
Interactions of Chromatin Context, Binding Site Sequence Content, and Sequence Evolution in Stress-Induced p53 Occupancy and Transactivation
It is well established that p53 binds DNA elements near p53 target genes to regulate the response to cellular stress. To assess factors influencing binding to response elements and subsequent gene expression, we have analyzed 2932 p53-occupied response elements (p53REs) in the context of genome-wide chromatin state, DNA accessibility and dynamics, and considered roles for binding-sequence specificity and evolutionary conservation. While p53 occupancy level shows little apparent direct relationship to gene expression change, after grouping expressed genes by their chromatin status at baseline, a relationship between occupancy of p53REs and gene expression change emerged. Analysis of p53RE sequences demonstrated that p53 occupancy was strongly correlated with sequence similarity to p53RE consensus, but surprisingly, was inversely correlated with nucleosome accessibility (DHS) and evolutionary conservation. These data revealed a systematic interaction between p53RE content and chromatin context that affects both quantitative p53 occupancy and the induced transactivation response to exposure. Moreover, this interaction appears to have been tuned via evolutionary events involving transposable elements, which strongly bind p53, but in only a few instances affect gene expression levels. Models of p53-regulated gene expression response that consider both chromatin state and sequence context may prove useful in guiding strategies for cancer prevention or therapy.
Published in the journal:
. PLoS Genet 11(1): e32767. doi:10.1371/journal.pgen.1004885
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004885
Summary
It is well established that p53 binds DNA elements near p53 target genes to regulate the response to cellular stress. To assess factors influencing binding to response elements and subsequent gene expression, we have analyzed 2932 p53-occupied response elements (p53REs) in the context of genome-wide chromatin state, DNA accessibility and dynamics, and considered roles for binding-sequence specificity and evolutionary conservation. While p53 occupancy level shows little apparent direct relationship to gene expression change, after grouping expressed genes by their chromatin status at baseline, a relationship between occupancy of p53REs and gene expression change emerged. Analysis of p53RE sequences demonstrated that p53 occupancy was strongly correlated with sequence similarity to p53RE consensus, but surprisingly, was inversely correlated with nucleosome accessibility (DHS) and evolutionary conservation. These data revealed a systematic interaction between p53RE content and chromatin context that affects both quantitative p53 occupancy and the induced transactivation response to exposure. Moreover, this interaction appears to have been tuned via evolutionary events involving transposable elements, which strongly bind p53, but in only a few instances affect gene expression levels. Models of p53-regulated gene expression response that consider both chromatin state and sequence context may prove useful in guiding strategies for cancer prevention or therapy.
Introduction
Tumor suppressor p53 is activated in response to DNA damage and cellular stress signals and regulates the expression of target genes to elicit cell-growth arrest, DNA damage repair, or apoptosis to prevent the propagation of damaged or compromised cells [1], [2]. Understanding the regulatory logic of p53 is critical to understanding p53 biology in normal and tumor cells. The canonical p53 responsive element (p53RE) is composed of two decamers of RRRCWWGYYY, where R = purine, W = A or T and Y = pyrimidine, separated by a spacer of 0–21 nucleotides (nt), leading to millions of putative p53RE sites in the human genome [3]. About two hundred p53REs have been characterized in detail but p53 chromatin immunoprecipitation sequencing (ChIP-seq) experiments indicate there are thousands of p53 targets and numerous exposure-specific patterns of binding and transactivation [4]–[9]. These patterns have been variously attributed to sequence-specific binding [5], [6], p53 post-translational modifications [10], targeting coactivators/factors [11], post-transcriptional effects [12], as well as, chromatin status at the binding site [13]–[15]. However, the rules governing the sequence specificity and functional output of regulatory interactions between p53 and the genome are not yet fully understood.
A number of in vitro studies have clearly demonstrated that p53RE sequence variation, including polymorphisms and spacer length, affect p53 binding to DNA and subsequent transcriptional activation [16]–[20]. These studies show that p53REs with higher similarity to the p53RE consensus display stronger binding and those that contain spacer sequences larger than 1 nt between the decamers show dramatically reduced binding. These studies, however, did not consider the impact of the varied genomic chromatin contexts on p53 binding to response elements and evolutionary conservation of p53REs. In addition, p53 binding motifs occur frequently in primate-specific interspersed repeats, including retroviral long terminal repeats (LTRs [21]), short (SINE [22], [23], such as Alu), and long interspersed nuclear elements (LINEs [24]). Considering the potential role of transposition in the evolution of cis-regulatory elements [25], we ask if these recently evolved p53 binding sites have acquired sequence properties or chromatin contexts which might indicate if they are functionally suppressed or utilized.
Chromatin accessibility can be a primary determinant of transcription factor (TF) occupancy [26], [27]. For example, ligand-activated glucocorticoid receptor binding occurred with near absolute preference (∼95%) for binding sites located in accessible chromatin [26] and recent Encyclopedia of DNA Elements Project (ENCODE) reports [28] have supported this phenomenon for numerous other TFs. In contrast, Nili et. al. [14] described a different situation for the tumor suppressor protein p53, demonstrating that in the tumor cell line MCF7, p53 often binds genomic regions that have high existing nucleosome density and low chromatin accessibility. Other studies have suggested that stress-induced binding at specific CDKN1A (p21) p53REs is associated with chromatin structure at these locations [15]. These observations argue for further exploration of the role of chromatin-accessibility in regulating p53 access to DNA regulatory elements. Specific chromatin modifications at histone proteins correlate with TF binding, transcription initiation, elongation, enhancer activity and repression [29]. To elucidate how p53 RE sequences and local chromatin context interact to affect p53 binding and gene transactivation, we have taken advantage of a well-characterized lymphoblastoid cell-culture exposure model system [30]–[32]. The ENCODE project has generated Hidden Markov models (HMM) [32], [33] that annotate the human genome into distinct (15 or 7 HMM states) chromatin states. These models are based on combinatorial patterns of eight informative histone modification marks, Zinc finger CCCTC-binding factor (CTCF), the presence of DNase I hypersensitivity (DHS) and the spatial relationships with gene transcription across nine cell types. The ENCODE ChromHMM model describes the chromatin landscape in unstressed cells and highlights transcriptional regulatory elements reliably and robustly [32], [34]. We have integrated these ENCODE data sets into our experimental data with the aim of deconvoluting the roles for chromatin context/state, p53RE sequence, and p53 occupancy in the transactivation of p53 target genes.
Thus, in the present study we have examined genome-wide changes in p53 binding (ChIP-seq), H3K4me3 (ChIP-seq), gene expression, and DNase I hypersensitivity (DNase-seq) that were induced by the DNA-damaging, chemotherapeutic agent doxorubicin (DXR) in human lymphoblastoid cell lines (LCL). The goal was to assess these changes in the context of ENCODE chromatin states, and consider the role of p53RE sequence content and evolutionary conservation. We revealed several unique relationships, including inverse correlations between p53 occupancy and both chromatin accessibility and evolutionary conservation. Notably, our novel, integrated genome-wide analysis demonstrated that p53RE sequence content is highly correlated with genomic occupancy, and that chromatin state context strongly modulates the relationship between p53 occupancy level and changes in gene expression. Furthermore, the interaction between these features appears to have been shaped by evolutionary selective pressure, likely driven by transposable elements. Our study sheds new light on the complex interrelationship between chromatin state and p53RE sequence in p53 genomic occupancy, and suggests the importance of considering the interactions of sequence content and epigenetic factors in interpreting p53-mediated stress responses.
Results
Identification of genomic p53 binding sites and target genes in human lymphoblastoid cells
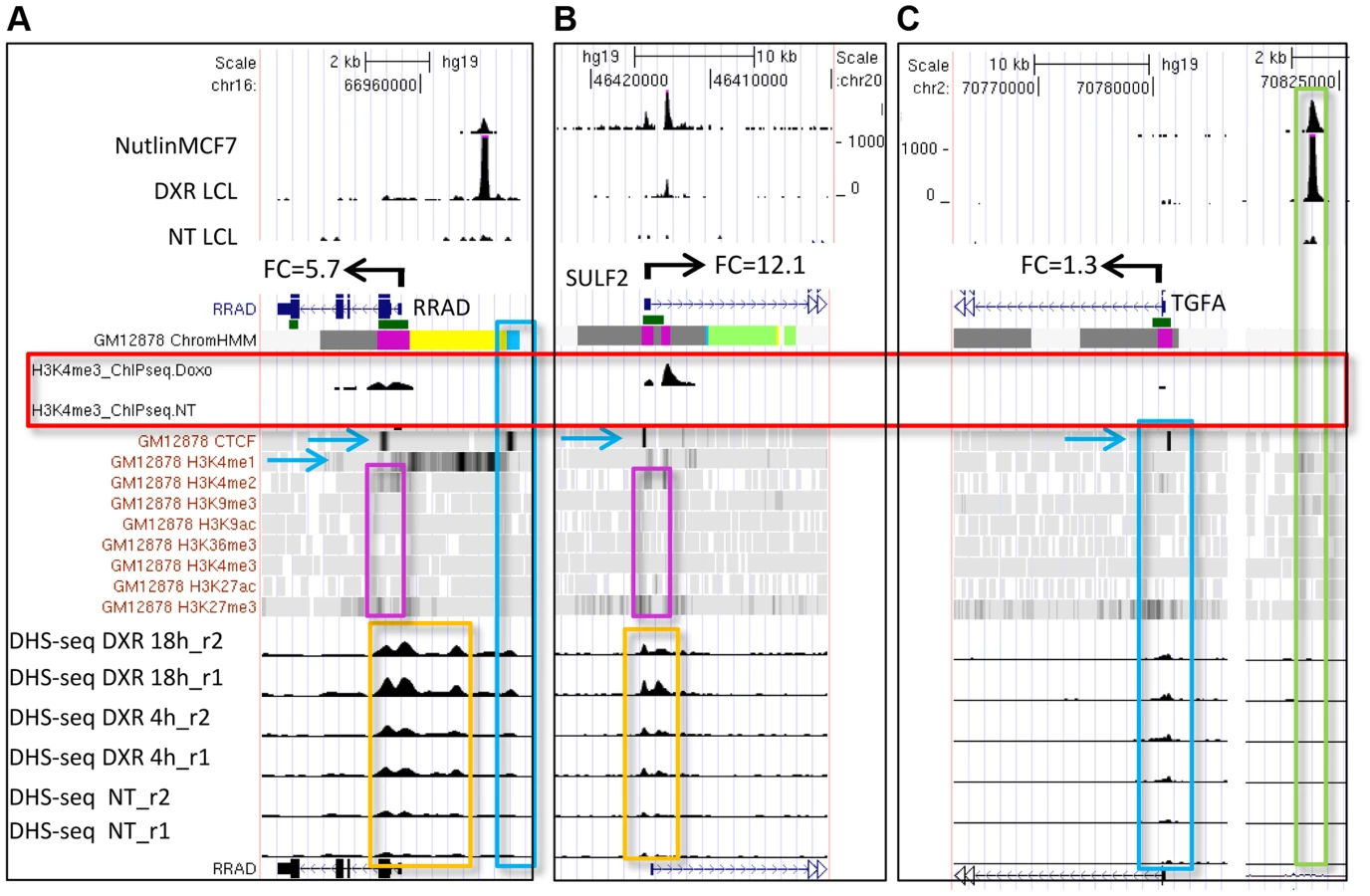
We carried out p53 activating treatments in LCLs (GM06993, GM11992, and GM12878) using a 0.3 µg/mL dose of DXR and prepared mRNA samples at 4 and 18 hrs of treatment. Chromatin for p53 and H3K4me3 ChIP-seq was prepared from samples collected at 18 hrs, a time point when both growth arrest and apoptosis-related p53 gene targets are occupied [35]. Following DXR-treated p53 ChIP-seq, we detected 6436 peaks at a 5% false discovery rate (FDR) using the QuEST Program [36]. Then, using a cut-off of 30 overlapping sequence reads, we classified 2932 p53 peaks in DXR-treated samples as high-confidence peaks, and with the same sequence read cutoff there were thirty p53 peaks evident under no treatment (NT) conditions. As in primary lymphocytes [37], we observed very limited p53 binding in the absence of stress and the DXR treatment greatly increased the occupancy of p53 at known and newly identified response elements. Among these high-confidence peaks, 91% (2664/2932) have putative p53REs, and 122 of them overlapped known p53 binding sites (Table 1, S1 Fig.). All further analyses focused on these high confidence p53 peaks and the 2415 genes that were associated with them by proximity. Fig. 1 displays a UCSC genome browser view of a sample of these data integrated with ENCODE data from GM12878 cells. Tracks A, B, F, H, I, are experimental data from LCLs generated in our lab for the present study. Tracks A–E display p53 ChIP-seq peaks in the PGPEP1 (Pyroglutamyl-Peptidase I)-GDF15 (Growth Differentiation Factor 15) region from this study and two other independent studies [7], [8] using MCF7 cells and IMR90 cells. Many p53 peaks are highly reproducible across multiple experimental conditions (orange box), while other binding locations are specific to treatment and tissue as noted by others [8]. Comparing the present data with three other published p53 ChIP-seq datasets we determined that 69% of 2932 peaks were observed in at least one other independent study. Among the high-confidence peaks unique to this study, 91% (822/903) were observed as significant peaks in an independent p53 ChIP-Seq experiment in LCLs. Histone H3 lysine 4 tri-methylation (H3K4me3, tracks H and I) is present at the PGPEP1 actively transcribed promoter (red box) but very low at the GDF15 promoter (track I, purple box), which displays insulating CTCF marks (CTCF, track J, also blue arrows, blue box) in the untreated cells.


To provide a multifaceted view of how the chromatin landscape impacts p53-occupied genes and to explore if p53-inducible genes are actively suppressed at baseline, we have used the ENCODE Hidden Markov Models (two versions, ChromHMM15 [33] and ChromHMM7 Combined [32], which are based on ChIP-seq measurements for multiple histone modifications, CTCF ChIP-seq, DNaseI-Seq, and FAIRE-seq as measured in nine cell lines, including the LCL GM12878, under basal conditions. In Fig. 1, the ChromHMM7 track (G) summarizes the chromatin characteristics of this region of the genome. For example, the red box highlights the promoter region of the p53 inducible gene, PGPEP1. Numerous histone modifications and DHS align at this position, and the ChromHMM7 track displays red indicative of an active promoter (State 1). In the same genomic region within the orange box where p53 peaks were detected, the ChromHMM track displays a strong DHS peak and multiple histone modifications (tracks J) particularly H3K27ac, H3K4me1 and H3K4me2 indicative of an active, distal enhancer. We used the ChromHMM7 Combined model (ENCODE website) to classify p53 occupied regions and genes as: 1) active promoter (e.g., PGPEP1, active histone marks present: H3K4me2, H3K4me3, H3K27ac, H3K9ac at>93% frequency); 2) promoter flanking region; 3) enhancer (H3K4me1 at>96% frequency); 4) weak enhancer/open chromatin; 5) insulator CTCF enriched; 6) transcribed; 7) repressed, including polycomb and heterochromatin. Some examples of known characterized p53 target genes and their associated HMM states are listed in Table 1.
Stress-induced H3K4me3 changes correlate with gene expression changes for p53-responsive genes
The GDF15 gene had very low expression under no treatment conditions with very little H3K4me3 at its TSS and was strongly induced following p53 activation with the increase of H3K4me3 marks (Fig. 1, purple box). We examined these phenomena across all p53 occupied genes. Of the 2932 DXR-induced p53-occupied regions, 1697 genes had detectable gene expression levels using the Affymetrix Exon Array 1.0 either under no treatment (NT) conditions or after treatment. We grouped expression values into deciles based on baseline H3K4me3 levels at TSS (ordered from low to high, Fig. 2A), we observed a very striking, linear relationship over time (NT, 4 hr, 18 hr after DXR treatment). Expression values for untreated cells (black bars) display a strong linear trend for increasing mean gene expression values with increasing H3K4me3 decile. Following DXR treatment (open bar = 4 hr, gray bar = 18 hr), the group of genes with low initial H3K4me3 levels (deciles D1–D3, left side of graph) show the largest change in expression. Thus highly inducible p53-regulated genes typically do not display activating H3K4me3 marks in unstressed cells. We performed regression analysis on the DXR-induced change in H3K4me3 modification with the DXR-induced change in gene expression which showed a significant correlation (Fig. 2B, r2 = 0.41, P<0.0001). As mentioned, the highly-induced p53 gene GDF15 displayed both low H3K4me3 and low expression (Fig. 1, purple box) at baseline. However, GDF15 also displayed insulating CTCF binding (blue box) at TSS. We tested if all low H3K4me3 DXR-induced p53 genes were enriched with repressive chromatin marks at the TSS or gene body relative to down-regulated genes, and determined that highly-inducible p53 genes were characterized by having preexisting chromatin marks indicating insulated (CTCF), polycomb (PcG/H3K27me3), or repressive heterochromatin status (trend test, P<0.0001, Fig. 2C). Examples of known p53 genes displaying repressive chromatin marks are listed in Table 1.

Stress-induced gene expression correlates with steady-state chromatin state (ChromHMM) at TSS
Fig. 3A demonstrates the distribution of p53 peaks among ChromHMM states and the median distance of the binding locations to the nearest TSS. While the genome of GM12878 cells can be segmented by ChromHMM into promoters (∼1%), enhancers (∼15%), transcribed (∼18%) and repressed regions (∼64%) [33], [38] (S1A Fig.), it is quite clear that p53 binding was highly enriched for promoters, enhancers, and transcribed regions. Most characterized p53RE sites occur in active promoters (S1B Fig.; for examples, see Table 1). While most binding in active promoters was proximal to transcription start sites (TSS), it is notable that 42% of binding locations (those in enhancers, CTCF regions, transcribed regions) have median distances that are at least 32 Kb distal from a TSS, and many binding sites located in repressed regions are much further from a TSS (Fig. 3A). These distal p53 binding locations found in enhancers and repressed chromatin were reproducible in other published p53 ChIP-seq datasets;>76% of these peaks were observed in at least one other experiments (S1 Table).

We also used the ENCODE ChromHMM15 at the TSS to classify the state of the gene nearest to the p53 ChIP-seq peak (illustrated as in Fig. 1 lower panel and Fig. 3B) to identify “poised” promoter regions that carry both repressive (H3K27me3) and activating (H3K4me2/3) marks, and these genes on average showed>3-fold induction (S2B Fig.). Fig. 4A–B displays two genes, RRAD (Ras-Related Associated with Diabetes) and SULF2 (Extracellular Sulfatase 2) with H3K27me3 and H3K4me2 marks (purple boxes), no H3K4me3 marks were present at the TSS, and expression levels were low with no treatment. Following treatment with the DNA damaging agent DXR, H3K4me3 marks appeared (red box) and for each of these poised genes, mRNA expression increased dramatically along with increases in DHS at 4 and 18hrs (orange boxes). However, not all genes follow this pattern. While TGFA (Transforming Growth Factor, alpha) (Fig. 4C) had similar H3K4me2, H3K27me3, and CTCF marks indicative of a poised promoter at baseline and also displayed a very large, upstream p53 binding peak, only minimal H3K4me3 and gene expression changes were observed. Thus p53 occupancy had little effect on TGFA transactivation, although there was some effect on DHS at the location of CTCF binding (blue box), suggesting other factors may be important for TGFA regulation. The patterns observed for chromatin accessibility change (DHS tracks) for individual genes were notable. Peaks of DHS were co-located at sites of H3K4me3 change (red box vs orange box) and also appeared to align with pre-existing H3K4me1/2 and/or CTCF binding patterns (Figs. 4A-C, blue arrows, blue boxes). Among genes that displayed pre-existing heterochromatin marks at the location of their p53 binding peak (e.g. Fig. 4C, TGFA; green box), it appeared that p53 binding had minimal detectable effect on nucleosome accessibility (DHS) at some binding sites, even when examined at greater magnification (S3 Fig.).
Comparing the level of p53 occupancy (ChIP-seq peak density) among ChromHMM states for all peaks (n = 2932), we observed significantly higher levels of occupancy for p53 peaks located in distal enhancers relative to promoters and other states (S4A Fig.). However, the result of grouping peaks by ChromHMM at the TSSs of nearby genes indicated that the small group of genes with insulating CTCF at their TSS displayed the highest p53 occupancy (S4B Fig.). The effect of insulated CTCF binding near the TSS prior to treatment was dramatic and this group of genes showed the greatest average change in gene expression (Fig. 5A). Overall p53 peak density displayed a very modest, but significant, correlation with change in gene expression (r2 = 0.06, P <0.0001, S4C Fig.). However, the more correlated patterns observed for peak density and change in gene expression after stratifying genes by ChromHMM state at the TSS (Figs. 5B, S4D), suggested that chromatin status modifies the relationship between binding/occupancy and transactivation.

p53 response element sequence content interacts with chromatin context (content versus context)
Because recent investigations have suggested that prior chromatin state (before treatment) could predict the occupancy of inducible transcription factor binding [27], [33], [39], we sought to examine the ChromHMM defined state at the location of p53 occupancy and asked the extent to which p53 occupancy level was due to specific p53RE sequence content at the binding site or due to the open-chromatin accessibility (context) at binding locations prior to exposures to DXR. S5A Fig. displays the sequence logo for 103 experimentally verified p53REs that make up the p53 consensus and it closely matches the most enriched motif among p53 ChIP-seq peaks identified by de novo motif discovery [40] in this study (Fig. 6A). The most enriched motifs were determined for peaks in each ChromHMM state (S5B–D Fig.) and as expected, each ChromHMM state contained enriched motifs similar to the p53 consensus. However, among peaks lacking full canonical p53REs, p53RE half-sites (decamers) were common (90%, 239/267, Data File S1). The CTCF motif was enriched among peaks associated with down-regulated genes at open promoter state (S5B Fig.). For individual binding sites, we identified putative response elements and considered the calculated binding strengths in relation to chromatin state and other features such as DHS or evolutionary conservation. GADD45A (Growth arrest and DNA-damage-inducible, alpha) and BCL2A1 (BCL2-related protein A1) (S5E–F Fig.) are examples of how these parameters vary considerably across p53REs and other examples are provided in Table 1. We predicted p53RE binding strength using a position weight matrix (PWM) model and a second model based on in vitro measurements of p53 binding (Binding PWM, S6A Fig. [16], [31]) and then compared p53RE strength across ChromHMM states (Figs. 6B, S6C). Of interest, mean PWM scores for the p53RE nearest the peak maximum were relatively high in enhancers, consistent with high occupancy at enhancers; the mean PWM scores in repressed chromatin regions were highest. However, the PWMs for the p53REs located in open promoters were much lower. Among p53 peaks associated with genes having open promoters, we observed that both mean PWM and mean peak density were lower among down-regulated genes relative to up-regulated genes (Fig. 6C), and that there was a distinct DNA motif difference as well. Occupancy by p53 was more frequent among induced genes (69% vs 31%; up-regulated genes vs down-regulated genes; Fig. 6C). About 85% (270/317, fold change <0.8) of down-regulated genes displayed active promoters (Fig. 6C) and were highly expressed at baseline (S2A Fig.). Using the functional annotation program DAVID we examined Gene Ontology (GO) annotations of up and down-regulated genes among all chromatin states (Data File S1). The analysis revealed that p53 was the most likely regulator of these genes and that the most significant GO term is “regulation of cell death”. The enrichment for “DNA damage response” and “induction of apoptosis” was particularly strong when we compared highly occupied p53REs vs less occupied p53REs (Data File S1, DAVID analysis).

The p53 consensus RE is also characterized by a variable spacer region of 1–21 nt between the two decamer half-sites, and since the PWM is calculated independently of the variable spacer, these represent independent features. The length of the p53RE spacer is known to reduce the affinity of p53 for binding to its consensus sequence and transactivation [41]. While spacers longer than 3 nt were relatively rare, the weak p53REs that were located in active promoters were more likely to have spacers and have larger spacers (S6B Fig.), which was associated with weaker binding sequences (Fig. 6B). The negatively correlated patterns of spacer length and PWM strength (S6B–D Fig.) were further evidence that these weak p53REs may be functional. Thus, p53REs located in active promoters were weaker by two distinct criteria, while elements in more distant locations in enhancers and in repressed chromatin states appear to be significantly stronger.
Negative correlation of both p53RE PWM score and occupancy with chromatin accessibility (DNase 1 HS)
To directly visualize the relationship between p53RE binding strength score (PWM), occupancy, and chromatin accessibility, we ordered the dataset by computed PWM scores from low to high, and grouped data into deciles (Fig. 7). We first tested the relationship between PWM values and the frequency of spacers between the half-sites (Fig. 7A, spacers of various lengths shown by stacked bars) and observed spacers to be more common in p53REs with weaker PWM scores (consistent with S6B, S6D Figs.). A surprising result was that p53 peaks containing the strongest PWM scores proved to be far distant from TSSs (Fig. 7B, decile D10) while the weakest p53REs were <1 kb from the TSS. Particularly notable was the linear relationship between p53RE binding strength (PWM score) and p53 occupancy (peak density, Fig. 7C). To assess if this observation was unique only to DXR-treated LCLs, we accessed p53 ChIP-seq data from 5-fluorouracil (5FU) treatment of MCF7 breast cancer epithelial cells [8], calculated p53RE PWM scores within p53 ChIP-seq peaks (S7A Fig.), and compared PWM with ChIP-seq density (S7B Fig.). A similar linear relationship between PWM score and peak density was observed in MCF7 cells. By comparing these results with other published p53 ChIP-seq datasets [7]–[9] we determined that p53 ChIP-seq peak density was also highly significantly correlated with reproducibility of p53 peaks across multiple experimental systems (Data File S1). ChIP-seq peaks containing the weakest p53RE PWMs had lower occupancy and were associated with the highest DHS scores in untreated cells (Fig. 7D). This observation was also confirmed in MCF7 cells (S7C Fig.). Thus we observe that open promoters that were characterized by the presence of pre-existing accessible chromatin (DHS) were permissive for p53 binding to the weakest elements. Conversely, the p53 binding sites with the highest PWMs and highest occupancy tend to be in the most distal p53 binding locations (∼60 kb from a TSS and in heterochromatin), most of these sites display very low DHS, and these observations were independent of cell type.

Evolutionary conservation at p53 binding sites varies with chromatin state and RE binding strength, and is influenced by the presence of transposable elements
Gene regulatory regions, including promoters and enhancers typically display high evolutionary conservation, presumably due to negative selection pressure to preserve functional DNA elements [42]–[44]. However, we have previously noted that evolutionary conservation varied greatly among well-characterized functional p53REs, with some human elements displaying little homology with other mammals [45], and we wondered if p53RE conservation varies by chromatin state. Grouping p53REs by their ChromHMM state and plotting their average PhyloP score across the 20nts of the RE dramatically demonstrates this phenomenon (Fig. 8A). Response elements located in promoters and enhancers were highly conserved relative to those in CTCF enriched, transcribed, or repressed regions. When we ordered p53REs by PWM decile we revealed an inverse correlation between p53 occupancy and evolutionary conservation of the peak region, either by mouse-human percent identity (Fig. 8B) or by PhyloP score (S8A Fig.). These analyses demonstrate that more highly conserved p53REs are relatively weak p53REs and display lower occupancy relative to poorly conserved sequences.

Some primate-specific transposable elements (TEs) are known to contain p53REs [21], [22], [24]. To evaluate their influence on p53RE conservation scores, we analyzed the overlap of occupied p53REs (full sites with spacer ≤3) with repeat elements categorized in Repeat Masker (http://www.repeatmasker.org/faq.html). We observed that 33% (970 of 2932) of p53-occupied sites were embedded in distinct families of repeat elements, with a significant enrichment for the endogenous retroviral elements MER61, LTR10B1, and MLT1H, as well as others (Data File S1). Most TE-associated p53 ChIP-seq peaks (82.7%; 803/970) were also observed in independent experimental datasets (Data File S1). The presence of TE-associated p53REs strongly affected conservation scores among HMM states (Figs. 8A, S8B–C). For example, TE frequency was inversely associated with PhyloP scores, being high in heterochromatin, intermediate in enhancers, low in promoters (S8B, C Fig.). As expected, average PhyloP values were significantly lower among all TE-associated p53REs relative to non-TE p53REs (Fig. 8C). Surprisingly, when we examined PhyloP values for individual families of TEs, we observed Alu and L2 family p53REs were dramatically negative compared to all others (Fig. 8C). We considered if chronological age of the TE might explain this pattern but this is not the case because Alu repeats are among the oldest of TE families while L2 is among the youngest [46]. Negative PhyloP values are predicted to be fast evolving and may be related to recent evolutionary selection as suggested by Pollard, et al [47] and this would be consistent with the p53 pathway acquiring new primate specific functions.
To test if TE-associated p53REs affect sequence content (PWM, Fig. 8D) and occupancy (p53 ChIP peak density, Fig. 8E), we compared TE-associated p53REs with that of non-repeat p53REs. We observed significantly higher PWM scores in all TE-associated p53REs, with several families particularly high (MER61, MLT1H, LTR10B; Fig. 8D) and the p53 ChIP-seq peak density plot displays a similar, correlated pattern (Fig. 8E). Thus the relationship between PWM and ChIP-seq peak density that we observed previously among all p53 peaks (Fig. 7C), holds among families of repeats (S8D Fig.). If these peaks were functionally associated with nearby genes we would expect treatment-induced changes in H3K4me3 peak density at the TSS. Fig. 8F shows that genes nearby TE-associated p53REs display less average H3K4me3 changes than nonTE-associated genes. Notable exceptions to this phenomenon include genes nearby L2 (66 genes) and MIR families (46 genes) of TEs which showed significantly higher levels of DXR-induced gene expression (Fig. 8F). Two known p53 genes (DDR1, p53RE in L2a; EPHA2, p53RE in MIRb) are among these groups. The p53REs that overlap L2 family repeats seemed unusual in that they show both strongly negative PhyloP values (Fig. 8C) and display changes in H3K4me3 at nearby TSS locations (Fig. 8F). We explored several of these L2-associated p53 binding sites in more detail and browser views of two strongly negative PhyloP p53REs that also show altered H3K4me3 at the TSS following p53 activation by DXR are shown in S9A–B Figs. (H3K4me3 data in Data File S2). In addition, four L2-associated p53REs with negative PhyloP values that only show DHS changes at the location of the binding site are given in S9C–F Fig.
Discussion
We assessed genome-wide in vivo p53 occupancy, H3K4me3, DHS and gene expression in human lymphoblastoid cells treated with the DNA-damaging chemotherapeutic agent DXR. Previous studies of functional regulatory variation indicate that LCLs are responsive to a variety of treatments [28], including p53-activating, DNA-damaging agents like DXR [16], [31] and ionizing radiation [48]. On a genome-wide scale, we quantified the relationship between dynamic, stress-induced changes in gene expression and changes in H3K4me3 (Figs. 2A–B, 4A–B), a mark present on the nucleosomes flanking nucleosome-free regions that coincide with active TSSs [49], [50]. Notably, the group of genes with the greatest change in expression appear to be actively repressed at baseline (Figs. 2C, 4A–B), either via CTCF insulation, polycomb marks or the presence of heterochromatin. Strikingly, when we stratify the 1697 genes that display mRNA expression by the ChromHMM state at their TSS, we observe a linear trend between patterns of quantitative p53 occupancy (peak density) and fold change gene expression (Fig. 5B). Based on single-gene experimental models of p53 occupancy and transactivation, this relationship is not unexpected, but the clarity achieved by considering chromatin state on a genome-wide scale is dramatic (comparing S4C Fig. with Fig. 5B). Thus, chromatin state models created under steady-state conditions, such as the ENCODE ChromHMM, were highly predictive of genome-wide p53-mediated responses, and can help elucidate the mechanisms that enable those responses. This interesting finding should be examined in other inducible gene expression pathways and other cell types.
While greater than 50% of well-studied p53-regulated genes have binding sites proximal to a TSS, only about 25% of p53 binding was near a TSS in our study (Fig. 3B). Binding sites located in distal enhancer elements (∼34-kb from a TSS) comprised 26% of peaks, while another ∼30% were located in regions of heterochromatin even more distal to a TSS (median of 70 kb from a TSS). Approximately 80% of these distal sites in repressed chromatin were also identified in other genome-wide studies of p53 binding [6]–[9]; however, most of these sites remain functionally uncharacterized. Studies utilizing long distance chromatin capture techniques (HiC, 3C) under p53-activated conditions will be needed to confirm distal interactions between p53 binding and transactivation. Nevertheless, we did observe a clear relationship between p53 occupancy of p53REs located in repressed chromatin regions, and inducible transactivation of many genes within repressed regions (e.g., TGFA, ANK1, GDNF, CYP4F3).
In our study 91% (2664/2932) of the high confidence p53 occupied regions contained sequences similar to the motif created from 103 known p53 binding sites (S5A Fig.). We asked if p53 binding sequences located in different chromatin functional regions might display characteristic differences, and we observed that among the 592 peaks occurring in active promoters, the most prominent motif closely matched the p53 consensus (S5B Fig., p = 3.9×10−63). Interestingly, the next most common motif in peaks located in active promoters closely resembled the CTCF motif (p = 4.8×10−27) and this motif was present more commonly in down-regulated genes. However, these p53-related CTCF motifs were not associated with the CTCF insulator function as these regions displayed high nucleosome accessibility and the genes had high average expression levels. In contrast, genes classified as CTCF insulated by the ENCODE HMM model had low basal expression level but high inducibility (Fig. 5A–B). Among the most highly induced genes, we observed that prior to treatment, there was enrichment of repressive chromatin features such as CTCF, polycomb (PcG-H3K27me3) or heterochromatin (Fig. 2C). Genes with poised promoters carrying both H3K27me3 and H3K4me2 at the TSS (Figs. 4A–C, S2C) were an important group among these inducible genes and include a number of known p53 response genes (RRAD, TGFA, PERP, GPR87, RNF144B, TNFSF10A).
Espinosa and Gomes [13], [51] have hypothesized that loss of CTCF insulation activity could allow for inducible expression of some poised p53 genes. DXR induced changes in DHS that co-locate precisely with CTCF binding in untreated cells are consistent with this notion. Akdemir et. al. [52] proposed that changes in H3K27me3 status at poised genes via a mechanism of p53-JMJD3/UTX interaction could drive the conversion of poised genes to actively expressed genes. However, in the present experiment, the expression of these chromatin remodeling factors were not detectable. We did observe that changes in H3K4me3 were strongly confluent with altered DHS patterns and consistent with transcriptional activation of some poised p53 genes like SULF2 following treatment. The detailed mechanism drives the release of these specific genes from their poised state is an area of future study. Down-regulated genes had quite different characteristics; most down-regulated genes (85%) had open promoters (Fig. 6C) and displayed high expression at baseline (S2A Fig.). Interestingly, both the density of p53 binding, and the calculated p53RE PWM score associated with down-regulated genes were significantly lower compared with p53REs from induced genes (p<0.0001, Fig. 6C). Nikulenkov et. al. [8] observed a similar lower peak density among down-regulated genes. Wang et. al. [19] analyzed a small number of p53REs and suggested there was a preferred motif for down-regulated genes that is different from up-regulated genes. We also detected differences in the most enriched motif between the two groups of sequences. Specifically at nucleotides 10 and 11 in the p53RE motif, “TG” was present in the up-regulated REs and a “CA” motif occurred in the down-regulated genes (Fig. 6C). Consistent with this altered motif, the p53REs in down-regulated genes have a weaker match with the canonical sequence and lower mean PWM (Fig. 6C). However, this change is unlikely to be a general feature of p53REs because among all chromatin states many up-regulated genes contain the “CA” motif at nucleotides 10, 11. Pathway and functional analysis of each chromatin state by DAVID analysis showed that p53 is the most likely regulator of these genes and that the most significant GO terms include “regulation of cell death” and “response to DNA damage stimulus”.
The relative importance of DNA sequence content and chromatin landscape in determining a transcription factor's genomic binding has been subjected to considerable experimental exploration [28], [53]–[55], including several with p53 [14]–[16], [56]. Chromatin accessibility determines the binding of glucocorticoid hormone receptor [26] and STAT1 [57]. However, in the present study, we detected that a large fraction (29%) of p53 binding occurred at distal, repressed chromatin regions. The quantitative level of p53 occupancy was high in these regions of high intrinsic nucleosome occupancy, which is consistent with the findings of Nili et. al [14]. Thus, p53 occupancy and inducible gene expression occurred in regions of high nucleosome density indicated by existing H3K27me3 marks, heterochromatin, or low DHS. Notably, at the level of individual binding sites (e.g., upstream of TGFA), high levels of p53 occupancy can occur with minimal local displacement of nucleosomes (Figs. 4C, S3; as detected by DHS-seq). However, for poised genes marked with both H3K27me3 and H3K4me2 (Fig. 4A–C), p53 occupancy was associated with nucleosome accessibility changes at the TSS, but not necessarily at the p53 binding site. In these cases, as gene expression was induced, we observed the open DHS region spreading from the TSS, and the DHS pattern resembles the DXR-induced H3K4me3 pattern.
A particularly notable result is the relationship between quantitative occupancy and binding strength of the p53RE sequence relative to consensus. Modeling p53RE binding strength using a statistical approach (PWM), or with an experimental binding-based model (binding PWM, S6A Fig.), we observed genome-wide correlations between binding strength and occupancy (peak density). This relationship was further validated using p53 ChIP-seq data generated from a different laboratory using a different cell type and treatment (S7A–C Fig.). Thus, p53RE sequence content strongly affects occupancy which is in agreement with several in vitro studies that reported single nucleotide changes in the p53RE sequence or the spacer length alter p53 binding, transactivation of constructs or gene expression [16], [20], [41]. Recently a single nucleotide variation in a p53RE in the KITLG gene was demonstrated to alter p53 binding, transactivation of KITLG and be strongly associated with cancer [58]. However, our present study is the first genome-wide study to demonstrate the quantitative relationship between sequence content and genome-wide occupancy levels.
Surprisingly, p53RE PWM scores and occupancy were strongly negatively associated with chromatin accessibility and evolutionary conservation (see Figs. 7D, 8B, S7C) at the binding site. That is, more conserved, relatively weak p53REs were observed to be found in accessible chromatin regions. Conversely, many strong p53REs with low conservation scores were located in heterochromatic regions with very low DHS. The p53REs located within enhancers displayed relatively high levels of conservation, PWM score, occupancy and accessibility. The contrast in levels of conservation and chromatin accessibility among p53REs is driven by the presence of primate-specific repeat elements of viral transposon origin that are characterized by low conservation, high PWM, high occupancy but low accessibility. This high occupancy, low accessibility and minimal nucleosome displacement observation is consistent with the Cui et. al [22] hypothesis that p53REs are accessible on the external surface of nucleosomes. Cui et. al. described this for Alu-associated p53REs; however, our data suggests that it may be a general feature of p53-occupied REs in heterochromatin, particularly those in retroviral-associated TEs (e.g. MLT, LTR, MER). Over many millions of years of evolution transposable elements have rearranged primate genomes [46], and the presence of a multitude of p53 binding sites in various TEs [21]–[24] likely is related to an evolutionary mechanism for creating p53REs from TE sequences. Our present study uniquely reveals the interrelationships between TEs, p53RE sequence content, occupancy, and chromatin state.
There are several interesting and important questions concerning the functional nature of these TE-associated p53REs. Are these TEs actively suppressed (as a genome defense) and can they be reactivated in tumors or in normal cells following genomic stress like DNA damage? Leonova et. al. suggest that TEs are epigenetically suppressed and p53 is involved in transcriptional silencing of them [59]. We observed that TE-associated p53REs were significantly stronger REs and were more highly occupied than non-repeat p53REs; however, most reside in repressive, closed chromatin. In addition, this occupancy did not typically translate into induced H3K4me3 levels at nearby gene TSSs or changes in gene expression. This would be consistent with suppression of these TE-associated p53REs by p53 binding. Also, one may ask, what are the characteristics of p53REs located in TEs that would allow them to be co-opted (exaptation) and used to regulate the expression of nearby genes [60], [61]? It is unknown which features of chromatin or changes in chromatin structure might facilitate the utilization or exaptation of transposon-derived p53REs (e.g., domestication of transposons). While this question cannot be easily answered with the present data, it is intriguing that we observe some p53REs that overlap with TE-repeats (Alu and L2) display strongly negative PhyloP values suggesting the possibility of recent evolutionary selection [47]. In addition, if we consider that p53 occupancy of TE-associated p53REs is sometimes accompanied by altered DHS, H3K4me3 or gene expression at nearby genes (as shown in S9 Fig.), it seems likely that many of them are functionally utilized.
Thus it is possible that both suppression and exaptation have occurred with many strong, newly-evolved TE-associated p53REs being actively suppressed via chromatin state but a small number of them are exapted and brought into the p53 regulatory network for use under stress conditions. These observations imply a potential functional link between the variation in genomic p53RE sequence, chromatin state and gene regulation, which has co-evolved over time. As pointed out by Leonova et. al. [59], characterizing the p53-mediated deregulation of transposable elements in tumors could be useful in clinical cancer diagnoses. Understanding how all of these variables interact and impact the p53-regulated response to DNA damage in primary and tumor cells will be important both for developing strategies for prevention of cancer in healthy individuals and for understanding outcomes in cytotoxic therapy for cancer.
Materials and Methods
Cell culture
HapMap (http://hapmap.ncbi.nlm.nih.gov/) CEU human lymphoblastoid cell lines GM06993, GM11992, and GM12878 (Coriell Cell Repositories, Camden, NJ) were cultured in RPMI 1640 medium supplemented with 15% heat-inactivated fetal bovine serum (Life Technologies) and 1X antibiotics/antimycotics (Life Technologies). Cells were incubated at 37°C with 5% CO2. During experiments, cells were grown in petri dishes and treated with 0.3 µg/ml doxorubicin (Sigma, St. Louis, MO) for ChIP-seq, DNase I hypersensitivity, and gene expression.
Gene expression microarray analysis
GM12878 cells were cultured in triplicate and were either untreated or treated with 0.3 µg/ml doxorubicin for 4 and 18 hrs. Total RNAs was extracted using total RNA Miniprep kit (QIAGEN) with DNase 1 digestion. Total RNA was subjected to the standard cDNA synthesis, dye labeling and hybridization as per Affymetrix's protocols for the Human Exon ST 1.0 array (Affymetrix, Santa Clara, CA), and processed at the NIEHS Microarray Core. Exon expression data was analyzed through Affymetrix Expression Console using gene-level RMA summarization and sketch-quantile normalization methods. The t-test was used to calculate the significance of gene expression change. All the Affymetrix microarray data used here have been deposited into the GEO database (GSE51709). DXR induced gene expression array data was validated in a separate cell line using the Illumina gene expression array platform (Data File S1), as well as RT-PCR for selected genes. For replication by qRT-PCR, the cDNA was generated using the First-Strand Synthesis system (Life Technologies). For each biological replicate, target genes were amplified in triplicate using FAM probes designed to span exon junctions for each target gene and PCR Master Mix (Life Technologies). Studies investigating the relationship between gene expression in lymphoblast cell lines and in vitro p53 binding have been reported previously ([16], [31]).
Chromatin Immunoprecipitation-Sequencing (ChIP-Seq)
Lymphoblastoid cells were seeded in 15-cm dishes and treated with 0.3 µg/mL doxorubicin or vehicle water for 18 hrs. Chromatin from 100×106 cells was used for each p53 ChIP-seq experiment. ChIP for p53 was carried out essentially as described in the Agilent Mammalian ChIP-on Chip protocol Version 20.0 (Agilent, Santa Clara, CA) using mouse monoclonal antibody DO-7 (BD Biosciences, San Jose, CA) or a non-specific rabbit IgG conjugated to secondary Dynal magnetic beads (Life Technologies). H3K4me3 ChIP was carried out with the H3K4me3 antibody (EMD Millipore #07-473, Billerica, MA). ChIP and input DNA were quantified using Qubit (Invitrogen). NIH Intramural Sequencing Center (NISC, Bethesda, MD) and the National Center for Genome Resources (NCGR, Santa Fe, NM) created the libraries using the standard Illumina ChIP-seq protocol. ChIP or input libraries were sequenced using Illumina Genome Analyzer IIx. For validation of occupancy, p53 ChIP-PCR was performed in triplicate using Sybr primers designed to span p53RE peaks in promoters of p53 target genes on a 7900 HT real-time PCR machine (Applied Biosystems).
DNase I hypersensitivity mapping
Genome-wide mapping of DNase I-hypersensitive sites (DHS) was carried out on 2 replicates of untreated and DXR-treated GM12878 cells for 4 and 18 hrs. For DHS profiling, intact nuclei were treated with DNase I, and the DNase I-hypersensitive fraction was analyzed by sequencing as previously described [62]. Data from two biological replicates under each experimental condition are displayed as tracks in the UCSC genome browser in Figs. 4, S3, S9. For analysis of DHS relative to ENCODE genome wide chromatin state, we downloaded ENCODE/OpenChrom (Duke University, Durham, NC) GM12878 DHS data (Fig. 1, track K) [28], [33]. These data (DHS signal intensity) were averaged across p53 binding regions, transformed (log2 +1), grouped by PWM decile and plotted versus p53 peak density (Figs. 7D and S7C).
p53 ChIP-seq, and H3K4me3 ChIP-seq data analysis
p53 ChIP-seq, H3K4me3 ChIP-seq, and input DNA sequence reads generated from the Illumina GAIIx were aligned against the human reference sequence (GRCh37p5, or hg19, June 2011) using the Burrows-Wheeler Alignment (BWA) Tool (Li et al., 2008). The uniquely mapped short reads were used to identify regions of the genome with significant enrichment in p53-associated DNA sequences. The peak detection was performed by QuEST 2.4 software [36] using the ‘Transcription factor binding site’ setting (bandwidth of 30 bp, region size of 300 bp) or the “Histone-type ChIP” setting (bandwidth of 100 bp, region size of 1000 bp) for H3K4me3 ChIP-seq, and the ‘stringent peak calling’ parameters (corresponding to 50-fold ChIP to input enrichment for seeding the regions and 3-fold ChIP enrichment for extending the regions).
We also used three published studies of p53 ChIP-seq from Nutlin-treated MCF7 cells [8], 5FU-treated MCF7 cells [8], 5FU-treated IMR90 cells [7], Nutlin and DXR-treated Human osteosarcoma U2OS cells [9], which deposited the raw reads at the NCBI SRA database. All sequence reads were aligned against the human reference sequence (GRCh37p5, or hg19, June 2011) using the Burrows-Wheeler Alignment (BWA) Tool (Li et al., 2008). For these published studies the peak detection was also performed by QuEST 2.4 software [36] using the ‘Transcription factor binding site’ setting (bandwidth of 30 bp, region size of 300 bp) for p53 ChIP-seq and the ‘stringent peak calling’ parameters. In Fig. 1 our H3K4me3 ChIP-seq tracks were prepared using the MACS program in order to display comparability with ENCODE tracks.
De novo motif discovery
MEME Suite [40] was used for the de novo motif analysis of the genomic DNA sequences identified by ChIP-seq experiments. Briefly, repeat-masked DNA sequence for each peak was trimmed to 100 bp, centered to the maximal signal of original peak. The MEME algorithm was then applied to identify the top 10 most enriched motifs with a length from 6 to 25.
Searching for putative p53REs using the position weight matrix
Position weight matrix (PWM) score was used to computationally estimate the binding strength to p53. The p53 PWM model was built on 103 published, experimentally validated p53RE sequences from the literature (NCBI PUBMED database), by converting nucleotide frequency values to a position weight matrix score as described [63]. Next the PWM score for any putative p53RE was calculated by summing the individual matrix values that correspond to the observed nucleotide at each position in that site. The potential p53REs in a genomic sequence were detected by sliding a window along the input sequence, considering the spacer in p53REs [64]. Briefly, at each chromosomal position, PWM score calculations were performed on DNA sequences with 20–23 nucleotides, corresponding to p53REs with a spacer of 0, 1, 2, and 3, respectively. For DNA sequences longer than 20, the first 10 nucleotides and the last 10 nucleotides were concatenated first to make a sequence of 20 nucleotides. For fast searching, a pattern composed of at least 3 of 4 core C and G nucleotides in the p53RE consensus was applied before PWM calculation. In addition, the same calculations were performed on the reversed complimentary sequences. In total, at each chromosomal position, eight calculations were performed. At a given location a putative p53RE was reported based on the following three criteria: (i) the sequence matches the search pattern; and (ii) the PWM score is above the PWM score threshold; and (iii) it contains the shortest spacer and has the largest PWM score. A second computational analysis based on measured binding in an in vitro binding assay, termed a binding PWM, was determined using a method described in Noureddine et. al. [31].
Analyses of ENCODE ChIP-Seq Datasets and ChromHMM state for GM12878 cells
Datasets and sample information for histone modifications and CTCF binding for the GM12878 cell line were from the ENCODE project (http://genomes.ucsc.edu) via the laboratory of Bradley Bernstein (Broad Institute) [34], at the Massachusetts General Hospital/Harvard Medical School. The chromatin state segmentation was produced in Manolis Kellis's Computational Biology group at the Massachusetts Institute of Technology [33], [54]. We created analysis files based on 2932 p53 ChIP-seq peaks that had≥30 overlapping sequence reads (Data File S1) and 2415 genes associated with these peaks (many genes had multiple peaks, Data File S2). We merged ENCODE data (ChromHMM15 and ChromHMM7 Combined models, PhyloP, DHS) into our data analysis files that included ChIP-seq peak density (p53, H3K4me3), gene expression (intensity at each time point, fold change). Examples of these data are displayed in Fig. 1. In the various analyses we grouped p53 peaks (or genes) based on the H3K4me3 status of nearby genes, the ChromHMM7 Combined status at p53 peaks or at the TSS of nearby genes, or on the computationally calculated binding strength (PWM). ChromHMM7 Combined model failed to call states for some regions and the ChromHMM15 was used in these cases. ChromHMM15 states 14 and 15 (repetitive) were dropped from analysis as no p53 ChIPseq peaks were uniquely mapped to these regions. We also specifically looked at expression levels of genes with “poised” promoters carrying H3K4me1/2 marks and H3K27me3 marks (ChromHMM15 state 3). To visualize linear trends related to H3K4me3 or PWM scores we used decile analysis in which PWM values were ordered from low to high and grouped into deciles. We then evaluated other parameters relative to these linear trends. Regression, t-tests, and other statistics were calculated using Graphpad Prism (GraphPad Software).
Evolutionary conservation analysis
To evaluate the evolution constraint on p53 binding sites, we utilized the placental mammal basewise conservation scores (PhyloP, phylogenetic p-values) downloaded from the UCSC genome browser website. These scores are computed from the PHAST package (http://compgen.bscb.cornell.edu/phast/) for multiple alignments of 45 vertebrate genomes to the human genome. The phyloP scores measure acceleration (faster evolution than expected under neutral drift) as well as conservation (slower than expected evolution). Sites predicted to be conserved are assigned positive scores, while sites predicted to be fast-evolving are assigned negative scores. The absolute values of the scores represent log p-values under a null hypothesis of neutral evolution. The sequence alignment and human-mouse conservation score were analyzed as previously described [45]. Transposon element repeats cataloged by Repeat Masker were accessed at UCSC genome browser website and aligned with p53REs to determine overlap.
Data access
Sequence reads for ChIP-seq experiments are deposited in the NCBI SRA database (p53 GSE46991, H3K4me3 GSE51713) and the NCBI Gene Expression Omnibus (GEO: http://www.ncbi.nlm.nih.gov/geo). Gene expression data are available at GEO under accession number GSE51709.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Gomez-LazaroM, Fernandez-GomezFJ, JordanJ (2004) p53: twenty five years understanding the mechanism of genome protection. Journal of physiology and biochemistry 60 : 287–307.
2. SchetterAJ, HarrisCC (2012) Tumor suppressor p53 (TP53) at the crossroads of the exposome and the cancer genome. Proceedings of the National Academy of Sciences of the United States of America 109 : 7955–7956.
3. RileyT, SontagE, ChenP, LevineA (2008) Transcriptional control of human p53-regulated genes. Nature reviews Molecular cell biology 9 : 402–412.
4. ChanC, WangY, ChowPK, ChungAY, OoiLL, et al. (2013) Altered Binding Site Selection of p53 Transcription Cassettes by Hepatitis B Virus X Protein. Molecular and cellular biology 33 : 485–497.
5. SmeenkL, van HeeringenSJ, KoeppelM, van DrielMA, BartelsSJ, et al. (2008) Characterization of genome-wide p53-binding sites upon stress response. Nucleic acids research 36 : 3639–3654.
6. WeiCL, WuQ, VegaVB, ChiuKP, NgP, et al. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124 : 207–219.
7. BotchevaK, McCorkleSR, McCombieWR, DunnJJ, AndersonCW (2011) Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell cycle 10 : 4237–4249.
8. NikulenkovF, SpinnlerC, LiH, TonelliC, ShiY, et al. (2012) Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell death and differentiation 19 : 1992–2002.
9. MenendezD, NguyenTA, FreudenbergJM, MathewVJ, AndersonCW, et al. (2013) Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic acids research 41 : 7286–7301.
10. SmeenkL, van HeeringenSJ, KoeppelM, GilbertB, Janssen-MegensE, et al. (2011) Role of p53 serine 46 in p53 target gene regulation. PloS one 6: e17574.
11. AndrysikZ, KimJ, TanAC, EspinosaJM (2013) A genetic screen identifies TCF3/E2A and TRIAP1 as pathway-specific regulators of the cellular response to p53 activation. Cell reports 3 : 1346–1354.
12. FreemanJA, EspinosaJM (2013) The impact of post-transcriptional regulation in the p53 network. Brief Funct Genomics 12 : 46–57.
13. GomesNP, EspinosaJM (2010) Disparate chromatin landscapes and kinetics of inactivation impact differential regulation of p53 target genes. Cell cycle 9 : 3428–3437.
14. Lidor NiliE, FieldY, LublingY, WidomJ, OrenM, et al. (2010) p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome research 20 : 1361–1368.
15. MillauJF, BandeleOJ, PerronJ, BastienN, BouchardEF, et al. (2011) Formation of stress-specific p53 binding patterns is influenced by chromatin but not by modulation of p53 binding affinity to response elements. Nucleic acids research 39 : 3053–3063.
16. BandeleOJ, WangX, CampbellMR, PittmanGS, BellDA (2011) Human single-nucleotide polymorphisms alter p53 sequence-specific binding at gene regulatory elements. Nucleic acids research 39 : 178–189.
17. JordanJJ, MenendezD, IngaA, NoureddineM, BellDA, et al. (2008) Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS genetics 4: e1000104.
18. TomsoDJ, IngaA, MenendezD, PittmanGS, CampbellMR, et al. (2005) Functionally distinct polymorphic sequences in the human genome that are targets for p53 transactivation. Proceedings of the National Academy of Sciences of the United States of America 102 : 6431–6436.
19. WangB, XiaoZ, RenEC (2009) Redefining the p53 response element. Proceedings of the National Academy of Sciences of the United States of America 106 : 14373–14378.
20. VeprintsevDB, FershtAR (2008) Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic acids research 36 : 1589–1598.
21. WangT, ZengJ, LoweCB, SellersRG, SalamaSR, et al. (2007) Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proceedings of the National Academy of Sciences of the United States of America 104 : 18613–18618.
22. CuiF, SirotinMV, ZhurkinVB (2011) Impact of Alu repeats on the evolution of human p53 binding sites. Biology direct 6 : 2.
23. ZemojtelT, KielbasaSM, ArndtPF, BehrensS, BourqueG, et al. (2011) CpG deamination creates transcription factor-binding sites with high efficiency. Genome biology and evolution 3 : 1304–1311.
24. HarrisCR, DewanA, ZupnickA, NormartR, GabrielA, et al. (2009) p53 responsive elements in human retrotransposons. Oncogene 28 : 3857–3865.
25. SchmidtD, SchwaliePC, WilsonMD, BallesterB, GoncalvesA, et al. (2012) Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 148 : 335–348.
26. JohnS, SaboPJ, ThurmanRE, SungMH, BiddieSC, et al. (2011) Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature genetics 43 : 264–268.
27. Guertin MJ, Lis JT (2010) Chromatin landscape dictates HSF binding to target DNA elements. PLoS genetics 6.
28. ThurmanRE, RynesE, HumbertR, VierstraJ, MauranoMT, et al. (2012) The accessible chromatin landscape of the human genome. Nature 489 : 75–82.
29. HonGC, HawkinsRD, RenB (2009) Predictive chromatin signatures in the mammalian genome. Human molecular genetics 18: R195–201.
30. SmirnovDA, MorleyM, ShinE, SpielmanRS, CheungVG (2009) Genetic analysis of radiation-induced changes in human gene expression. Nature 459 : 587–U120.
31. NoureddineMA, MenendezD, CampbellMR, BandeleOJ, HorvathMM, et al. (2009) Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS genetics 5: e1000462.
32. DunhamI, KundajeA, AldredSF, CollinsPJ, DavisCA, et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489 : 57–74.
33. ErnstJ, KheradpourP, MikkelsenTS, ShoreshN, WardLD, et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473 : 43–49.
34. RamO, GorenA, AmitI, ShoreshN, YosefN, et al. (2011) Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147 : 1628–1639.
35. RoblesAI, WangXW, HarrisCC (1999) Drug-induced apoptosis is delayed and reduced in XPD lymphoblastoid cell lines: possible role of TFIIH in p53-mediated apoptotic cell death. Oncogene 18 : 4681–4688.
36. ValouevA, JohnsonDS, SundquistA, MedinaC, AntonE, et al. (2008) Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nature Methods 5 : 829–834.
37. ShakedH, ShiffI, Kott-GutkowskiM, SiegfriedZ, HauptY, et al. (2008) Chromatin immunoprecipitation-on-chip reveals stress-dependent p53 occupancy in primary normal cells but not in established cell lines. Cancer research 68 : 9671–9677.
38. HoffmanMM, ErnstJ, WilderSP, KundajeA, HarrisRS, et al. (2013) Integrative annotation of chromatin elements from ENCODE data. Nucleic acids research 41 : 827–841.
39. Guertin MJ, Lis JT (2012) Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Current opinion in genetics & development.
40. Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Menlo Park, California: AAAI Press.
41. Jordan JJ, Menendez D, Inga A, Nourredine M, Bell D, et al.. (2008) Noncanonical DNA Motifs as Transactivation Targets by Wild Type and Mutant p53. PLoS genetics 4.
42. VernotB, StergachisAB, MauranoMT, VierstraJ, NephS, et al. (2012) Personal and population genomics of human regulatory variation. Genome research 22 : 1689–1697.
43. NephS, VierstraJ, StergachisAB, ReynoldsAP, HaugenE, et al. (2012) An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489 : 83–90.
44. GbadegesinMA (2012) Transposable elements in the genomes: parasites, junks or drivers of evolution? African journal of medicine and medical sciences 41 Suppl: 13–25.
45. HorvathMM, WangX, ResnickMA, BellDA (2007) Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS genetics 3: e127.
46. GiordanoJ, GeY, GelfandY, AbrusanG, BensonG, et al. (2007) Evolutionary history of mammalian transposons determined by genome-wide defragmentation. PLoS computational biology 3: e137.
47. PollardKS, HubiszMJ, RosenbloomKR, SiepelA (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome research 20 : 110–121.
48. JenKY, CheungVG (2005) Identification of novel p53 target genes in ionizing radiation response. Cancer research 65 : 7666–7673.
49. GuentherMG, LevineSS, BoyerLA, JaenischR, YoungRA (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130 : 77–88.
50. OzsolakF, SongJS, LiuXS, FisherDE (2007) High-throughput mapping of the chromatin structure of human promoters. Nature biotechnology 25 : 244–248.
51. GomesNP, EspinosaJM (2010) Gene-specific repression of the p53 target gene PUMA via intragenic CTCF-Cohesin binding. Genes & development 24 : 1022–1034.
52. AkdemirKC, JainAK, AlltonK, AronowB, XuX, et al. (2014) Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res 42 : 205–223.
53. DunhamI, KundajeA, AldredSF, CollinsPJ, DavisCA, et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489 : 57–74.
54. ErnstJ, PlastererHL, SimonI, Bar-JosephZ (2010) Integrating multiple evidence sources to predict transcription factor binding in the human genome. Genome research 20 : 526–536.
55. Pique-RegiR, DegnerJF, PaiAA, GaffneyDJ, GiladY, et al. (2011) Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome research 21 : 447–455.
56. LiM, HeY, DuboisW, WuX, ShiJ, et al. (2012) Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Molecular cell 46 : 30–42.
57. RobertsonAG, BilenkyM, TamA, ZhaoY, ZengT, et al. (2008) Genome-wide relationship between histone H3 lysine 4 mono - and tri-methylation and transcription factor binding. Genome research 18 : 1906–1917.
58. Zeron-MedinaJ, WangX, RepapiE, CampbellMR, SuD, et al. (2013) A Polymorphic p53 Response Element in KIT Ligand Influences Cancer Risk and Has Undergone Natural Selection. Cell 155 : 410–422.
59. LeonovaKI, BrodskyL, LipchickB, PalM, NovototskayaL, et al. (2013) p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proceedings of the National Academy of Sciences of the United States of America 110: E89–98.
60. MicaleL, LoviglioMN, ManzoniM, FuscoC, AugelloB, et al. (2012) A fish-specific transposable element shapes the repertoire of p53 target genes in zebrafish. PloS one 7: e46642.
61. HudaA, Marino-RamirezL, JordanIK (2010) Epigenetic histone modifications of human transposable elements: genome defense versus exaptation. Mobile DNA 1 : 2.
62. SongL, ZhangZ, GrasfederLL, BoyleAP, GiresiPG, et al. (2011) Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome research 21 : 1757–1767.
63. WassermanWW, SandelinA (2004) Applied bioinformatics for the identification of regulatory elements. Nature reviews Genetics 5 : 276–287.
64. WangX, TomsoDJ, ChorleyBN, ChoHY, CheungVG, et al. (2007) Identification of polymorphic antioxidant response elements in the human genome. Human molecular genetics 16 : 1188–1200.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 1
Nejčtenější v tomto čísle
- The Global Regulatory Architecture of Transcription during the Cell Cycle
- A Truncated NLR Protein, TIR-NBS2, Is Required for Activated Defense Responses in the Mutant
- Proteasomes, Sir2, and Hxk2 Form an Interconnected Aging Network That Impinges on the AMPK/Snf1-Regulated Transcriptional Repressor Mig1
- The SWI2/SNF2 Chromatin Remodeler BRAHMA Regulates Polycomb Function during Vegetative Development and Directly Activates the Flowering Repressor Gene