
Sumoylation Influences DNA Break Repair Partly by Increasing the Solubility of a Conserved End Resection Protein
Proper repair of DNA lesions is crucial for cell growth and organism development. Both the choice and capacity of DNA repair pathways are tightly regulated in response to environmental cues and cell cycle phase. Recent work has uncovered the importance of protein modifications, such as phosphorylation and sumoylation, in this regulation. Sumoylation is known to be critical for the efficient repair of highly toxic DNA double-strand breaks in both yeast and humans, and this is partly mediated by influencing DNA end resection. However, it has been unclear for which resection factor sumoylation is important, how sumoylation influences specific attributes of the relevant targets, and how this modification is coordinated with phosphorylation-based regulation. Here, we provide exciting new insights into these issues by revealing that 1) a conserved end resection factor is a SUMO target relevant to this process, 2) this regulation favors a specific repair pathway, 3) sumoylation collaborates with phosphorylation to promote protein solubility, and 4) sumoylation influences DNA repair via an “ensemble effect” that entails simultaneous small alterations of multiple substrates. Our work reveals both a novel mechanism and a general principle for SUMO-mediated regulation of DNA repair.
Published in the journal:
. PLoS Genet 11(1): e32767. doi:10.1371/journal.pgen.1004899
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004899
Summary
Proper repair of DNA lesions is crucial for cell growth and organism development. Both the choice and capacity of DNA repair pathways are tightly regulated in response to environmental cues and cell cycle phase. Recent work has uncovered the importance of protein modifications, such as phosphorylation and sumoylation, in this regulation. Sumoylation is known to be critical for the efficient repair of highly toxic DNA double-strand breaks in both yeast and humans, and this is partly mediated by influencing DNA end resection. However, it has been unclear for which resection factor sumoylation is important, how sumoylation influences specific attributes of the relevant targets, and how this modification is coordinated with phosphorylation-based regulation. Here, we provide exciting new insights into these issues by revealing that 1) a conserved end resection factor is a SUMO target relevant to this process, 2) this regulation favors a specific repair pathway, 3) sumoylation collaborates with phosphorylation to promote protein solubility, and 4) sumoylation influences DNA repair via an “ensemble effect” that entails simultaneous small alterations of multiple substrates. Our work reveals both a novel mechanism and a general principle for SUMO-mediated regulation of DNA repair.
Introduction
Efficient and accurate genome repair requires regulatory mechanisms that adjust DNA repair levels and pathway usage depending on the cellular context. For example, in response to increased lesion loads, DNA repair pathways are upregulated [1]–[3]. Additionally, DNA double-strand breaks (DSBs) are repaired by either homologous recombination (HR) or non-homologous end joining (NHEJ) depending on the cell cycle stage [4], [5]. The regulatory changes in these situations occur rapidly, involve many targets, and are reversible [1]–[3]. They are often enabled by protein modifications that reversibly add modifier groups to multiple targets. The best-illustrated example of this is protein phosphorylation mediated by the DNA damage checkpoint and cyclin-dependent kinases, which occurs within minutes of changes in repair needs and affects hundreds of protein targets (e.g. [6]–[9]).
More recently, another protein modification, sumoylation, has emerged as a key regulator of genome repair (reviewed in [10]–[13]). However, many important details of how sumoylation influences DNA repair have yet to be elucidated. For example, sumoylation is important for DSB repair in humans and yeast partly by promoting DNA end resection [14]–[18]. Yet, it has been unclear for which resection factor(s) sumoylation is relevant, how sumoylation influences specific attributes of such target(s), and how this modification is coordinated with phosphorylation-based regulation.
To address these questions, we used budding yeast as a model system to examine the sumoylation of a conserved DNA end resection factor, Sae2. Sae2 collaborates with the Mre11-Rad50-Xrs2 (MRX) complex in processing multiple kinds of DSBs, including those with clean ends and ends capped with proteins or hairpin structures (reviewed in [19], [20]). Sae2 and MRX can remove the capping structure and 100–300 bp of single-stranded DNA from DSBs in a process called end clipping [21]–[29]. This first stage of DSB end resection is followed by long-range resection via parallel pathways mediated by the Exo1 exonuclease and the Sgs1/Dna2 helicase-nuclease pair [22], [24]. End clipping commits DSB repair to HR, as resected DNA ends are poor substrates for NHEJ. This commitment point is tightly regulated in conjunction with cell cycle phase [30]–[32], as NHEJ is more beneficial in G1 when sister chromatids are absent, whereas recombination constitutes more faithful repair during S and G2 phases when the synthesized sister chromatids provide accurate repair templates.
Previous studies have shown that kinases confer cell cycle-dependent regulation of end clipping [31]–[37]. Both S phase cyclin-dependent kinase (CDK) and DNA damage checkpoint kinases phosphorylate Sae2 to promote end clipping [33], [36], [37]. This is achieved at least partly by dynamically increasing Sae2 protein solubility [37]. This form of regulation is critical as Sae2 is predominantly present as inactive aggregates in G1, presumably to limit resection in this phase [36], [37]. Upon treatment with DNA damaging agents in S phase, phosphorylation of Sae2 facilitates the rapid release of active monomeric and dimeric forms from the inactive aggregates to promote end clipping, and thus HR [37].
Here, we show that the conserved sumoylation of Sae2 also promotes its functions in the processing and repair of multiple kinds of DSBs. Interestingly, like phosphorylation, sumoylation also increases the levels of soluble Sae2. We show that the two different modifications act in synergy to achieve a stronger effect on Sae2 function. Moreover, we present evidence that sumoylation of MRX also favors resection, suggesting that the coordinated sumoylation of multiple substrates leads to greater biological consequences.
Results
Sae2 sumoylation occurs on a single lysine in its self-association domain
We and others recently reported that five proteins involved in DNA end resection are sumoylated upon DNA damage in budding yeast [17], [18]. Here we examined the sumoylation of the Sae2 protein. The Sae2 sumoylated form migrates ∼20 kDa higher than the unmodified form upon SDS-PAGE, as expected from the typical up-shift caused by mono-sumoylation (Fig. 1A). The sumoylated form can be preferentially detected by an antibody against the SUMO moiety (Fig. 1A and [17]). In addition, this modification was abolished by the simultaneous removal of both the homologous Siz1 and Siz2 SUMO ligases, but not of either single ligase (Fig. 1A). These results validate Sae2 sumoylation and indicate that the Siz ligases redundantly sumoylate Sae2.

To evaluate the functional consequences of Sae2 sumoylation, we identified the lysine that is targeted for sumoylation. Sae2 possesses two sumoylation consensus motifs, ψKxE/D, where ψ is a large hydrophobic acid [38], [39]. Mutating one of these sites, K97, to arginine abolished its sumoylation in vivo (Fig. 1B). This residue is conserved among Sae2 orthologs in closely related Saccharomyces species (Fig. S1A). To confirm that K97 is the SUMO conjugation site, Sae2 was co-expressed with sumoylation enzymes in E. coli to enable its sumoylation (see Methods). A higher-migrating form of Sae2 in the purified protein prep was specifically eliminated by treatment with the desumoylase Ulp1, indicating that it is the sumoylated form of Sae2 (Fig. S1B). Consistent with the in vivo finding, Sae2-K97R mutant protein was not sumoylated in vitro (Fig. 1C). Together, our in vivo and in vitro data indicate that lysine 97 is the bona fide SUMO conjugation site on Sae2. K97 is located within the N terminal domain that is important for self-association in several organisms (Fig. 1D and [37], [40]–[42]).
Sae2 orthologs are also sumoylated
As conserved modification of a protein in different organisms is indicative of functional importance, we examined whether Sae2 orthologs that share DNA resection functions are also targeted for sumoylation. To this end, we subjected recombinant Sae2 orthologs, namely fission yeast Ctp1 and human CtIP, to sumoylation in E. coli using reconstituted fission yeast and mammalian SUMO conjugating systems, respectively. Both SpCtp1 and hCtIP exhibited a slow migrating modified form only upon co-expression of SUMO and conjugating enzymes (Fig. 1E–1F), suggesting that they can be sumoylated. The conserved sumoylation of Sae2 orthologs supports the notion that this modification can be functionally relevant.
Sae2 sumoylation facilitates processing of complex DSB ends
Next, we investigated whether abolition of Sae2 sumoylation affects its functions by studying the sae2-K97R allele. sae2-K97R did not affect Sae2 protein levels in normal growth conditions or after genotoxin treatment at either 30°C or 37°C, excluding an effect of sumoylation on general protein stability (Fig. 2A and S1C Fig.). We then tested whether sae2-K97R affects the processing and repair of complex DNA ends, such as those capped by hairpin structures or covalently linked with proteins, using established assays. To query hairpin repair in vivo, we measured recombination that requires removal of hairpins formed through inverted Alu sequences at DSBs [27], [43]. Consistent with a previous report, deleting SAE2 greatly reduced the recombination rate measured in this assay (Fig. 2B and [27], [43]). sae2-K97R showed a 2-fold reduction (Fig. 2B), suggesting a moderate deficiency of Sae2 function in hairpin removal.
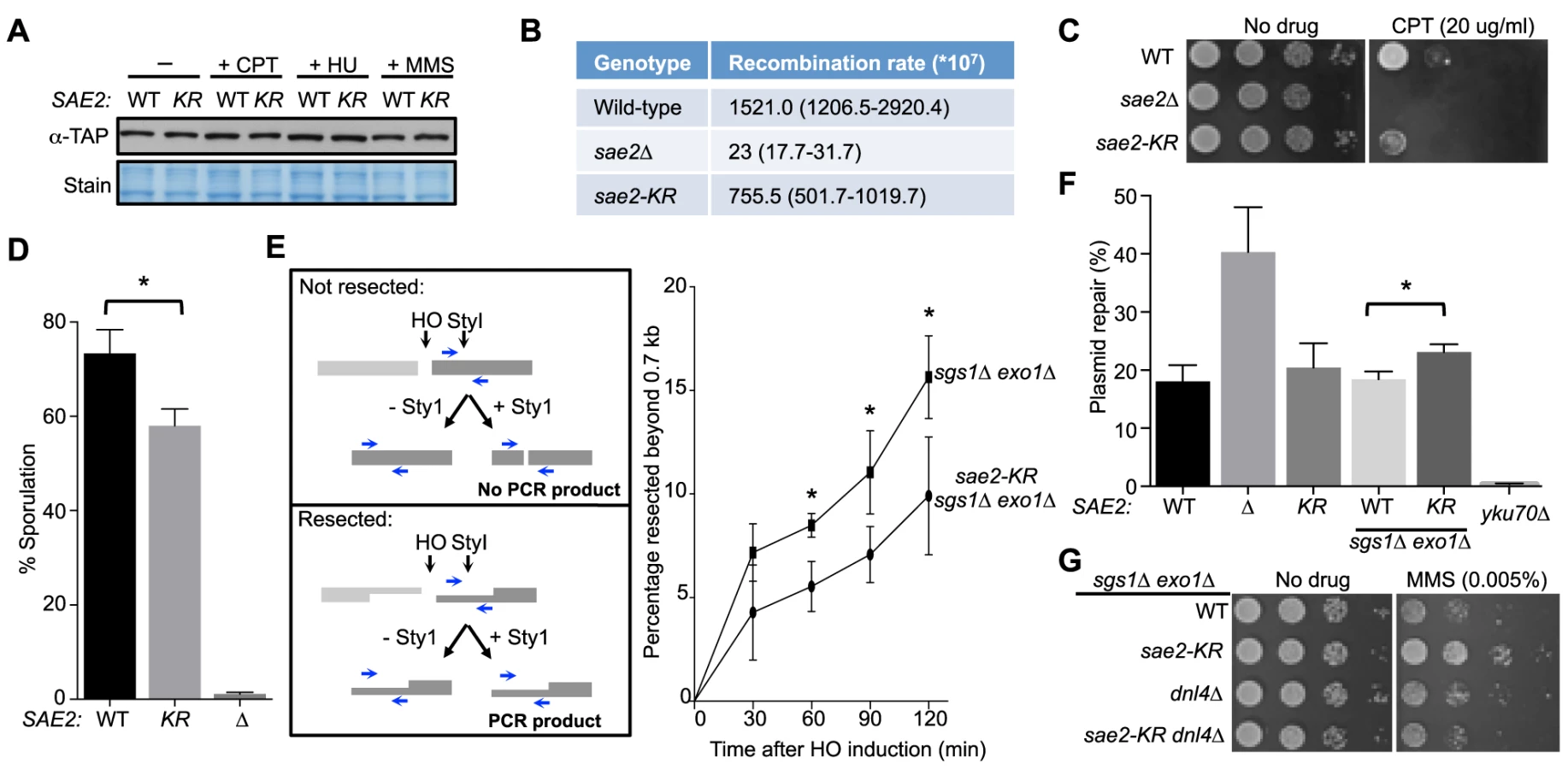
To examine processing of DSB ends that are covalently linked with proteins, we first examined Sae2-mediated processing of DSBs capped with Top1, which are induced upon camptothecin (CPT) treatment [29], [44]. Consistent with previous reports, sae2Δ cells were sensitive to CPT (Fig. 2C and [29], [44]). sae2-K97R cells exhibited an increase in sensitivity to CPT at 37°C (Fig. 2C), suggesting that sumoylation of Sae2 contributes to CPT repair. Consistent with this, Sae2 sumoylation is induced by CPT treatment and elevated temperature (S1D Fig.). Second, we examined sporulation efficiency, as Sae2-mediated removal of Spo11 conjugated to DSB ends in meiosis is required for sporulation (Fig. 2D and [28], [45]–[47]). sae2-K97R homozygous diploid cells exhibited a reproducible 20% reduction in this assay, indicating a moderate defect (Fig. 2D, p<0.05). Taken together, these results show that sae2-K97R is partially defective in the processing and repair of complex DSBs.
Sae2 sumoylation promotes DNA end clipping
We proceeded to assess whether Sae2-mediated end clipping of clean DSB ends is affected by sae2-K97R. In yeast, end clipping can be directly assayed at DSBs induced by the endonuclease HO at the MAT locus [22]. As shown previously, because end clipping is an intermediate stage in end resection, it can be best monitored when the downstream extensive resection step is blocked by removing Sgs1 and Exo1 [22]. Both qPCR - and Southern blot-based assays can be used to assess Sae2 function in this setting. The two assays take advantage of the fact that single-stranded DNA generated by resection is resistant to restriction enzyme digestion. In the qPCR-based assay, PCR products amplified using primers flanking a StyI site located 700 bp distal to the DSB are compared between digested and undigested samples (Fig. 2E, left panel, and [24], [30], [48]). PCR products from a control locus, ADH1, are used for normalization (see Methods). Using this assay, we found that sae2-K97R exhibited 60–80% of the wild-type level of resection in a time course of 120 minutes in the sgs1Δ exo1Δ background (Fig. 2E, right). The lethality of sae2Δ sgs1Δ exo1Δ prevents comparison of sae2-K97R defects with sae2Δ in this setting [22].
In the Southern blot assay, end clipping products run as a smear of bands below the HO cut (unprocessed) fragment, and both types of bands are detected by a radio-labeled probe specific to a sequence flanking the DSB (S2A Fig. and [22]). In sgs1Δ exo1Δ cells, the intensity of the smear moderately increased as the unprocessed fragment diminished with time (S2A–S2C Fig.), signifying the progress of end clipping [22]. Introducing the sae2-K97R mutation reduced end clipping efficiency, as seen by the persistence of the unprocessed fragment and decreased intensity of the smear below (S2A–S2C Fig.). Quantification of three independent strains indicated a reduction of up to 50% in the fraction of end clipping products among total cut fragments (Fig. S2C). Taken together, both the qPCR - and Southern blot-based assays show that lack of Sae2 sumoylation impairs end clipping of clean DSBs.
Sae2 sumoylation suppresses NHEJ in the sgs1Δ exo1Δ background
As end clipping disfavors NHEJ, its impairment would promote NHEJ [36], [49], [50]. Accordingly, a prediction of the observed end clipping defect in sae2-K97R sgs1Δ exo1Δ cells compared with sgs1Δ exo1Δ cells (Fig. 2E and S2A–S2C Figs.) is that the former should have higher NHEJ levels. Indeed, we detected an ∼30% increase in NHEJ in the triple mutant compared with the double, using a standard chromosomal NHEJ assay (S2D Fig.). As this assay was performed side-by-side with the Southern blot-based resection assay, equal efficiency of HO cleavage between genotypes was confirmed (S2A Fig.). We also used a well-established plasmid-based NHEJ assay in which cells are transformed with linearized or undigested plasmid DNA, and survival on selective media serves as a readout for successful repair by NHEJ [51]. sae2-K97R again exhibited a moderate increase in this assay in the sgs1Δ exo1Δ background, while its effect in the SGS1 EXO1 background was not statistically significant (Fig. 2F).
We noticed that sae2-K97R sgs1Δ exo1Δ showed more resistance to the DNA damaging agent methyl methanesulfonate (MMS) than sgs1Δ exo1Δ (Fig. 2G). This is in contrast to the inviability of sae2Δ sgs1Δ exo1Δ [22]. One interpretation is that moderate reduction of end clipping in the absence of extensive resection allows more NHEJ, thus better survival, whereas complete loss of resection confers lethality even with endogenous levels of DNA damage. Supporting this idea, the higher MMS resistance of sae2-K97R sgs1Δ exo1Δ depends on the NHEJ factor Dnl4 (Fig. 2G). These findings and the increased NHEJ seen for sae2-K97R are consistent with this mutant's impairment in end resection (Fig. 2E and S2A–S2C Fig.).
Sumoylation and phosphorylation of Sae2 contribute separately to DNA damage resistance
As phosphorylation of Sae2 is required for its resection function and DNA damage resistance [33], [36], [37], we asked whether sae2-K97R interferes with this modification. The phosphorylated forms of Sae2 manifest as slower migrating bands, and mutating two main Mec1/Tel1 phosphorylation sites and an adjacent serine, namely S249, S278 and T279 (sae2-3A, [33], [37]) results in the loss of the top bands (Figs. 1D and 3A). We found that Sae2-K97R exhibited a similar mobility shift as its wild-type counterpart (Fig. 3A), suggesting that sumoylation of Sae2 does not interfere with its phosphorylation.

To further elucidate the interplay between the two modifications, we assayed the sumoylation levels of Sae2 phosphorylation mutants. Neither sae2-3A nor sae2-S267A, which abrogates CDK-mediated phosphorylation, affected Sae2 sumoylation (Fig. 3B–3C). Consistent with this, mec1Δ cells exhibited normal levels of Sae2 sumoylation (Fig. 3D), despite being deficient for Sae2 phosphorylation [33]. Together, these results show that sumoylation of Sae2 does not require its phosphorylation.
As phosphorylation and sumoylation of Sae2 occur independently, and both contribute to Sae2 function, we examined whether their biological effects are additive. We found that combining the K97R and 3A mutations, or the K97R and S267A mutations, resulted in greater sensitivity to MMS and CPT compared to mutants that were defective for only one modification (Fig. 3E–3F). These results indicate that sumoylation and phosphorylation of Sae2 make separate contributions to DNA damage resistance.
Sumoylation of Sae2 increases the levels of soluble Sae2
We proceeded to examine how sumoylation of Sae2 affects its function. As shown recently, an important means of regulating Sae2 by protein modification is through increasing its solubility [37]. Sae2 is primarily in inactive aggregate forms in G1, whereas the soluble and active fraction of Sae2 increases upon entering S phase in the presence of DNA damage [37]. Phosphorylation of Sae2 by Mec1 and S-CDK promotes this increase [37]. Considering the genetic interaction between the two types of Sae2 modifications, we examined if sumoylation also alters the levels of soluble Sae2.
Using an established solubility assay, we examined G1-arrested cells after release into 0.03% MMS [37]. Consistent with our genetic data, levels of soluble Sae2-K97R-3A protein were significantly lower than that of Sae2-3A after cells were released from G1 (Fig. 4A). We also observed a similar but smaller decrease in levels of soluble Sae2-K97R when compared with that of wild-type Sae2 (Fig. 4B). In this case, the level of soluble Sae2-K97R protein is decreased by ∼25% compared to wild-type in a cell cycle-independent manner, suggesting that sumoylation by itself also affects Sae2 solubility. This reduction is less severe than that of Sae2-3A alone, which showed an S phase-specific decrease in Sae2 soluble levels by up to 50%, compared with wild-type Sae2 (Fig. 4C). These results suggest that while both phosphorylation and sumoylation promote Sae2 solubility, the former has a stronger effect. As the solubility difference between Sae2-3A and Sae2-K97R mutants is only 25%, yet their MMS sensitivities differ greatly, it is possible that the Sae2-3A has additional defects besides solubility.

Evidence supporting a role for MRX sumoylation in DSB processing
Because sae2-K97R exhibited mild resection defects and genotoxin sensitivity (Fig. 2 and S2 Fig.), and not to the level that has been reported for SUMO E2 (e.g. [17]), we reasoned that sumoylation likely wields a strong influence on this process by additionally targeting other factor(s), such as MRX. As mapping sumoylation sites on the three subunits of MRX proved difficult, we devised a genetic strategy to reduce MRX sumoylation. It has been shown that the catalytic domain of the de-sumoylating enzyme Ulp1 (UD) when fused with a protein can lead to the targeted removal of SUMO conjugated to the protein or its interactors [52]. A fusion with mutations of four residues required for catalytic activity and SUMO interaction (UD*) was used to control for the effect of tagging with this domain [52].
MRX physically interacts with the Ku complex, which arrives at DSBs concomitantly with MRX [53], [54]. We tested if fusing the UD domain to the Ku70 subunit (YKU70-UD) can specifically decrease MRX sumoylation. To this end, we introduced the YKU70-UD or YKU70-UD* constructs at the endogenous YKU70 locus with its native promoter. As shown in Fig. 5A, sumoylation of all three subunits of MRX was either abolished or strongly reduced in cells expressing YKU70-UD compared with YKU70-UD* control cells. To assess the specificity of desumoylation, we examined proteins that arrive at DSBs around the same time as MRX [53], [55]. YKU70-UD did not affect the sumoylation of Sae2, or the Ku-interacting protein Lif1 (Fig. 5B). In addition, sumoylation of the downstream recombination proteins Rad1 and Saw1 was not affected by YKU70-UD (Fig. 5B). Together, these results suggest that YKU70-UD can limit MRX sumoylation with good specificity.

We then examined whether YKU70-UD has a phenotype indicative of defective MRX-mediated resection. As MRX deficiency can exacerbate sae2Δ sensitivity to DNA damaging agents in certain contexts [44], we tested whether reducing MRX sumoylation by YKU70-UD causes a similar phenotype. Indeed, we found that YKU70-UD worsened the MMS sensitivity of sae2Δ cells, while YKU70-UD* conferred suppression (Fig. 5C). The latter effect is likely due to the tag's interference with Ku function, such as in inhibiting HR by Exo1 exclusion [44], [56], [57]. That YKU70-UD is additive with sae2Δ suggests that the defects caused by reduced MRX sumoylation overrides any suppression conferred by defective Ku function. This in turn suggests the possibility that reduced MRX sumoylation impairs its resection function. To test this idea, we examined resection dynamics in YKU70-UD vs. YKU70-UD* cells. In both qPCR - and Southern blot-based assays, DSB resection was decreased by 15–20% in YKU70-UD cells compared to YKU70-UD*, most obviously at early time points (Fig. 5D–5E and S3A–S3B Fig.), suggesting that sumoylation of MRX facilitates resection.
To assess if the sumoylation of MRX and Sae2 independently promotes resection, we measured end resection in YKU70-UD sae2-K97R cells. As shown in Fig. 5E, sae2-K97R further compromised resection in YKU70-UD, but not YKU70-UD*, cells. The moderate additivity in resection defects did not result in exacerbation of YKU70-UD's MMS sensitivity by sae2-K97R (Fig. 5C), likely because it is insufficient to confer MMS sensitivity or YKU70-UD exerts compensatory effects due to impaired Ku function. Taken together, our results suggest that sumoylation of MRX, in addition to that of Sae2, contributes to resection.
Discussion
Regulation of DNA repair pathway levels and capacity in response to cell cycle changes and lesion loads is important for genome maintenance and damage resistance. Despite recent progress, many forms and targets of regulation are not yet identified or understood. One of the most highly regulated DNA repair proteins is the end resection factor, Sae2. Its solubility is tightly controlled such that its activity and other functions are limited in G1 phase and increased in S phase under damage conditions [37]. We reveal here that regulation of Sae2 solubility is partly mediated by sumoylation. We also show that sumoylation collaborates with checkpoint-dependent phosphorylation in facilitating Sae2 function in end clipping. Furthermore, we provide evidence that sumoylation promotes DNA end resection by additionally targeting the MRX nuclease. This work reveals a novel mechanism of SUMO-mediated regulation of DNA repair and uncovers an example wherein sumoylation and phosphorylation, as well as multiple sumoylation events, collaborate to promote nuclease function (see model in Fig. 5F).
Proteins that cleave DNA are double-edged swords, and unscheduled DNA cleavage has to be minimized. In general, upstream constraints such as limiting recruitment to lesions can restrict the activity of downstream factors (e.g. [58]–[61]). As Sae2 is one of the first resection proteins to arrive at DSBs without any known recruiters [55], it is not surprising that it is subjected to other forms of regulation. Our findings and previous reports strongly suggest that Sae2 regulation can be achieved by different protein modifications that collaborate to ensure timely availability of the active forms of the protein [37], [62], [63].
Thus far, regulation of protein solubility by sumoylation has been reported only for proteins involved in neuronal diseases (e.g. [64]–[66]). We now provide the first example wherein sumoylation regulates the solubility of a DNA metabolism protein. As protein aggregation is a widespread phenomenon caused by high intrinsic aggregation potential of the protein (e.g. [67], [68]), it is conceivable that SUMO-mediated protein solubilization is a general effect. Such an effect by SUMO may be similar to its promotion of solubility in recombinant protein applications [69], [70]. Thus, in addition to the previously proposed glue effect of sumoylation in bridging interactions in complexes [18], [71], [72], sumoylation can also have the opposite effect of “anti-glue” to disperse proteins from aggregates. As sumoylation occurs in the Sae2 self-association domain and at a region of high aggregation potential (S4A Fig. and [41], [73]), steric or charge changes conferred by sumoylation in these regions can disfavor aggregation.
Several independent analyses show that lack of Sae2 sumoylation moderately reduces end resection and increases NHEJ (Fig. 2B–2F and S2A–S2D Fig.). The correlation of these effects with changes in the levels of soluble Sae2 suggests that the decreased availability of active Sae2 can at least partly account for the end resection defects and NHEJ increase, though other possibilities such as defective DSB recruitment cannot be excluded. As the resection defect of sae2-K97R is less severe than that of the SUMO E2 mutant, sumoylation of additional resection factors also likely matters. Indeed, reduction of MRX sumoylation also impairs resection, and in a manner additive with sae2-K97R (Fig. 5). Although a thorough examination of MRX sumoylation awaits mapping of sumoylation sites on all three subunits, these results suggest that sumoylation achieves a large biological effect by simultaneously inducing small changes in multiple substrates (“ensemble effect”). This suggestion is consistent with the observations that several dozen repair proteins are sumoylated upon DNA damage [17], [18], [74], and individual non-sumoylatable mutants usually exhibit only mild phenotypes (e.g. [75]–[77]). We propose that the ensemble effect model is common in DNA repair regulation and other processes to confer robustness to a system. We also note that the usefulness of this strategy is also seen for other protein modifications (e.g. [32], [62]), and that as in the case of sumoylation, the effects of a particular modification are unique to the substrate, rather than conforming to a general mechanism (e.g. [8], [78]–[80]).
In summary, our work provides strong evidence for a new role for sumoylation in regulating DNA repair and its collaboration with phosphorylation-based regulation. Considering that only a few sumoylated substrates in DNA repair have been examined in detail thus far, future studies on additional substrates and the interplay between sumoylation and other forms of regulation will greatly expand our knowledge of how repair pathway levels and choice are determined in cells.
Materials and Methods
Yeast strains and genetic manipulations
Strains used are listed in Table 1. Only one strain per genotype is shown for simplicity, but at least two strains per genotype were tested in each assay. Standard yeast protocols were used for strain generation, growth and medium preparation. As siz1Δ siz2Δ results in amplification of the 2 micron plasmid [81], strains with siz1Δ siz2Δ mutations were cured of the plasmid as described [82].

Protein preparation and detection of sumoylated proteins
Detection of the sumoylated form of Sae2 was performed as described previously [17]. In brief, log phase cultures were treated with 0.3% methyl methanesulfonate (MMS, Sigma-Aldrich) or 50 ug/ml camptothecin (CPT, Sigma-Aldrich) or at 37°C for 2 h. Cells were lysed by bead beating under denaturing conditions, and TAP-or HA-tagged proteins were immunoprecipitated. These were then washed and eluted with loading dye, followed by SDS-PAGE and western blotting with antibodies against SUMO [83], the protein A portion of the TAP tag (Sigma-Aldrich) or HA (12CA5). We note that as the Fc portion of the SUMO antibody interacts with the Protein A part of TAP, it also detects the unmodified protein, but more strongly so for the sumoylated form because of additional high affinity for SUMO. Protein preparation for detecting Sae2 phosphorylation and protein levels was performed as described [33]; DNA damage treatment was performed as above.
Assessment of soluble Sae2 protein levels
Assay was performed essentially as described [37] except that all Sae2 constructs were expressed from its own chromosomal locus. In brief, G1-arrested cells were released into 0.03% MMS and samples were harvested at the indicated time points for protein and FACS analyses. Upon complete cell lysis by bead beating and removal of DNA by DNaseI treatment, cell extract was centrifuged at high speed (14k rpm for 30 min) to separate the soluble fraction from the insoluble. The soluble fractions were analyzed by SDS-PAGE and western blotting against the tag. The housekeeping protein Adh1 was used as loading control as its levels are invariant during the time course. FACS analyses show proper arrest and release for all the strains examined. We note that Sae2 is sumoylated in both G1 and S phases during this procedure (S4B Fig.). To assess Sae2 soluble forms in different strains, two spore clones of each genotype were examined in at least two independent tests. For quantification, we compared solubility between the two strains for each time point. In brief, we first determined the soluble Sae2 protein level relative to loading control for each strain at each time point, and then calculated the ratio between the genotypes to represent it in Fig. 4. The student's t test statistical analysis was performed for “Sae2 protein level relative to loading control” between the two genotypes from 6 repeats (2 trials with 3 spores).
In vitro sumoylation assay
Both GST-tagged Sae2 and hCtIP were sumoylated in E. coli by co-expression with E1 (Aos1-Uba2), E2 (Ubc9) and SUMO-1 [84] (the pT-E1E2S1 plasmid was a gift from Dr. Hisato Saitoh). Plasmids for expression of GST-tagged Sae2 and hCtIP are derivatives of pGEX-4T1 and were a gift from Dr. Stephen Jackson [36], [85]. The plasmid expressing GST-Sae2K97R (pRS72) was made by site-directed mutagenesis of pGEX-4T1-Sae2. Plasmids were transformed into BL21 (DE3) cells, and single colonies were used to inoculate overnight cultures of LB (plus 100 ug/ml ampicillin or 25 ug/ml ampicillin and 17 ug/ml chloramphenicol), which were incubated at 30°C with shaking at 250 rpm. These starter cultures were used to inoculate fresh LB (with ampicillin plus chloramphenicol added as required) cultures to an OD600 of 0.1, which were then grown at 30°C with shaking at 250 rpm to an OD600 of 1.2. The temperature was then lowered to 25°C and 250 uM IPTG added to induce expression of the proteins. The cultures were incubated for another 16 h at 25°C before harvesting by centrifugation. The cell pellet was resuspended in PBS (pH 7.3) supplemented with 1 mM PMSF and 5 mM DTT. After sonication and centrifugation (47000×g at 4°C for 20 min), the soluble protein fraction was loaded onto an equilibrated Glutathione Sepharose Fast Flow column (GE Healthcare Life Sciences). The column was washed with 10 column volumes of PBS and the GST-Sae2 and GST-Sae2-SUMO proteins eluted with 2 column volumes of 50 mM Tris-HCl (pH 8.0), 30 mM reduced glutathione. Proteins were dialyzed against 20 mM Tris, pH 8.0 containing 150 mM NaCl at 4°C. To perform SUMO cleavage reactions, recombinant purified Ulp1 (10 nM) [86] was added to 5 uM partially purified GST-Sae2/GST-Sae2-SUMO and incubated at 23°C for 30 min in buffer containing 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% Tween-20, and 2 mM DTT. The proteins were separated on a 10% SDS PAGE gel and analyzed on a western blot probed with an anti-GST antibody (GE Healthcare Life Sciences). 3HA-tagged SpCtp1 was sumoylated in E. coli BL21 (DE3) cells by co-expression with the S. pombe E1 (Rad31+Fub2), E2 (Hus5 fused to a 6×His-tag) and SUMO (Pmt3GG). The 6×His-Hus5 is fused to 3HA-SpCtp1 and Pmt3GG is tagged with GST. Full details of the plasmid constructs will be provided elsewhere. Transformed BL21 (DE3) cells were cultured and proteins were isolated as described above.
Inverted Alu recombination assay
These were performed as described [43]. Single colonies were picked from streakouts and allowed to grow for 3 days. Each single colony was resuspended in 0.25 ml water by vortexing (0th dilution) and ten-fold serial dilutions were prepared. 100 ul of the 5th dilution was plated onto complete medium, while 100 ul of the 2nd dilution (or 0th dilution for sae2Δ cells) was plated onto medium lacking lysine. Successful recombination by processing Alu-generated hairpin DSBs generates LYS+ colonies. Fourteen colonies were analyzed in this manner for each genotype, and the recombination rate was calculated by fluctuation analysis.
DSB resection assays
Both qPCR - and Southern blot-based assays were performed as described [17], [48]. For both assays, a DSB at the MAT locus was introduced by galactose-induced expression of the HO endonuclease throughout the time course either in asynchronous (Figs. 2E and 5D, S2A–S2C and S3A–S3B Figs.), or G2-arrested cultures (Fig. 5E). Samples were collected at the indicated time points. Genomic DNA was isolated and an aliquot was subjected to digestion with XbaI and StyI. For Southern blot-based method, digested DNA was subjected to native agarose gel electrophoresis, transferred to Hybond XL (GE Healthcare) membranes, and hybridized with radiolabeled DNA probes. Quantification of intensities of bands on the Southern blots was done using ImageGauge. DSB end resection at each time point was calculated as the ratio of the signal intensity at that time point to that at the first time point after HO induction. Note that as Sae2 sumoylation was strongly increased at 37°C (S1D Fig.), sae2-K97R phenotype in the above assays (except Fig. 5E) was examined at this temperature.
qPCR-based resection assay was performed as described [48]. In brief, 150 ng of genomic DNA isolated as above was subjected to restriction enzyme digestion with StyI or mock-digested in a reaction volume of 15 ul. DNA was diluted by addition of 55 ul of ice-cold dH2O. 8.8 ul of the diluted DNA was used for each qPCR reaction in a total volume of 20 ul. Primer sequences are specified in [48]. PCRs were performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) with the Bio-Rad DNA Engine Chromo 4 system and corresponding software (Opticon). All reactions were amplified using the following program: 95°C for 10 min, 40 cycles of (95°C for 15 s, followed by 58°C for 60 s), and melting curve 10 min. Reactions were set up in triplicates for all primer pairs and the resulting average threshold cycle (Ct) value was used for calculation. The percentage of DNA resected to 0.7 kb in HO-cut DNA was calculated by x = 200/{(1+2ΔCt)*f}, where ΔCt = Ct,digestion−Ct,mock, and f is the fraction cut by HO as quantified by Southern blot analysis. All Ct values were corrected for DNA concentrations by comparing with values for amplification at the ADH1 locus. For both resection assays, at least two spore clones of each genotype were examined in two or more trials.
NHEJ assays
The analysis of chromosomal NHEJ levels was performed as previously described [87]. DSB induction was induced for 1.5 h, was performed side-by-side with the resection assay (compare cleavage efficiency at 1.5 h). DSBs were induced in cells that cannot repair the break by HR and rely on NHEJ for repair; thus, NHEJ proficiency can be discerned by comparing the numbers of colonies that survive transient DSB induction. Plasmid-based NHEJ assay was performed by transforming either 1 ng of undigested or 20 ng of BamHI-digested pRS416 plasmid carrying URA3 into competent cells, and plating on medium lacking uracil. For yku70Δ control cells, 100 ng of digested plasmid was used for transformation. Successful NHEJ repair results in ligation of the linearized plasmid and thus growth on -URA medium. Transformation efficiency was calculated as the number of colonies on -URA medium divided by the amount of DNA transformed. NHEJ repair for each genotype was calculated as the ratio of transformation efficiencies of digested to undigested samples. For both NHEJ assays, at least two spore clones of each genotype were examined in two or more independent trials.
Other methods
Spot assays were performed as described previously [17]. Briefly, log phase cells were diluted 10-fold and spotted onto YPD media with or without CPT or MMS. Plates were incubated at 30°C (Fig. 5C) or 37°C (Figs. 2D, 2G and 3E–3F), and photographed after 24–72 h. At least two spore clones of each genotype were examined in two or more independent trials. Sporulation assay was performed essentially as described [36]. Diploid SK1 cells were grown overnight in YPD medium, washed twice with warm sporulation medium, and left in sporulation medium for 36 h at 30°C. The percentage of sporulated cells was determined by light microscopy.
Supporting Information
Zdroje
1. CicciaA, ElledgeSJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40 : 179–204.
2. MarechalA, ZouL (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5.
3. PoloSE, JacksonSP (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25 : 409–433.
4. SymingtonLS, GautierJ (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45 : 247–271.
5. ChapmanJR, TaylorMR, BoultonSJ (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47 : 497–510.
6. HerzbergK, BashkirovVI, RolfsmeierM, HaghnazariE, McDonaldWH, et al. (2006) Phosphorylation of Rad55 on serines 2, 8, and 14 is required for efficient homologous recombination in the recovery of stalled replication forks. Mol Cell Biol 26 : 8396–8409.
7. SmolkaMB, AlbuquerqueCP, ChenSH, ZhouH (2007) Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA 104 : 10364–10369.
8. ChenX, NiuH, ChungWH, ZhuZ, PapushaA, et al. (2011) Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol 18 : 1015–1019.
9. MatosJ, BlancoMG, MaslenS, SkehelJM, WestSC (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147 : 158–172.
10. CremonaCA, SarangiP, ZhaoX (2012) Sumoylation and the DNA Damage Response. Biomolecules 2 : 376–388.
11. JacksonSP, DurocherD (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49 : 795–807.
12. UlrichHD (2014) Two-way communications between ubiquitin-like modifiers and DNA. Nat Struct Mol Biol 21 : 317–324.
13. JentschS, PsakhyeI (2013) Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu Rev Genet 47 : 167–186.
14. MaedaD, SekiM, OnodaF, BranzeiD, KawabeY, et al. (2004) Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair 3 : 335–341.
15. MorrisJR, BoutellC, KepplerM, DenshamR, WeekesD, et al. (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462 : 886–890.
16. GalantyY, BelotserkovskayaR, CoatesJ, PoloS, MillerKM, et al. (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462 : 935–939.
17. CremonaCA, SarangiP, YangY, HangLE, RahmanS, et al. (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Mol Cell 45 : 422–432.
18. PsakhyeI, JentschS (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151 : 807–820.
19. PaullTT (2010) Making the best of the loose ends: Mre11/Rad50 complexes and Sae2 promote DNA double-strand break resection. DNA Repair (Amst) 9 : 1283–1291.
20. MimitouEP, SymingtonLS (2011) DNA end resection–unraveling the tail. DNA Repair (Amst) 10 : 344–348.
21. LengsfeldBM, RattrayAJ, BhaskaraV, GhirlandoR, PaullTT (2007) Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 28 : 638–651.
22. MimitouEP, SymingtonLS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455 : 770–774.
23. ClericiM, MantieroD, LucchiniG, LongheseMP (2005) The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem 280 : 38631–38638.
24. ZhuZ, ChungWH, ShimEY, LeeSE, IraG (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134 : 981–994.
25. NicoletteML, LeeK, GuoZ, RaniM, ChowJM, et al. (2010) Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat Struct Mol Biol 17 : 1478–1485.
26. CannavoE, CejkaP (2014) Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514 : 122–125.
27. LobachevKS, GordeninDA, ResnickMA (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108 : 183–193.
28. NealeMJ, PanJ, KeeneyS (2005) Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436 : 1053–1057.
29. DengC, BrownJA, YouD, BrownJM (2005) Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics 170 : 591–600.
30. ZierhutC, DiffleyJF (2008) Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27 : 1875–1885.
31. IraG, PellicioliA, BalijjaA, WangX, FioraniS, et al. (2004) DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 : 1011–1017.
32. AylonY, LiefshitzB, KupiecM (2004) The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23 : 4868–4875.
33. BaroniE, ViscardiV, Cartagena-LirolaH, LucchiniG, LongheseMP (2004) The functions of budding yeast Sae2 in the DNA damage response require Mec1 - and Tel1-dependent phosphorylation. Mol Cell Biol 24 : 4151–4165.
34. FalckJ, FormentJV, CoatesJ, MistrikM, LukasJ, et al. (2012) CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep 13 : 561–568.
35. ZhangY, ShimEY, DavisM, LeeSE (2009) Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair (Amst) 8 : 1235–1241.
36. HuertasP, Cortes-LedesmaF, SartoriAA, AguileraA, JacksonSP (2008) CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455 : 689–692.
37. FuQ, ChowJ, BernsteinKA, MakharashviliN, AroraS, et al. (2014) Phosphorylation-regulated transitions in an oligomeric state control the activity of the Sae2 DNA repair enzyme. Mol Cell Biol 34 : 778–793.
38. RodriguezMS, DargemontC, HayRT (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem 276 : 12654–12659.
39. SampsonDA, WangM, MatunisMJ (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J Biol Chem 276 : 21664–21669.
40. DubinMJ, StokesPH, SumEY, WilliamsRS, ValovaVA, et al. (2004) Dimerization of CtIP, a BRCA1 - and CtBP-interacting protein, is mediated by an N-terminal coiled-coil motif. J Biol Chem 279 : 26932–26938.
41. KimHS, VijayakumarS, RegerM, HarrisonJC, HaberJE, et al. (2008) Functional interactions between Sae2 and the Mre11 complex. Genetics 178 : 711–723.
42. YouZ, ShiLZ, ZhuQ, WuP, ZhangYW, et al. (2009) CtIP links DNA double-strand break sensing to resection. Mol Cell 36 : 954–969.
43. LobachevKS, StengerJE, KozyrevaOG, JurkaJ, GordeninDA, et al. (2000) Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J 19 : 3822–3830.
44. FosterSS, BalestriniA, PetriniJH (2011) Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol 31 : 4379–4389.
45. PrinzS, AmonA, KleinF (1997) Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics 146 : 781–795.
46. KeeneyS, KlecknerN (1995) Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA 92 : 11274–11278.
47. McKeeAH, KlecknerN (1997) A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics 146 : 797–816.
48. ChenH, LisbyM, SymingtonLS (2013) RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell 50 : 589–600.
49. LeeK, LeeSE (2007) Saccharomyces cerevisiae Sae2 - and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 176 : 2003–2014.
50. DengSK, GibbB, de AlmeidaMJ, GreeneEC, SymingtonLS (2014) RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat Struct Mol Biol 21 : 405–412.
51. BoultonSJ, JacksonSP (1996) Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15 : 5093–5103.
52. AlmedawarS, ColominaN, Bermudez-LopezM, Pocino-MerinoI, Torres-RosellJ (2012) A SUMO-dependent step during establishment of sister chromatid cohesion. Curr Biol 22 : 1576–1581.
53. WuD, TopperLM, WilsonTE (2008) Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics 178 : 1237–1249.
54. PalmbosPL, DaleyJM, WilsonTE (2005) Mutations of the Yku80 C terminus and Xrs2 FHA domain specifically block yeast nonhomologous end joining. Mol Cell Biol 25 : 10782–10790.
55. LisbyM, BarlowJH, BurgessRC, RothsteinR (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118 : 699–713.
56. MimitouEP, SymingtonLS (2010) Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29 : 3358–3369.
57. HangLE, LopezCR, LiuX, WilliamsJM, ChungI, et al. (2014) Regulation of Ku-DNA association by Yku70 C-terminal tail and SUMO modification. J Biol Chem
58. LiF, DongJ, PanX, OumJH, BoekeJD, et al. (2008) Microarray-based genetic screen defines SAW1, a gene required for Rad1/Rad10-dependent processing of recombination intermediates. Mol Cell 30 : 325–335.
59. GuzderSN, SommersCH, PrakashL, PrakashS (2006) Complex formation with damage recognition protein Rad14 is essential for Saccharomyces cerevisiae Rad1-Rad10 nuclease to perform its function in nucleotide excision repair in vivo. Mol Cell Biol 26 : 1135–1141.
60. TohGW, SugawaraN, DongJ, TothR, LeeSE, et al. (2010) Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Repair 9 : 718–726.
61. ShimEY, ChungWH, NicoletteML, ZhangY, DavisM, et al. (2010) Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. Embo J 29 : 3370–3380.
62. RobertT, VanoliF, ChioloI, ShubassiG, BernsteinKA, et al. (2011) HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471 : 74–79.
63. LafranchiL, de BoerHR, de VriesEG, OngSE, SartoriAA, et al. (2014) APC/CCdh1 controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J
64. SteffanJS, AgrawalN, PallosJ, RockabrandE, TrotmanLC, et al. (2004) SUMO modification of Huntingtin and Huntington's disease pathology. Science 304 : 100–104.
65. JanerA, WernerA, Takahashi-FujigasakiJ, DaretA, FujigasakiH, et al. (2010) SUMOylation attenuates the aggregation propensity and cellular toxicity of the polyglutamine expanded ataxin-7. Hum Mol Genet 19 : 181–195.
66. KrumovaP, MeulmeesterE, GarridoM, TirardM, HsiaoHH, et al. (2011) Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol 194 : 49–60.
67. Fernandez-EscamillaAM, RousseauF, SchymkowitzJ, SerranoL (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22 : 1302–1306.
68. PawarAP, DubayKF, ZurdoJ, ChitiF, VendruscoloM, et al. (2005) Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol 350 : 379–392.
69. ZuoX, LiS, HallJ, MatternMR, TranH, et al. (2005) Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli. J Struct Funct Genomics 6 : 103–111.
70. MalakhovMP, MatternMR, MalakhovaOA, DrinkerM, WeeksSD, et al. (2004) SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5 : 75–86.
71. LinDY, HuangYS, JengJC, KuoHY, ChangCC, et al. (2006) Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell 24 : 341–354.
72. ShenTH, LinHK, ScaglioniPP, YungTM, PandolfiPP (2006) The mechanisms of PML-nuclear body formation. Mol Cell 24 : 331–339.
73. Conchillo-SoleO, de GrootNS, AvilesFX, VendrellJ, DauraX, et al. (2007) AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 8 : 65.
74. SilverHR, NissleyJA, ReedSH, HouYM, JohnsonES (2011) A role for SUMO in nucleotide excision repair. DNA Repair 10 : 1243–1251.
75. SarangiP, BartosovaZ, AltmannovaV, HollandC, ChavdarovaM, et al. (2014) Sumoylation of the Rad1 nuclease promotes DNA repair and regulates its DNA association. Nucleic Acids Res 42 : 6393–6404.
76. SarangiP, AltmannovaV, HollandC, BartosovaZ, HaoF, et al. (2014) A Versatile Scaffold Contributes to Damage Survival via Sumoylation and Nuclease Interactions. Cell Rep 9 : 143–152.
77. SacherM, PfanderB, HoegeC, JentschS (2006) Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol 8 : 1284–1290.
78. TomimatsuN, MukherjeeB, Catherine HardebeckM, IlchevaM, Vanessa CamachoC, et al. (2014) Phosphorylation of Exo1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 5 : 3561.
79. Gallo-FernandezM, SaugarI, Ortiz-BazanMA, VazquezMV, TerceroJA (2012) Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res 40 : 8325–8335.
80. MorinI, NgoHP, GreenallA, ZubkoMK, MorriceN, et al. (2008) Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J 27 : 2400–2410.
81. BurgessRC, RahmanS, LisbyM, RothsteinR, ZhaoX (2007) The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol Cell Biol 27 : 6153–6162.
82. TsalikEL, GartenbergMR (1998) Curing Saccharomyces cerevisiae of the 2 micron plasmid by targeted DNA damage. Yeast 14 : 847–852.
83. ZhaoX, BlobelG (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci USA 102 : 4777–4782.
84. UchimuraY, NakamuraM, SugasawaK, NakaoM, SaitohH (2004) Overproduction of eukaryotic SUMO-1 - and SUMO-2-conjugated proteins in Escherichia coli. Anal Biochem 331 : 204–206.
85. SartoriAA, LukasC, CoatesJ, MistrikM, FuS, et al. (2007) Human CtIP promotes DNA end resection. Nature 450 : 509–514.
86. MossessovaE, LimaCD (2000) Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell 5 : 865–876.
87. MooreJK, HaberJE (1996) Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 16 : 2164–2173.
88. ZhaoX, MullerEG, RothsteinR (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2 : 329–340.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 1
Nejčtenější v tomto čísle
- The Global Regulatory Architecture of Transcription during the Cell Cycle
- A Truncated NLR Protein, TIR-NBS2, Is Required for Activated Defense Responses in the Mutant
- Proteasomes, Sir2, and Hxk2 Form an Interconnected Aging Network That Impinges on the AMPK/Snf1-Regulated Transcriptional Repressor Mig1
- The SWI2/SNF2 Chromatin Remodeler BRAHMA Regulates Polycomb Function during Vegetative Development and Directly Activates the Flowering Repressor Gene