with 30 mg of labeled cRNA, washed, stained, and scanned as described in the Affymetrix Manual. Statistical analyses were performed using the R and Bioconductor statistical packages. All the genes integrated in the analysis shown in Figure 5 were differentially expressed by at least 2-fold with a p value <0.05. The raw data from the microarray experiment have been deposited in ArrayExpress (accession number E-MTAB-2740).
Statistics
Each experiment was repeated independently a minimum of three times (unless otherwise indicated), error bars represent the standard error of the mean of replicate experiments (unless otherwise indicated). Statistical significance was determined using Student's t test or log–rank test on GraphPad Prism, and P values of <0.05 = *, <0.01 = ** and <0.001 = *** were considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LemaitreB, Miguel-AliagaI (2013) The digestive tract of Drosophila melanogaster. Annu Rev Genet 47 : 377–404.
2. BuchonN, BroderickNA, LemaitreB (2013) Gut homeostasis in a microbial world: insights from Drosophila melanogaster. Nat Rev Microbiol 11 : 615–626.
3. LeeKA, LeeWJ (2014) Drosophila as a model for intestinal dysbiosis and chronic inflammatory diseases. Dev Comp Immunol 42 : 102–110.
4. FerrandonD (2013) The complementary facets of epithelial host defenses in the genetic model organism Drosophila melanogaster: from resistance to resilience. Curr Opin Immunol 25 : 59–70.
5. HaEM, LeeKA, ParkSH, KimSH, NamHJ, et al. (2009) Regulation of DUOX by the G alpha q-Phospholipase C beta-Ca2+ Pathway in Drosophila Gut Immunity. Developmental Cell 16 : 386–397.
6. LeeKA, KimSH, KimEK, HaEM, YouH, et al. (2013) Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila. Cell 153 : 797–811.
7. HaEM, LeeKA, SeoYY, KimSH, LimJH, et al. (2009) Coordination of multiple dual oxidase-regulatory pathways in responses to commensal and infectious microbes in Drosophila gut. Nat Immunol 10 : 949–957.
8. TzouP, OhresserS, FerrandonD, CapovillaM, ReichhartJM, et al. (2000) Tissue-specific inducible expression of antimicrobial peptide genes in Drosophila surface epithelia. Immunity 13 : 737–748.
9. BassetA, KhushRS, BraunA, GardanL, BoccardF, et al. (2000) The phytopathogenic bacteria Erwinia carotovora infects Drosophila and activates an immune response. Proc Natl Acad Sci U S A 97 : 3376–3381.
10. BuchonN, BroderickNA, PoidevinM, PradervandS, LemaitreB (2009) Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell Host Microbe 5 : 200–211.
11. Bosco-DrayonV, PoidevinM, BonecaIG, Narbonne-ReveauK, RoyetJ, et al. (2012) Peptidoglycan sensing by the receptor PGRP-LE in the Drosophila gut induces immune responses to infectious bacteria and tolerance to microbiota. Cell Host Microbe 12 : 153–165.
12. NeyenC, PoidevinM, RousselA, LemaitreB (2012) Tissue - and ligand-specific sensing of gram-negative infection in drosophila by PGRP-LC isoforms and PGRP-LE. J Immunol 189 : 1886–1897.
13. AmcheslavskyA, JiangJ, IpYT (2009) Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell 4 : 49–61.
14. JiangH, PatelPH, KohlmaierA, GrenleyMO, McEwenDG, et al. (2009) Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137 : 1343–1355.
15. BuchonN, BroderickNA, ChakrabartiS, LemaitreB (2009) Invasive and indigenous microbiota impact intestinal stem cell activity through multiple pathways in Drosophila. Genes Dev 23 : 2333–2344.
16. ChakrabartiS, LiehlP, BuchonN, LemaitreB (2012) Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe 12 : 60–70.
17. OpotaO, Vallet-GelyI, VincentelliR, KellenbergerC, IacovacheI, et al. (2011) Monalysin, a novel ss-pore-forming toxin from the Drosophila pathogen Pseudomonas entomophila, contributes to host intestinal damage and lethality. PLoS Pathog 7: e1002259.
18. JiangH, EdgarBA (2012) Intestinal stem cell function in Drosophila and mice. Curr Opin Genet Dev 22 : 354–360.
19. LemaitreB, GirardinSE (2013) Translation inhibition and metabolic stress pathways in the host response to bacterial pathogens. Nat Rev Microbiol 11 : 365–369.
20. CuendaA, RousseauS (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773 : 1358–1375.
21. QiM, ElionEA (2005) MAP kinase pathways. J Cell Sci 118 : 3569–3572.
22. CraigCR, FinkJL, YagiY, IpYT, CaganRL (2004) A Drosophila p38 orthologue is required for environmental stress responses. EMBO Rep 5 : 1058–1063.
23. CullyM, GenevetA, WarneP, TreinsC, LiuT, et al. (2010) A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol Cell Biol 30 : 481–495.
24. ShinzawaN, NelsonB, AonumaH, OkadoK, FukumotoS, et al. (2009) p38 MAPK-dependent phagocytic encapsulation confers infection tolerance in Drosophila. Cell Host Microbe 6 : 244–252.
25. DavisMM, PrimroseDA, HodgettsRB (2008) A member of the p38 mitogen-activated protein kinase family is responsible for transcriptional induction of Dopa decarboxylase in the epidermis of Drosophila melanogaster during the innate immune response. Mol Cell Biol 28 : 4883–4895.
26. HanSJ, ChoiKY, BreyPT, LeeWJ (1998) Molecular cloning and characterization of a Drosophila p38 mitogen-activated protein kinase. Journal of Biological Chemistry 273 : 369–374.
27. HanZQS, EnslenH, HuXD, MengXJ, WuIH, et al. (1998) A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Molecular and Cellular Biology 18 : 3527–3539.
28. ChenJ, XieC, TianL, HongL, WuX, et al. (2010) Participation of the p38 pathway in Drosophila host defense against pathogenic bacteria and fungi. Proc Natl Acad Sci U S A 107 : 20774–20779.
29. SeisenbacherG, HafenE, StockerH (2011) MK2-dependent p38b signalling protects Drosophila hindgut enterocytes against JNK-induced apoptosis under chronic stress. PLoS Genet 7: e1002168.
30. HaEM, OhCT, BaeYS, LeeWJ (2005) A direct role for dual oxidase in Drosophila gut immunity. Science 310 : 847–850.
31. SeongKH, LiD, ShimizuH, NakamuraR, IshiiS (2011) Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145 : 1049–1061.
32. GuptaS, CampbellD, DerijardB, DavisRJ (1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267 : 389–393.
33. RynesJ, DonohoeCD, FrommoltP, BrodesserS, JindraM, et al. (2012) Activating transcription factor 3 regulates immune and metabolic homeostasis. Mol Cell Biol 32 : 3949–3962.
34. Hai T (2006) The ATF Transcription Factors in Cellular Adaptive Response. In: Ma J, editor. Gene expression and regulation. New York: Springer, 2006. pp. 329–340.
35. WongtrakulJ, SukittikulS, SaisawangC, KettermanAJ (2012) Mitogen-activated protein kinase p38b interaction with delta class glutathione transferases from the fruit fly, Drosophila melanogaster. J Insect Sci 12 : 107.
36. OkamuraT, ShimizuH, NagaoT, UedaR, IshiiS (2007) ATF-2 regulates fat metabolism in Drosophila. Mol Biol Cell 18 : 1519–1529.
37. ShiversRP, PaganoDJ, KooistraT, RichardsonCE, ReddyKC, et al. (2010) Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet 6: e1000892.
38. RaingeaudJ, GuptaS, RogersJS, DickensM, HanJ, et al. (1995) Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem 270 : 7420–7426.
39. HoetzeneckerW, EchtenacherB, GuenovaE, HoetzeneckerK, WoelbingF, et al. (2011) ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med 18 : 128–134.
40. LuD, ChenJ, HaiT (2007) The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem J 401 : 559–567.
41. LeeSH, BahnJH, WhitlockNC, BaekSJ (2010) Activating transcription factor 2 (ATF2) controls tolfenamic acid-induced ATF3 expression via MAP kinase pathways. Oncogene 29 : 5182–5192.
42. ZmudaEJ, QiL, ZhuMX, MirmiraRG, MontminyMR, et al. (2010) The roles of ATF3, an adaptive-response gene, in high-fat-diet-induced diabetes and pancreatic beta-cell dysfunction. Mol Endocrinol 24 : 1423–1433.
43. GottarM, GobertV, MichelT, BelvinM, DuykG, et al. (2002) The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 416 : 640–644.
44. InoueH, TatenoM, Fujimura-KamadaK, TakaesuG, Adachi-YamadaT, et al. (2001) A Drosophila MAPKKK, D-MEKK1, mediates stress responses through activation of p38 MAPK. EMBO J 20 : 5421–5430.
45. SekyrovaP, BohmannD, JindraM, UhlirovaM (2010) Interaction between Drosophila bZIP proteins Atf3 and Jun prevents replacement of epithelial cells during metamorphosis. Development 137 : 141–150.
46. VodovarN, VinalsM, LiehlP, BassetA, DegrouardJ, et al. (2005) Drosophila host defense after oral infection by an entomopathogenic Pseudomonas species. Proc Natl Acad Sci U S A 102 : 11414–11419.
47. Miller JH (1972) Experiments in molecular genetics. New York: Cold Spring Harbor Laboratory Press.
48. GreenspanP, MayerEP, FowlerSD (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100 : 965–973.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 9
Nejčtenější v tomto čísle
- Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals
- Nipbl and Mediator Cooperatively Regulate Gene Expression to Control Limb Development
- Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel
- Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation