
Connecting Replication and Repair: YoaA, a Helicase-Related Protein, Promotes Azidothymidine Tolerance through Association with Chi, an Accessory Clamp Loader Protein
During the replication of the cell’s genetic material, difficulties are often encountered. These problems require the recruitment of special proteins to repair DNA so that replication can be completed. The failure to do so causes cell death or deleterious changes to the cell’s genetic material. In humans, these genetic changes can promote cancer formation. Our study identifies a repair protein that is recruited to problem sites by interactions with the replication machinery. These interactions provide a means by which the cell can sense, respond to and repair damage that interferes with the completion of DNA replication.
Published in the journal:
. PLoS Genet 11(11): e32767. doi:10.1371/journal.pgen.1005651
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005651
Summary
During the replication of the cell’s genetic material, difficulties are often encountered. These problems require the recruitment of special proteins to repair DNA so that replication can be completed. The failure to do so causes cell death or deleterious changes to the cell’s genetic material. In humans, these genetic changes can promote cancer formation. Our study identifies a repair protein that is recruited to problem sites by interactions with the replication machinery. These interactions provide a means by which the cell can sense, respond to and repair damage that interferes with the completion of DNA replication.
Introduction
All cells must balance ongoing DNA synthesis with repair reactions that are necessary to overcome problems in replication. Persistent, unreplicated single-strand gaps in DNA may be caused by lesions or by DNA structures that impede DNA polymerization in either the replication template or the nascent strand. Filling of single-strand DNA (ssDNA) gaps (a process termed “post-replication gap repair”) can be accomplished by translesion DNA synthesis, involving specialized translesion polymerases (reviewed in [1]), by template-switching to enlist an undamaged sister-strand as template [2, 3] or by homologous recombination (reviewed in [4]). Recruitment of DNA processing enzymes including nucleases, helicases and topoisomerases to a persistent gap, signaled by the presence of single-strand DNA binding protein (SSB), may also aid repair [5]. If left unrepaired, single-strand gaps are converted to potentially lethal double-strand breaks (DSBs) by endonucleolytic cleavage or by convergence of a new replication fork into the incompletely replicated region.
To study the process of ssDNA gap repair in E. coli, we have employed the nucleoside analog 3’ azidothymidine (AZT) [6], which acts as a chain terminator when incorporated into DNA by DNA polymerases. AZT arrests replication and causes single-strand gaps to accumulate in vivo, as evident by the cellular accumulation of foci of SSB protein. In addition, AZT promotes induction of the SOS DNA damage response, dependent on the RecFOR mediator proteins that promote RecA recombinase binding to single-strand gaps in DNA [6]. In E. coli, the genetic requirement for RecFOR is diagnostic for the formation of ssDNA gaps in adjoining duplex DNA and is distinct from single-strand DNA caused by resection of DSBs, which require RecBCD for processing and RecA loading (reviewed in [4]). E. coli cells can tolerate a certain level of AZT monophosphate incorporation, which appears to be excised from the 3’ nascent strain by Exonuclease III [6], a 3’ to 5’ exonuclease acting on duplex DNA from a nick or gap [7]. AZT also appears to elicit recombination via the RecAFOR pathway and to produce DSBs at some level, which are repaired via the alternative RecABCD recombination pathway [6].
To identify new functions that aid in AZT tolerance, we performed a genetic screen for expression-dependent suppression of AZT-sensitive phenotypes of several mutant strains. A plasmid library representing tac-promoter expressed E. coli open reading frames (ORFs) [8] was screened in pools for the ability to increase plating efficiency in the presence of normally-toxic levels of AZT. Two ORFs were recovered repeatedly: one encoding the putative Fe-S helicase, yoaA, and the other encoding an accessory protein of the replisome clamp loader (χ), holC. Interestingly, these proteins have been reported to interact physically by pulldowns of affinity-tagged bait proteins, followed by mass spectrometric analysis of interacting peptides [9]. We confirm this interaction in this study. The association of a DNA helicase with a replisome component provides a potential way to target a repair factor to a stalled replication fork.
In E. coli, the bulk of DNA replication is catalyzed by the DNA polymerase III holoenzyme, which participates in a plethora of protein interactions that regulate its activity and processivity (reviewed in [10]). The DNA polymerase III core complex (α, dnaE; θ, holE; and ε, dnaQ) is tethered to its template strand by the processivity clamp, β (encoded by dnaN gene). Loading and unloading of the clamp is facilitated by a clamp-loader complex, consisting of three subunits encoded by dnaX (τ and/or γ), and one each encoded by holA (δ) and holB (δ’). This clamp loader complex binds through τ to both the fork helicase protein, DnaB, and to the α subunit of the core DNA polymerase III complex. Two additional proteins, χ (encoded by holC) and ψ (encoded by holD), act in a dimeric complex as accessory proteins to the clamp loader [11]. Completing the cycle of interactions, ψ binds to γ or τ of the clamp loader [12] and χ binds to single-strand DNA binding protein, SSB, [13], that coats ssDNA revealed as fork unwinding proceeds.
The function of the accessory clamp loader proteins, χ and ψ, has been somewhat unclear since the core complex ([τ/γ]3δδ’) is sufficient to load and unload β clamps. However, a number of functions have been suggested by biochemical studies. χψ may aid assembly of the core clamp loader complex by increasing the affinity of τ/γ with δ and δ’ [14]. χ assists DNA polymerization on SSB-coated substrates [15] and promotes 5’ strand displacement [16]; the SSB-χ interaction may also aid the stability of Polymerase III on its template. χ promotes the handoff of primers from primase to PolIII [17]. ψ may also enhance clamp-loader activity and enforce an order to clamp assembly [18]. In E. coli, χ is nonessential (although mutants are quite sick and accumulate suppressors), and ψ’s essentiality can be suppressed by mutations that prevent the induction of the SOS response or by loss of translesion polymerases and the cell division inhibitor controlled by the SOS response [19]. The non-essentiality of χ and ψ are consistent with the hypothesis that χ and ψ are not obligatory for replication but replication in their absence causes ssDNA gaps to accumulate in the fork. This could result from defects in replication and/or the inability to elicit repair of gaps. Single molecule fluorescence microscopy of labeled replisome components suggests that χψ complexes are in excess of core polymerase complexes [20]. Therefore, χψ could be recruited independently through the SSB-χ interaction and provide a stand-alone, polymerase-independent function.
Our genetic studies reported here implicate the previously uncharacterized YoaA protein in the repair of replication forks and AZT tolerance. YoaA is related in protein sequence to a known E. coli Fe-S helicase of the Rad3/XPD family [21], DinG [22–24]. In addition, we show that a network of interactions is required for AZT tolerance: YoaA with χ, χ with SSB and ψ with χ. Our work suggests that persistent gaps in DNA accumulate SSB, which recruits YoaA. We propose that YoaA’s unwinding activity permits the proofreading exonuclease of Pol III to remove AZT from the nascent strand. Additionally, our work illustrates a new strategy by which replisome interactions enlist proteins directly to facilitate gap repair.
Results
Genetic suppressor screen and AZT-sensitivity phenotypes
To identify new repair factors, we performed a genetic screen for expression-dependent suppressors of the AZT sensitivity (AZTs) of various mutants defective in repair functions or their regulatory pathways. A pNTR - mobile plasmid library expressing each E. coli ORF (under the tac promoter) was introduced by conjugation into various AZTs strains and those isolates that conferred increased tolerance to AZT were selected. We screened for suppressors of the following genetic mutants with different defects in repair: xthA (AZT excision), recB (DSB repair), lexA3 (SOS regulatory response), relA (stringent regulatory response), rpoS (general stress response), parE-ts (Topoisomerase IV). Two ORFs were found repeatedly as suppressors in multiple screens: yoaA, an ORF of unknown function predicted to encode an Fe-S helicase, and holC, the χ subunit of the replication clamp loader complex. Both ORFs were isolated as suppressors of xthA and parE-ts; holC was isolated as a suppressor of recB and yoaA as a suppressor of relA, rpoS and lexA3. (Please note that the suppressor screen was not performed to saturation, so the failure to isolate an ORF from any particular strain does not mean it is not a suppressor.) Our interest in these suppressors was piqued by the fact that χ is a component of the replisome and has a reported physical interaction with the yoaA-encoded protein (b1808), documented in a high-throughput study of E. coli protein interactions [9].
The ability of holC and yoaA to suppress the extreme AZT-sensitivity of several strains was determined by reintroduction of the mobile plasmids by DNA transformation. Plasmid-borne expression of either holC or yoaA dramatically increased tolerance to chronic AZT exposure (Fig 1) in a number of genetic backgrounds, including lexA3 (defective in the SOS response), recA (defective in the SOS response and homologous recombination) and xthA (defective in exonuclease III). Expression of holC or yoaA improved AZT tolerance even in wild-type strains, as judged by colony size on AZT-containing medium (Fig 1), although holC or yoaA expression did not alter plating efficiency (i. e. the number of colonies that form). We hypothesize that holC and yoaA overexpression reduce the cellular burden of AZT by assisting in the removal of AZT-monophosphate from DNA. Expression of holD, encoding ψ, a partner to χ in the accessory clamp loader complex, or holA or holB, the δ and δ’ components of the clamp loader, did not alter AZT tolerance in the wild-type background (see S1 Fig). Expression of dinG, a DNA helicase and paralog of yoaA, was toxic and therefore could not be tested for suppressor activity (see S1 Fig).

Knockout mutants of yoaA and its paralog dinG were tested for effects on tolerance of AZT chronic exposure. Mutants in yoaA were sensitive to AZT, whereas mutants in dinG were sensitive only to very high concentrations of AZT. The double yoaA dinG mutant exhibited strong synergistic sensitivity to AZT (Fig 2). Sensitivity of both yoaA and yoaA dinG strains could be complemented by pNTR plasmid-expressed yoaA (Fig 3). These data indicate that YoaA plays a key role in AZT tolerance in wild-type E. coli cells, with DinG providing a partial backup function.

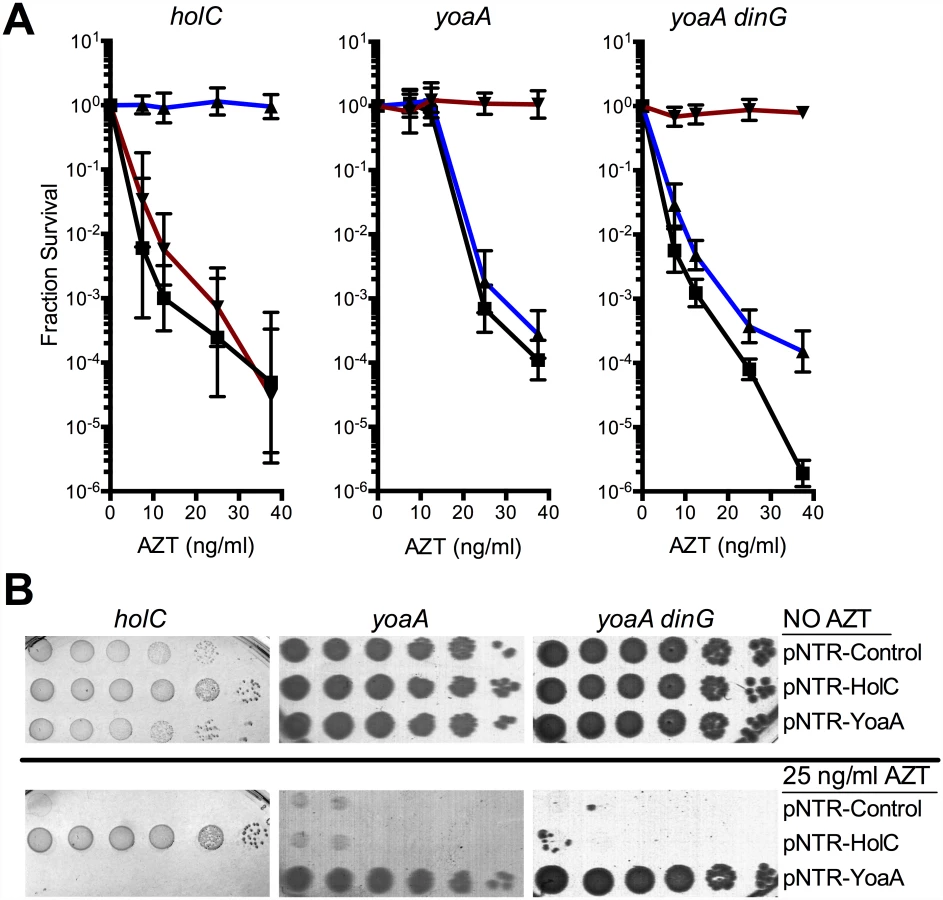
Genetic and physical interaction of YoaA and χ
Because of the reported interaction between the YoaA and χ proteins, we tested whether yoaA suppression required holC and whether holC required yoaA (Fig 3). An E.coli strain with a deletion mutation in yoaA was strongly sensitive to AZT, with plating efficiency several orders of magnitude below wild-type strains (Fig 2). This phenotype was fully complemented by plasmid-expressed yoaA but holC expression did not suppress the phenotype (Fig 3). Likewise, a holC mutant poorly tolerates AZT exposure (Fig 2) and is complemented by plasmid-expressed holC but not suppressed by yoaA (Fig 3). This stands in contrast to other AZT-sensitive strains that were strongly suppressed by both yoaA and holC expression (Fig 1). The codependence of holC and yoaA suppression indicates that a YoaA/χ complex mediates tolerance to AZT and that the formation or effectiveness of this complex is enhanced by increased expression of either the YoaA or χ component. Interestingly, we note that the holC mutant, as assayed by plating efficiency, was more sensitive to AZT than the yoaA mutant; in fact, this increased sensitivity was ameliorated by loss of yoaA (Fig 2)—that is, functional YoaA leads to AZT-sensitivity in holC mutant strains. In addition, yoaA mutations appear to suppress the poor growth phenotype of the holC strain. Introduction of a YoaA-expressing plasmid into this yoaA holC strain re-sensitized it to AZT (S2 Table). These data indicate that YoaA function is, in some way, deleterious in the absence of χ, but not when χ is present.
A previous study employed mass spectrometry to identify E. coli proteins that interacted with 1000 TAP-tagged essential or conserved proteins [9]. This included holC-TAP, which copurified with a number of other proteins in the replisome and with YoaA (designated as “b1808” in that study). This study did not detect YoaA as an interactor with any other member of the clamp loader complex (including ψ, holD; γ/τ dnaX; δ, holA; or δ’, holB), suggesting that the χ:YoaA interaction might be direct. We sought to confirm this interaction by two means: by protein pulldown assays and by yeast two-hybrid analysis. We expressed a His6-tagged χ (HolC) protein (pCA24N-holC; [25]) in E. coli AG1 and immobilized the protein on a Ni-NTA resin. Extracts of E. coli BL21/DE3 expressing a biotin-binding domain (BBD)-YoaA fusion protein were then applied to the His6-HolC bound resin. Protein from input, wash and bound fractions were resolved by SDS-PAGE and subjected to Western blot analysis with Neutravidin-conjugated horseradish peroxidase to detect BBD-YoaA (Fig 4A). BBD-YoaA was indeed detected in the bound fraction (Fig 4A, lane 9), indicating a physical interaction, albeit possibly indirect, between χ and YoaA. A reciprocal pulldown experiment, using streptavidin-agarose resin to bind BBD-YoaA (bait) and a penta-His antibody to probe Western blots for His6-HolC (prey), likewise detected interacting His6-HolC. (S3 Fig). To detect a potential direct interaction, both holC and yoaA were subjected to yeast two-hybrid analysis (Fig 4B). An interaction between YoaA and HolC relative to controls was confirmed by the His phenotype of the strain. Because no other E. coli protein was expressed in the yeast assay strain, this latter result supports a direct physical interaction between χ and the YoaA protein.

Mutational analysis of YoaA and χ suppression of AZT-sensitivity
Based on its amino acid sequence and similarity to DinG, YoaA is a predicted to possess 5’ to 3’ DNA helicase activity and a Fe-S cluster (See S2 Fig). We mutated the pNTR-plasmid borne yoaA gene within several motifs including the Walker A box (K51A), Walker B/DEAH box (D225A) and putative Fe-S coordination site (C168A), all of which are predicted to affect helicase function. These plasmid alleles, and the wild-type yoaA control plasmid, were introduced into wild-type and yoaA mutant strains. We assayed these strains for AZT tolerance, with expression of the plasmid allele induced by IPTG (Fig 5). All three mutant alleles fail to complement the AZT sensitivity conferred by yoaA. In wild-type strains, although plating efficiency is not affected, the size of colonies formed on AZT medium was dramatically reduced by the three mutant alleles of yoaA, in comparison to yoaA+. These data support the notion that YoaA possesses ATPase activity essential to its genetic function (consistent with a helicase activity) and is likely also a Fe-S cluster containing protein, as is DinG.

The χ protein participates in two additional protein interactions: one with its partner ψ (HolD) and with SSB. We sought to determine whether these interactions were necessary for suppression of AZT sensitivity by holC expression. A previous study [26] identified χ residues essential for SSB interaction, including V117, R128 and Y131 (Fig 6A and 6B). We introduced the V117F, R128A and Y131L mutations demonstrated to abolish SSB interaction [26] into the pNTR-holC construct and assayed their ability, relative to holC+, to suppress AZT-sensitivity of wild-type or holCΔ strains. The SSB-binding-defective mutants had reduced ability to suppress the AZT-sensitivity of holC, as illustrated by both plating efficiency and colony size on AZT-containing media. These SSB-interaction mutants, when expressed in holC mutant strains did show increase in plating efficiency relative to the control plasmid (Fig 6A); however colonies formed on AZT-medium were small and slow-growing (Fig 6B). These mutants, particularly the V117F allele, also showed dominant reduction of AZT tolerance in wild-type strains, apparent by both plating efficiency (Fig 6A) and colony size (Fig 6B). Assuming that these substitutions do not affect protein folding or expression levels (which is consistent with their genetic dominance), this result confirms that the ability of χ to interact with SSB is critical to the mechanism that promotes AZT tolerance. Residues in χ that are required for its interaction with ψ have not been reported. However, the crystal structure of the χψ dimeric complex [11] suggests that χ-F64 is a good candidate since it appears buried in a hydrophobic pocket at the χ:ψ interface. We mutated pNTR-holC to carry a F64A mutation and showed that this mutation abolishes AZT tolerance promoted by pNTR-holC expression in both wildtype and holCΔ mutant strain backgrounds (Fig 6). To confirm that this mutation indeed affects the interaction between χ and ψ, we subjected holC and holC-F64A to yeast two-hybrid analysis with holD (Fig 4B). HolC and HolD exhibit an interaction comparable to “strong interaction” controls, with HolC-F64A significantly reducing the interaction. Therefore, suppression of AZT sensitivity by HolC expression appears to require an intact χψ complex.

Genetic interaction with exonucleases
In several bacterial groups including Gram-positives, the ortholog to YoaA/DinG helicase is fused to an N-terminal exonuclease domain from the DnaQ/ε 3’ exonuclease family [27]. Our prior genetic study of AZT tolerance in E. coli suggests that Exonuclease III, a dsDNA exonuclease that degrades a 3’ strand from nicks or gaps in DNA, is likely the enzyme that removes AZT monophosphate from the DNA chain [6]. Since YoaA and χ expression strongly suppressed AZT sensitivity of xthA mutant strains (Fig 1), we considered the possibility that χ recruitment of YoaA helicase to SSB-bound gaps might permit an alternative 3’ ssDNA exonuclease to remove AZT by unwinding the 3’ nascent strand from its template (Fig 7). In Gram-positive and other groups of bacteria (see Discussion below), this exonuclease is conveniently fused to the helicase polypeptide. This hypothesis predicts that loss of a 3’ exonuclease would weaken the ability of YoaA or HolC expression to promote AZT tolerance. E. coli possesses 8 members of the DnaQ exonuclease family including the proofreading activities for DNA polymerases I (polA), II (polB) and III (dnaQ), exonuclease I (xonA) and exonuclease X (exoX).

DnaQ itself emerged as a strong candidate for the YoaA-associated 3’ exonuclease. Suppression of AZT sensitivity by YoaA or HolC was strongly reduced by the mutD5 allele of the dnaQ gene (Fig 7). (The mutD5 allele of dnaQ is a T15I mutation adjacent to the catalytic glutamate in the Exo I motif of the protein [28]. This allele reduces exonuclease activity without affecting its association with the polymerase subunit α [29]; mutation of the catalytic glutamate is lethal [28].) Moreover, YoaA expression became somewhat toxic in mutD5 strains, evident by lower plating efficiency and smaller colonies of mutD5 strains, both on LB and LB AZT media. We explain the latter result by hypothesizing that single-strand DNA displaced by YoaA is deleterious when it fails to be degraded by DnaQ (see Discussion below).
Discussion
This work provides new evidence that the accessory proteins to the clamp loader complex play a role in coordination of DNA repair. Through a chain of protein interactions, the accessory clamp loader proteins χ and ψ may facilitate the binding of multiple repair and replication factors. Persistent unfilled gaps are likely to be bound by SSB, which recruits χψ. χ binds the YoaA helicase, which we hypothesize unwinds potentially damaged 3’ nascent ends such as those terminated by azidothymidine monophosphate. Our work suggests that YoaA permits ε, encoded by DnaQ, to degrade the 3’ nascent strand end. Whether this degradation occurs in the context of the fully assembled replisome, an αεθ Polymerase III core complex, or by a stand-alone ε subunit is not known. Through its ψ interaction with τ or γ, χψ recruits the clamp loader to the gap for ready assembly of β clamps at the site of the gap. These clamps may then bind repair factors such as MutLS, ligase, TLS polymerases or recruit DNA polymerase III to fill the gap. These interactions may aid rapid hand-off of repair intermediates, promoting maturation of a persistent gap into a repair substrate and then into a replicative complex. Association of YoaA with χ may also allow it to track with the fork, even in the absence of persistent SSB-bound gaps.
We define here a new potential DNA repair function, encoded by the previously uncharacterized gene yoaA, that aids in tolerance of the chain-terminating nucleoside, azidothymidine. We confirm by protein pull-down and yeast two-hybrid analysis that YoaA binds χ of the accessory clamp loader complex. Genetic analysis demonstrates that YoaA’s putative ATPase activity is important for its in vivo activity. YoaA is likely to function in DNA repair as a 5’ to 3’, Fe-S helicase, similar to its paralog, DinG. E. coli YoaA and DinG share 29% identity (see alignment in S2 Fig) over almost their entire length, including the four cysteines in the DinG Fe-S coordination site, C120, C194, C199, C205 [24], and helicase motifs. The dinG gene was discovered as a DNA damage-inducible gene, regulated by the LexA repressor as part of the SOS response [29–31]. DinG is a member of a large group of helicases, including the human excision repair factor XPD, and helicases FANCJ/BACH-1, RTEL1 and ChlR1/2 [21, 31]. Mutations affecting these human helicases are associated with a variety of human DNA repair-deficient genetic diseases (reviewed in [27]). The translocation direction of E. coli DinG on ssDNA is 5’ to 3’ and its helicase activity is somewhat structure-specific, with highest activity in the unwinding of bifurcated, fork-like structures. Both RNA:DNA and DNA:DNA hybrid molecules can be unwound by DinG [23].
DNA helicases may help avoid the formation of replication gaps or facilitate their subsequent repair by several means. Helicases may unwind DNA structures, such as hairpins or G - quadruplex structures, which impede polymerization or fork progression. They may displace bound proteins, including RNA polymerase, unwind stable RNA:DNA hybrids (“R-loops”) or disassemble protein complexes formed during repair. Helicases may promote template-switching reactions that function in repair. Helicases also process branched DNA structures that are considered to be intermediates in homologous recombination such as D-loops and Holliday junctions and therefore function in recombinational DNA gap repair. Mycobacterial DinG has been reported to unwind G-quadruplex structures, in vitro [32]. DinG is one of several helicases, including Rep and UvrD, that are required in E. coli to overcome problems associated with DNA replication clashes with strong transcription [33].
YoaA likely differs from DinG in that YoaA is associated with the replisome, through its χ interaction, although it remains possible that the two helicases possess specialized binding affinities or activities. We observed expression-dependent suppression of AZT sensitivity by YoaA probably because of the increased likelihood of association with the χψ complex and timely recruitment to gaps when intracellular levels of YoaA are increased. An increase in intracellular χ might also make YoaA recruitment more efficient. In the study that identified the YoaA:χ interaction, DinG was not detected as a replisome-associated polypeptide [9]. DinG’s weak AZT-sensitive mutant phenotype and genetic synergy with YoaA suggest that it can play a similar role to YoaA in repair, albeit more inefficiently. This inefficiency may result from an intrinsic difference in the binding or helicase activities of the DinG and YoaA or from the fact that YoaA is more efficiently recruited to gaps through its χ interaction. The sites within YoaA that interact with χ will be of interest, but are currently unknown. Our pull-down assays lead us to suspect that the C-terminus will be important since what we infer is a C-terminally truncated BBD-YoaA form, detected in our bacterial cell extracts by Western blot analysis, did not appear competent to interact with χ. Our genetic analysis supports the notion that YoaA interacts with the χψ complex rather than with χ alone in the absence of ψ. The consequences of χ:YoaA binding on χψ ‘s other interactions, such as to the clamp loader or to SSB, will be important topics for future investigation. We may find, for example, that YoaA interactions with χψ are in competition with the DnaX clamp-loader interaction, or alternatively, they may be neutral or even enhance each other’s recruitment.
A number of observations reported here support the idea that YoaA, when liberated from association with other factors, can be toxic. Mutants lacking χ are very sensitive to AZT exposure, a phenotype partially suppressed by loss of YoaA. In the absence of the χ targeting factor, YoaA may be deleterious to repair and cell survival. Additionally, induction of YoaA expression is toxic to cells with defective DNA Polymerase III proofreading (mutD5), particularly after AZT exposure. This toxicity may be related to genetic instability, the DNA damage response and/or toxic reactions promoted by accumulation of single-strand DNA. This situation may be analogous to the cold-sensitivity of ssDNA exonuclease mutants (lacking RecJ, ExoI, VII and X), in which toxicity is dependent on UvrD helicase function [34]. The toxicity of ssDNA may result from constitutive induction of the SOS response [35] and/or by stimulation of low-homology recombination reactions [36] or template-switching [2] that lead to genetic mutations or chromosome rearrangements. We propose that the physical interactions that target YoaA to the replication fork and promote the handoff of repair intermediates to replication functions act to control the potential toxicity of strand unwinding.
Many diverse bacterial genomes encode a putative helicase related to DinG and YoaA of E. coli, leading to the hypothesis that it evolved early in the history of life and plays an important role in bacterial cell fitness. Although E. coli and other γ-Proteobacteria encode two paralogs, many bacteria possess only a single member of this group. The β-Proteobacteria encode a single YoaA ortholog, with over 95% identity to E. coli YoaA and only about 40% to E. coli DinG. The α and δ-Proteobacteria encode a single protein more distantly related to both proteins (about 40% identical to YoaA and 30% to DinG). In Gram-positive, Firmicutes bacteria (such as Bacillus subtilis) and in the Thermus-Deinococcus, green nonsulfur and Fusobacteria groups, the YoaA/DinG related helicase is fused to an N-terminal domain with homology to the DnaQ/ε (DEDDh) family of 3’ exonucleases. Although these proteins have been termed in various databases as DinG orthologs, protein sequence alignments support the notion that they are more strongly related to E. coli’s YoaA protein than its DinG protein. S2 Fig shows the pairwise BLAST alignment between the B. subtilis protein and E. coli YoaA, compared to the alignment of E. coli (Eco) YoaA and DinG, showing multiple regions that align to YoaA but not DinG. This homology is apparent in the gapped regions shared by Bsu ε and Eco YoaA, relative to Eco DinG, particularly in the region between helicase motifs II and III. Overall identity of Bsu ε with Eco YoaA or DinG is 29% and 25%, respectively, with 10% or 14% gaps. The Bacillus protein does not retain the conserved cysteine residues shared by Eco YoaA and DinG that are required to form the Fe-S cluster in this helicase family and so is unlikely to be an Fe-S protein.
Our previous work implicated the 3’ dsDNA exonuclease, Exonuclease III, as an enzyme that removes AZT monophosphate (AZT-MP) from DNA in E. coli [6]: exonuclease III mutants are highly sensitive to normally sublethal concentrations of AZT and overproduction of the enzyme improves tolerance in wild-type strains. In this study, we found that mutants in the proofreading activity of DNA polymerases I or III show little or no sensitivity to AZT. This may be because AZT-MP is poorly proofread or, alternatively, because loss of proofreading is efficiently backed up by exonuclease III-mediated removal. We have no information about how efficiently AZT-MP can be removed in vitro from DNA by bacterial DNA polymerases. However, in both yeast and humans, AZT-MP is poorly proofread by DNA polymerase gamma, a DNA polymerase that readily incorporates AZT [37,38]. The work reported here suggests that YoaA DNA helicase activity aids removal of AZT-MP from 3’ termini by DnaQ, the proofreading subunit of DNA Pol III. When Pol III is bound to the paired template and nascent strand, AZT-MP may be poorly accessible to the exonuclease active site of the DnaQ because of AZT’s bulky 3’ azido group and/or because it pairs well with its template adenine residue. YoaA helicase may promote the dissociation of Pol III and/or unwind the nascent strain from its template (as shown in Fig 7), allowing it or a second enzyme to gain access to AZT at the 3’ nascent strand terminus.
Materials and Methods
Bacterial strains and growth conditions
E. coli K-12 strains isogenic to either the wild type MG1655 (F - rph-1) or wild type AB1157 background (S1 Text) were grown as previously described at 37° in Luria-Bertani (LB) medium, with 1.5% agar for plates [39]. With the exception of lexA3, parE-ts and mutD5, all mutant strains used in this study carried deletions of the indicated gene. LB medium was supplemented as necessary with antibiotics including ampicillin (Ap), chloramphenicol (Cm), kanamycin (Km), tetracycline (Tc), streptomycin (Sm) or gentamycin (Gm). Details of growth media and strain constructions are provided in the S1 Text.
Mobile plasmid genetic screen
Details of the screen can be found in the S1 Text. Briefly, screens were performed with MG1655 isogenic strains carrying mutations that confer increased AZT sensitivity, including xthA, recB, rpoS, lexA3, relA, parE-ts (grown at 30°). The plasmid library of pNTR-based mobile plasmids carrying E. coli ORFs [8] was obtained from the National Bioresource Project at the National Institute of Genetics in Japan, which were introduced into tester strains in pools and screened for suppression of AZT sensitivity. These plasmids are based on a ColE1 replicon, with a copy number of approximately 30 per cell.
Survival assays
Isolated mobile plasmids (S1 Table) were transformed into strains by electroporation [40]. For survival assays, strains were grown to OD595 0.3–0.7, serial diluted in 56/2 buffer, and then plated on LB agar plates supplemented with azidothymidine (AZT) as indicated. Plates were incubated at 37° for 24–36 hours. Total colony forming units (CFU) counts were obtained, normalized to the NO AZT counts, and then log10-transformed to obtain Fractional Survival. Averages and standard deviations were calculated from the log10-transformed Fractional Survival data, and plotted as a function of AZT dosage. Strains containing mobile plasmids pNTR-Control, pNTR-HolC, or pNTR-YoaA were plated on growth medium supplemented with Ap, and expression was induced by the addition of 1 mM IPTG. Because of day-to-day variation of the potency of AZT, assays illustrated in the figure panels were conducted in parallel.
Mutant alleles
Site-directed mutagenesis (Quikchange, Agilent Technologies) was used to construct mutant alleles of holC and yoaA on the pNTR mobile plasmids, as well as holC plasmid derived from pDONR221 by GATEWAY cloning (Life Technologies). Primers used to construct the site-directed mutant alleles are listed in S1 Table.
Yeast two-hybrid analysis
We used a commercially available yeast two-hybrid system (ProQuest, Life Technologies) to detect interactions between YoaA and HolC and between HolC and HolD when C-terminally fused to Gal4 DNA binding and activation domains. Details of the assays are provided in the Supplement. Readouts for transcriptional activation include URA3, HIS3 and lacZ expression in yeast strain MaV203. Controls include MaV203 “no interaction” strains containing pPC97 (no insert) + pPC86 (no insert); “weak interaction” strains containing pPC97-RB + pPC86-E2F1; “moderate interaction” strains containing pPC97-CH2S-dDP + pPC86-dE2F, and “strong interaction” strains containing pCL1 (encoding full-length GAL4) + pPC86. Primers used to construct GATEWAY cloned alleles of yoaA, holC, and holD are listed in S1 Table.
Protein extracts, pull-down assays and Western blot analysis to display YoaA and HolC protein interaction binding
Plasmid pSTL385 is a pET104.1-DEST based plasmid (Life Technologies) comprised of a N-terminal fusion of the biotin binding-domain (BBD) to yoaA, expressed from the T7 promoter. BBD-YoaA was expressed from E. coli B strain BL21 (DE3) (genotype fhuA2 lon ompT gal dcm hsdS λ DE3), grown in LB + Ap medium, and induced by the addition 1 mM IPTG for 2 hours. Plasmid pSTL386 is a pCA24N-based plasmid comprised of a N-terminal His fusion to holC under the lac promoter [41], which was transformed and expressed in strain AG1 (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1). The strain was grown in LB + Cm medium, induced by 1 mM IPTG for 2 hours. Cells of both overexpressing strains were harvested by centrifugation, resuspended and stored in Tris-sucrose buffer (50 mM Tris-HCl pH 7.5 10% sucrose) at -70°. Crude cell extracts were prepared by lysozyme lysis as described previously [42]. Pulldown of His6-HolC was performed from crude cell extracts with Ni-NTA agarose beads (Qiagen), equilibrated in wash buffer (50 mM Na2HPO4/NaH2PO4 pH 8.0, 500 mM NaCl, 20 mM imidazole). Resin was mixed with an equal volume of the crude cell extracts from the His6-HolC expressing strain, allowed to incubate at 20° for 1 hour and was then washed five times with wash buffer. YoaA-BBD crude lysate was similarly mixed with the His6-HolC bound resin, incubated and washed. Samples were eluted by mixing the resin 1 : 1 in 2x Laemmli sample buffer (120 mM Tris-HCI, pH 6.8, 4% SDS, 40% (w/v) glycerol, 0.02% bromophenol blue). Protein samples were subject to PAGE in 15% polyacrylamide gels and transferred to PVDF membrane transfer using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) and the methods provided by the manufacturer. The Western blot analysis was performed using the QIAexpress detection kit and protocol (Qiagen) and using detection of BBD-YoaA with a 1 : 10,000 dilution of NeutrAvidin Protein Horseradish Peroxidase Conjugated antibody (Pierce).
Supporting Information
Zdroje
1. Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Ann Rev Biochem. 2002; 71 : 17–50. 12045089
2. Goldfless SJ, Morag AS, Belisle KA, Sutera VA, Lovett ST. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol Cell. 2006;21 : 595–604. 16507358
3. Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair .2007;6 : 967–980. 17395553
4. Persky NS, Lovett ST. Mechanisms of recombination: lessons from E. coli. Crit Rev Biochem Mol Biol. 2008;43 : 347–370. doi: 10.1080/10409230802485358 19016098
5. Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL. SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol. 2008;43 : 289–318. doi: 10.1080/10409230802341296 18937104
6. Cooper DL, Lovett ST. Toxicity and tolerance mechanisms for azidothymidine, a replication gap-promoting agent, in Escherichia coli. DNA Repair. 2011;10 : 260–270. doi: 10.1016/j.dnarep.2010.11.007 21145792
7. Richardson CC, Lehman IR, Kornberg A. A deoxyribonucleic acid phosphatase-exonuclease from Escherichia coli. II. Characterization of the exonuclease activity. J Biol Chem. 1964;239 : 251–258. 14114851
8. Saka K, Tadenuma M, Nakade S, Tanaka N, Sugawara H, Nishikawa K, et al. A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res. 2005;12 : 63–68. 16106753
9. Butland G, Peregrin-Alvarez J, Li J, Yang W, Yang X, Canadien V, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433 : 531–537. 15690043
10. Kurth I, O'Donnell M. Replisome dynamics during chromosome duplication. EcoSal Plus. 2009. Available: http://www.asmscience.org/content/journal/ecosalplus/10.1128/ecosalplus.4.4.2 doi: 10.1128/ecosalplus.4.4.2
11. Gulbis JM, Kazmirski SL, Finkelstein J, Kelman Z, O'Donnell M, Kuriyan J. Crystal structure of the chi:psi sub-assembly of the Escherichia coli DNA polymerase clamp-loader complex. Eur J Biochem. 2004;271 : 439–449. 14717711
12. Xiao H, Dong Z, O'Donnell M. DNA polymerase III accessory proteins. IV. Characterization of chi and psi. J Biol Chem. 1993;268 : 11779–11784. 8505305
13. Kelman Z, Yuzhakov A, Andjelkovic J, O'Donnell M. Devoted to the lagging strand-the chi subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J. 1998;17 : 2436–2449. 9545254
14. Olson MW, Dallmann HG, McHenry CS. DnaX complex of Escherichia coli DNA polymerase III holoenzyme. The chi psi complex functions by increasing the affinity of tau and gamma for delta delta' to a physiologically relevant range. J Biol Chem. 1995;270 : 29570–29577. 7494000
15. Glover B, McHenry C. The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J Biol Chem. 1998;273 : 23476–23484. 9722585
16. Yuan Q, McHenry CS. Strand displacement by DNA polymerase III occurs through a tau-psi-chi link to single-stranded DNA-binding protein coating the lagging strand template. J Biol Chem. 2009;284 : 31672–31679. doi: 10.1074/jbc.M109.050740 19749191
17. Yuzhakov A, Kelman Z, O'Donnell M. Trading places on DNA—a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell. 1999;96 : 153–163. 9989506
18. Anderson S, Williams C, O'Donnell M, Bloom L. A function for the psi subunit in loading the Escherichia coli DNA polymerase sliding clamp. J Biol Chem. 2007;282 : 7035–7045. 17210572
19. Viguera E, Petranovic M, Zahradka D, Germain K, Ehrlich DS, Michel B. Lethality of bypass polymerases in Escherichia coli cells with a defective clamp loader complex of DNA polymerase III. Mol Microbiol. 2003;50 : 193–204. 14507374
20. Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328 : 498–501. doi: 10.1126/science.1185757 20413500
21. Koonin EV. Escherichia coli dinG gene encodes a putative DNA helicase related to a group of eukaryotic helicases including Rad3 protein. Nucleic Acids Res. 1993;21 : 1497. 8385320
22. Voloshin ON, Vanevski F, Khil PP, Camerini-Otero RD. Characterization of the DNA damage-inducible helicase DinG from Escherichia coli. J Biol Chem. 2003;278 : 28284–28293. 12748189
23. Voloshin ON, Camerini-Otero RD. The DinG protein from Escherichia coli is a structure-specific helicase. J Biol Chem. 2007;282 : 18437–18447. 17416902
24. Ren B, Duan X, Ding H. Redox control of the DNA damage-inducible protein DinG helicase activity via its iron-sulfur cluster. J Biol Chem. 2009;284 : 4829–4835. doi: 10.1074/jbc.M807943200 19074432
25. Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12 : 291–299. 16769691
26. Naue N, Fedorov R, Pich A, Manstein DJ, Curth U. Site-directed mutagenesis of the chi subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res. 2011;39 : 1398–1407. doi: 10.1093/nar/gkq988 20972214
27. White MF. Structure, function and evolution of the XPD family of iron-sulfur-containing 5'—>3' DNA helicases. Biochem Soc Trans. 2009;37 : 547–551. doi: 10.1042/BST0370547 19442249
28. Fijalkowska I, Schaaper RM. Mutants in the Exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc Natl Acad Sci USA. 1996;93 : 2856–2861. 8610131
29. Jonczyk P, Nowicka A, Fijałkowska IJ, Schaaper RM, Cieśla Z. In vivo protein interactions within the Escherichia coli DNA polymerase III core. J Bacteriol. 1998;180 : 1563–1566. 9515927
30. Lewis LK, Jenkins ME, Mount DW. Isolation of DNA damage-inducible promoters in Escherichia coli: regulation of polB (dinA), dinG, and dinH by LexA repressor. J Bacteriol. 1992;174 : 3377–3385. 1577702
31. Lewis LK, Mount DW. Interaction of LexA repressor with the asymmetric dinG operator and complete nucleotide sequence of the gene. J Bacteriol. 1992;174 : 5110–5116. 1629168
32. Thakur RS, Desingu A, Basavaraju S, Subramanya S, Rao DN, Nagaraju G. Mycobacterium tuberculosis DinG is a structure-specific helicase that unwinds G4 DNA: implications for targeting G4 DNA as a novel therapeutic approach. J Biol Chem. 2014;289 : 25112–25136. doi: 10.1074/jbc.M114.563569 25059658
33. Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 2010;29 : 145–157. doi: 10.1038/emboj.2009.308 19851282
34. Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci USA. 2001;98 : 6765–6770. 11381137
35. Hersh MN, Morales LD, Ross KJ, Rosenberg SM. Single-strand-specific exonucleases prevent frameshift mutagenesis by suppressing SOS induction and the action of DinB/DNA polymerase IV in growing cells. J Bacteriol. 2006;188 : 2336–2342. 16547019
36. Dutra BE, Sutera VA Jr., Lovett ST. RecA-independent recombination is efficient but limited by exonucleases. Proc Natl Acad Sci USA. 2007;104 : 216–221. 17182742
37. Eriksson S, Xu B, Clayton DA. Efficient incorporation of anti-HIV deoxynucleotides by recombinant yeast mitochondrial DNA polymerase. J Biol Chem. 1995;270 : 18929–18934. 7642550
38. Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J Biol Chem. 2001;276 : 40847–40857. 11526116
39. Miller J. A short course in bacterial genetics. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1992.
40. Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16 : 6127–6145. 3041370
41. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 : 2006 0008. 16738554
42. Lovett ST, Kolodner RD. Nucleotide sequence of the Escherichia coli recJ chromosomal region and construction of recJ-overexpression plasmids. J Bacteriol. 1991;173 : 353–364. 1987126
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 11
Nejčtenější v tomto čísle
- UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development
- Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis
- Genus-Wide Comparative Genomics of Delineates Its Phylogeny, Physiology, and Niche Adaptation on Human Skin
- Encodes Dual Oxidase, Which Acts with Heme Peroxidase Curly Su to Shape the Adult Wing