
The SMC Loader Scc2 Promotes ncRNA Biogenesis and Translational Fidelity
The structure of chromosomes contributes to the production of RNAs. Chromosome structure is maintained in part by an evolutionarily conserved group of proteins known as structural maintenance of chromosomes proteins. These proteins are loaded onto chromosomes by a second evolutionarily conserved protein complex known as Scc2-Scc4. The Scc2 component is often mutated in a human developmental disorder known as Cornelia de Lange syndrome. We find that Scc2 in budding yeast is important for the production of a group of RNAs known as snoRNAs. These RNAs play a critical role in ribosome production; without them the fidelity of ribosomes suffers. Ribosomes are the molecular machines that translate mRNAs to proteins. Scc2 is also important for the production of the RNA components of ribosomes. Our findings suggest Scc2 normally promotes the production of RNAs that support translational fidelity. We hypothesize that defects in protein synthesis may contribute to Cornelia de Lange syndrome.
Published in the journal:
. PLoS Genet 11(7): e32767. doi:10.1371/journal.pgen.1005308
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005308
Summary
The structure of chromosomes contributes to the production of RNAs. Chromosome structure is maintained in part by an evolutionarily conserved group of proteins known as structural maintenance of chromosomes proteins. These proteins are loaded onto chromosomes by a second evolutionarily conserved protein complex known as Scc2-Scc4. The Scc2 component is often mutated in a human developmental disorder known as Cornelia de Lange syndrome. We find that Scc2 in budding yeast is important for the production of a group of RNAs known as snoRNAs. These RNAs play a critical role in ribosome production; without them the fidelity of ribosomes suffers. Ribosomes are the molecular machines that translate mRNAs to proteins. Scc2 is also important for the production of the RNA components of ribosomes. Our findings suggest Scc2 normally promotes the production of RNAs that support translational fidelity. We hypothesize that defects in protein synthesis may contribute to Cornelia de Lange syndrome.
Introduction
Cohesion between sister chromatids generates the force that holds sister chromatids together until the onset of anaphase [1]. Cohesion is generated by cohesin, an evolutionarily conserved multi-subunit protein complex consisting of four core subunits: Smc1, Smc3, the α-kleisin subunit Mcd1/Scc1/Rad21, and the HEAT repeat-containing protein Scc3/SA1 or SA2. The complex forms a ring-like structure that entraps DNA [2, 3]. Smc1 and Smc3 belong to the structural maintenance of chromosome (SMC) ATPase superfamily [1, 4]. This family also includes subunits of the condensin and Smc5/6 complexes. Cohesin and condensin loading are facilitated by Scc2-Scc4 [5–7]. In addition to its role in chromosome segregation, cohesin promotes DNA damage repair [8] and regulates gene expression [9, 10].
It has been proposed that the human ortholog of SCC2, NIPBL, could regulate gene expression [11]. In budding yeast, Scc2 binds to Pol II transcribed genes encoding ribosomal proteins and small nuclear and nucleolar RNAs (snRNA and snoRNAs), Pol III transcribed genes encoding tRNAs and other noncoding RNAs, as well as pericentric domains [5]. These same regions are bound by condensin [5]. Defects in the association of Scc2 with these highly transcribed regions could potentially affect the expression of these genes. For instance, a recent report suggested Scc2 may help maintain a nucleosome-free region [12], which could potentially promote both SMC complex loading and gene expression. Since Scc2 target genes contribute to translation, a decrease in their expression may negatively affect translation.
Mutations in SCC2/NIPBL and genes encoding the cohesin subunits result in a developmental syndrome known as Cornelia de Lange syndrome (CdLS). The causative mutations are spread throughout the NIPBL gene, most often resulting in a partial loss of function [13]. How mutations in NIPBL cause developmental defects remains largely unknown. Examination of cells from affected CdLS probands suggests differential gene expression might cause the developmental defects observed in CdLS patients, rather than chromosome missegregation [14, 15]. Recent work using zebrafish to model CdLS is consistent with the idea that partial loss of function of nipbl reduces translation [16], but the molecular mechanisms are unclear.
In this study, we examined how the cohesin loader Scc2 regulates gene expression in budding yeast. We utilized a temperature-sensitive partial loss of function mutation in SCC2 (scc2-4) that has previously been shown to (1) reduce cohesin and condensin association with chromosomes at 37°C [5, 7], (2) delay DSB repair [8], and (3) disrupt nucleolar morphology at 30°C [5]. We found that the mutant protein is expressed at normal levels and displays a normal binding profile at centromeres, but has reduced association with genic regions including ribosomal DNA (rDNA), tDNAs, snoDNAs, and ribosomal protein genes. To examine the biological processes and RNAs regulated by Scc2, we performed RNA sequencing at permissive temperature. Consistent with a previous report [17], we observed differential expression of hundreds of genes. Gene expression signatures suggested both ribosome biogenesis and mitochondrial function would be impacted in the mutant and functional analysis confirms they are both negatively affected. One group of down-regulated genes were the H/ACA snoRNAs, which guide the site-specific pseudouridylaton of rRNAs, tRNAs, mRNAs, and noncoding RNAs [18–20]. The production of H/ACA snoRNAs appears to be facilitated by Scc2-dependent recruitment of the RNA Pol II-associated factor (Paf1) complex. The scc2-4 mutant showed defective rRNA production and modification with a mild reduction in global protein synthesis. More strikingly, translational fidelity was reduced as shown by decreased internal ribosomal entry site (IRES) usage, increased frameshifting, and decreased resistance to translational inhibitors. Our results in budding yeast strongly suggest that the Scc2 regulated gene expression program promotes translational fidelity.
Results
The scc2-4 mutant protein has reduced binding to genic regions
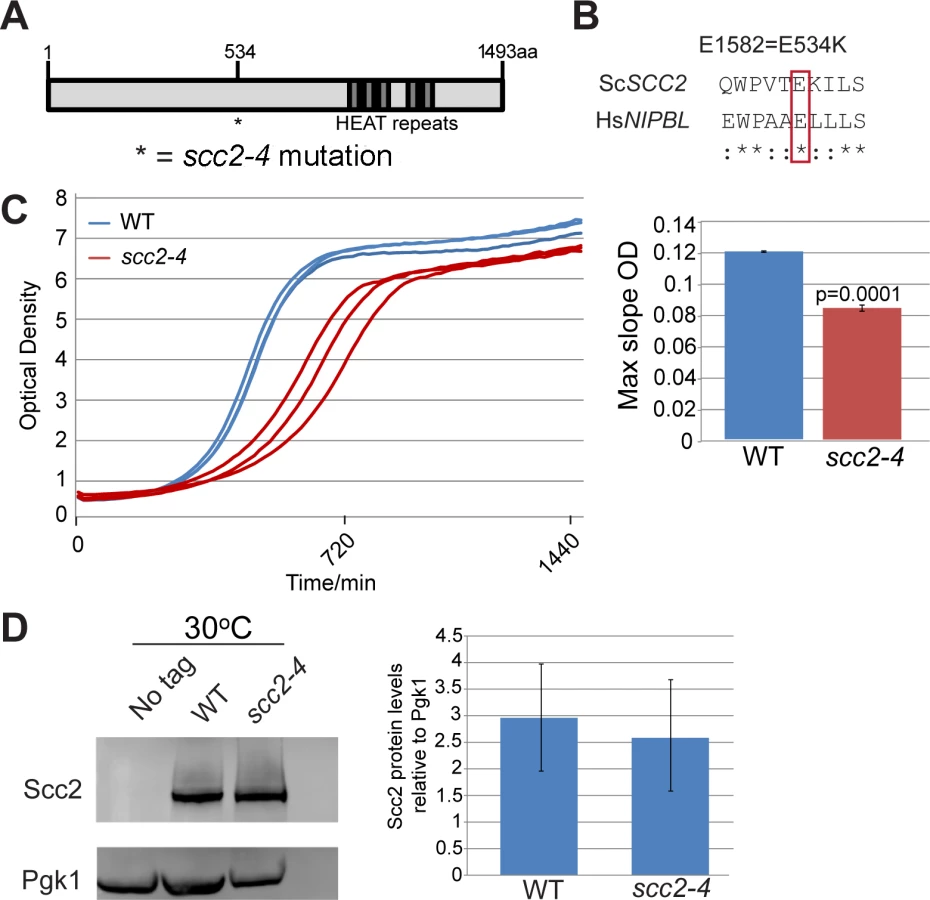
SCC2 is a large gene encoding a protein with several domains (Fig 1A). Human and budding yeast NIPBL/SCC2 do not align very well. However, the function of Scc2 in loading SMC complexes is evolutionarily conserved from yeast to humans. Evolutionarily conserved HEAT repeats are located at the C-terminus. By chemical mutagenesis, a temperature sensitive mutant was isolated (E534K), named scc2-4 [1]. The mutated amino acid is evolutionarily conserved from yeast to human (Fig 1B). The E534K mutation falls in a central region of the protein with unknown function. However, the surrounding amino acids are highly conserved. At 30°C, the scc2-4 mutant grows more slowly than WT (Fig 1C). Western blot analysis shows that the level of the mutant protein is similar to WT (Fig 1D).
In order to understand how the mutant Scc2 protein associates with the genome, we performed ChIP-seq. Strains were cultured in YPD+CSM (complete supplement mixture) at 30°C until mid-log phase, fixed in formaldehyde for 2 hrs, and chromatin was harvested for ChIP-seq. The basic pattern of binding for the WT Scc2 protein was similar to previous reports [5, 21]. Enrichment for different regions of the genome was further characterized using metagene plots (Fig 2). On the y-axis is the mean count per million (cpm) and on the x-axis is the position in base pairs. No apparent difference in centromere enrichment was observed for Scc2E534K when compared to the WT (Fig 2A). In contrast, we observed reduced enrichment for rDNA, snoDNAs (Box H/ACA and Box C/D), tDNAs, and the small and large ribosomal protein genes in the Scc2E534K ChIP (Fig 2). Thus, the mutation appears to compromise genic association without compromising centromere association.

Hundreds of genes are differentially expressed in the scc2-4 mutant
To investigate the biological processes regulated by Scc2, RNA sequencing was performed for WT and the scc2-4 mutant. Each strain was grown in triplicate, at 30°C in YPD+CSM to mid - log phase. The distribution of gene expression can be viewed in the MA plot, defined as a plot of log-intensity ratios (M-values) versus log-intensity averages (A-values) (Fig 3A). The log2 ratio of scc2-4/WT is shown on the y-axis and the geometric mean of the reads per kilobase of transcript per million reads mapped (RPKM) values on the x-axis. Using an adjusted p-value of 0.05 as criteria, there are 2644 differentially expressed genes, with 1285 up-regulated and 1359 down-regulated in scc2-4 mutant. Further applying a fold change threshold of 1.5 (corresponding to an absolute log2 value of 0.6) returns 823 up-regulated and 760 down-regulated genes for GO analyses and general comparisons. This is many more genes than are bound by Scc2, suggesting that many of the changes in gene expression are due to indirect effects. GO term analyses for up-regulated genes shows enrichment for genes involved in ribosome biogenesis and rRNA processing (Fig 3B and S1A and S1B Fig). The upregulation of ribosomal protein genes (RPs) and the processome in general suggests these mRNAs are not rate limiting in the scc2-4 mutant for ribosome biogenesis, although ribosome assembly does appear to be affected (see below). This upregulation is in direct contrast to our previous analysis of gene expression in the cohesin acetyltransferase mutant (eco1-W216G), which shows downregulation of these gene groups [22]. GO term analysis for the down-regulated genes shows enrichment for genes required for oxidative phosphorylation (Fig 3C and S1C and S1D Fig).

We further explored specific aspects of the gene expression profile in the scc2-4 mutant. The downregulation of genes involved in oxidative phosphorylation suggested the mutant might be hypersensitive to a drug that inhibits mitochondrial function, such as chloramphenicol which blocks protein synthesis by the mitochondrial ribosome. Consistent with their respective gene expression profiles, the scc2-4 mutant, but not the eco1-W216G mutant, showed very poor growth on plates with a sublethal dose of chloramphenicol (S2 Fig).
Another group recently published the gene expression profile of the scc2-4 mutant [17]. However, these experiments were conducted at non-permissive temperature whereas our experiments were all conducted under permissive conditions. The two data sets have some differences, as expected, but the gene expression profile from Lindgren et al suggests mitochondrial function and translation would be negatively impacted in the scc2-4 mutant, although this was not tested. However, they did show that cohesin binding locations were not significantly affected in the mutant background, helping to rule out this effect as an explanation for the differential gene expression.
Scc2 binds to genes that encode RNA components of ribonucleoprotein complexes such as NME1, a component of RNAse MRP that modifies ribosomal RNA, SCR1, part of the signal recognition complex, and SNR6, the U6 component of the spliceosome. The mutant protein binds less well to these regions, correlating with significantly lower expression in the mutant (p<0.05). Given the reduction in U6, we decided to analyze splicing in the mutant compared to WT. By measuring splicing efficiency (computing the mean nucleotide coverage over every spliced junction divided by the mean nucleotide coverage for reads falling within the exonic splicing unit), we were able to detect a modest but reproducible defect in splicing (S3 Fig). As a control we used the eco1-W216G mutant, where we did not find a splicing defect (S3 Fig).
Analysis of the correlation between genome-wide enrichment of Scc2-bound sequences and gene expression revealed contrasting findings. While loss of binding of Scc2 correlated with lower gene expression at certain regions of the genome (e.g. Box H/ACA, some Box C/D snoDNAs), there was increased expression at RP genes, and no correlation at other regions. Overall the gene expression pattern is likely the product of a combination of direct effects from changes in Scc2 binding and indirect effects. It is also possible that Scc2 may play different roles in gene expression at different groups of genes.
The scc2-4 mutant has defects in rRNA production and ribosome distribution
The reduced enrichment for the ribosomal DNA repeats in the scc2-4 mutant (Fig 2C), combined with our previous work demonstrating that cohesion is required to form a nucleolus [23] and produce normal levels of ribosomal RNA [22], drove us to examine rRNA production and nucleolar morphology in the scc2-4 mutant. rRNA synthesis was measured by pulse-labeling cells for 5 min with 3H-uridine. This approach is a reliable method for estimating RNA Pol I activity since most RNA synthesis during exponential growth is Pol I derived. Incorporation of uridine into total RNA was reduced by approximately 3-fold in the scc2-4 mutant (Fig 4A), consistent with the observed decrease in growth rate in the scc2-4 mutant. rRNA production was further investigated by pulse-labeling cells with 3H-methylmethionine for 5 min, chased with excess cold methionine for 5 min and extracting RNA. Incorporated 3H was quantified in 25S and 18 rRNAs (Fig 4B). rRNA is methylated cotranscriptionally in yeast, making this approach a reliable method for quantifying the production of the methylated forms of the 25S and 18S rRNAs [24]. By this method, we also observed a 3.6-fold reduction in the production of 25S rRNA (Fig 4B, right) and a 4-fold reduction in 18S rRNA production (Fig 4B, bottom right). We further examined the processing of the initial rRNA transcript into the 25S and 18S forms over time. We found no defect in the processing rate for 25S and 18S rRNAs in scc2-4 mutant (S4 Fig). In summary, the scc2-4 mutant has reduced rRNA production.

Nucleolar morphology has been reported to be aberrant in the scc2-4 mutant based on visualization of the nucleolar protein Net1 fused to GFP [5]. We extended this observation using electron microscopy (S5 Fig). The dense staining nucleolar material did not adopt the normal crescent shaped structure in the mutant, consistent with the idea that reduced binding of Scc2 and reduced loading cohesin and/or condensin at the rDNA could result in a failure to gather the rDNA repeats into a normal nucleolar structure.
Ribosomes are partially assembled in the nucleolus and transported into the nucleus and then cytoplasm to be fully assembled and engaged in translation. Since mutation in SCC2 results in differential expression of messages involved in ribosome biogenesis, including ribosomal protein genes and genes involved in processing and assembling ribosomes, we further investigated ribosome production in the scc2-4 mutant. GFP tagged protein components of the large (Rpl25) and small (Rps2) ribosomal subunits were expressed in the WT and scc2-4 mutant strains and examined by microscopy. Rather than the normal even distribution in the cytoplasm, we observed accumulation of ribosomal proteins in the scc2-4 mutant in the nucleus/nucleolus that suggested that ribosome assembly/export was affected (Fig 5A and 5D). Using flow cytometry, we found higher mean fluorescence for both 40S and 60S reporters in the scc2-4 mutant (Fig 5B and 5E). We further analyzed the data by generating a cumulative distribution function for each sample, calculating the distance between samples, and testing for statistical significance using the Kolmogorov—Smirnov (KS) test, see Materials and Method and [22] (Fig 5D and 5F). The KS test was statistically significant for both the 40S and 60S subunit reporters in the scc2-4 mutant compared to WT. This analysis suggests ribosome assembly may be deficient in the mutant. The defects in nucleolar morphology, rRNA production, and ribosome distribution in the scc2-4 mutant are quite similar to those reported for the cohesin acetyltransferase mutant, suggesting cohesion is important for these aspects of nucleolar structure and function.

Downregulation of H/ACA snoRNAs inhibits site-specific pseudouridylation in the scc2-4 mutant
One notable group of Scc2 bound genes that was down-regulated in the scc2-4 mutant was the box H/ACA snoRNA genes (Fig 6A). Some box C/D RNAs were also downregulated, but not all. Box H/ACA snoRNA genes stand alone whereas box C/D snoRNA genes lie in the introns of ribosomal protein genes which may make them subject to the regulation of their host genes. The differential expression of snoRNAs was not observed in the eco1-W216G mutant [22]. These small nucleolar rRNAs guide site-specific modification of rRNAs and other RNAs. Box C/D RNAs guide the methylation of RNAs in the context of a ribonucleoprotein particle. The box H/ACA snoRNAs are also part of ribonucleoprotein particles (snoRNPs) along with the essential proteins (Nhp2, Cbf5, Nop10 and Gar1) that together catalyze site-specific pseudouridylation of RNAs. Pseudouridylation is important for RNA stability and interactions with other RNAs and proteins [25]. Mutation of the CBF5 component of this snoRNP leads to reduced pseudouridylation of rRNA and reduced translational fidelity [26, 27]. These defects are thought to contribute to cases of dyskeratosis congenita caused by mutations in DKC1, the human ortholog of CBF5.

Downregulation of box H/ACA snoRNAs could reduce pseudouridylation of RNAs. To test this idea, rRNA was isolated from WT and the scc2-4 mutant and the distribution of pseudouridines was examined by the CMC-primer extension method. Mapping of pseudo-sites on rRNA revealed downregulation of pseudouridylation at positions Ψ1003 (guided by SNR5) and Ψ2258/60 (guided by SNR191) (Fig 6B). Reduced pseudouridylation may derive from the reduced expression of Box H/ACA snoRNAs observed in the scc2-4 mutant.
Paf1 and Scc2 show co-dependent recruitment at snoDNAs
snoRNAs are transcribed by RNA polymerase II. The Tbf1 transcription factor has been identified at snoRNA genes [28]. We examined whether the enrichment for snoRNA genes was compromised in ChIPs performed for either of these two baits in the scc2-4 mutant. The qPCR signal for snoRNA genes for both Tbf1 and RNA Pol II (all forms) is similar in WT and scc2-4 mutant samples (S6 Fig), suggesting defective localization of these proteins cannot account for the lower levels of snoRNAs observed in the mutant. The Paf1 complex, which is comprised of Paf1, Ctr9, Leo1, Rtf1, and Cdc73, is important for transcription elongation. [29–31]. The Paf1 complex has been shown to contribute to the production of snoRNAs [29, 32]. To assess whether Paf1 recruitment was affected, we tagged Paf1 at its endogenous locus and examined the enrichment for snoRNA genes by ChIP. In contrast to RNA Pol II and Tbf1, enrichment for snoDNAs (Box C/D and H/ACA snoRNAs) in the Paf1 ChIP was significantly reduced in the scc2-4 mutant (Fig 7A). In addition to the snoDNAs, enrichment for rDNA and RP genes was also reduced in Paf1 ChIP in the scc2-4 mutant (S7 Fig). Given the global decrease in Paf1 recruitment, we next asked about mRNA and protein levels of Paf1 in the scc2-4 mutant. While mRNA levels for the subunits of the Paf1 complex were not significantly affected in our RNA seq data, we observed less Paf1 protein in the scc2-4 mutant by Western blot (Fig 7B). The protein level of Ctr9, another member of the Paf1 complex was also reduced (Fig 7C), implying that the scc2-4 mutation compromises the steady state level of Paf1 complex subunits. Interestingly, deletion of PAF1 caused a reduction in protein levels of Scc2 (Fig 7B). Therefore, the simplest explanation for the decreases in ChIP/qPCR signal is that Paf1 and Scc2 depend on each other for stable expression.

The Paf1 complex has previously been shown to promote transcription elongation by RNA Pol I [24]. We explored whether Scc2 recruitment to snoDNAs, rDNA, and RP genes was affected in the absence of Paf1. Deletion of PAF1 resulted in reduced enrichment for snoDNAs in Scc2 ChIP (Fig 7D), implying that Scc2 and Paf1 are dependent on each other for full recruitment at these genes. While enrichment for the rDNA and RP genes was compromised in the Paf1 ChIP in the scc2-4 mutant (S7A and S7B Fig), deletion of PAF1 did not affect enrichment for these loci in the Scc2 ChIP (S7C and S7D Fig), suggesting that the low recruitment of Scc2E534K cannot be attributed to less Paf1. In addition, PAF1 deletion did not compromise Scc2 binding to other Paf1 target genes, implying that the requirement of Paf1 for Scc2 recruitment was unique to snoDNAs. These results suggest that Paf1 and Scc2 cooperate uniquely in their co-recruitment to snoDNAs (Fig 7E).
The scc2-4 mutant has poor translational fidelity
Since mutation of SCC2 affects rRNA biogenesis, we examined actively translating ribosomes by polysome analysis. Polysome analysis of WT and scc2-4 mutant strains showed nearly identical profiles (S8A Fig). To more carefully investigate the effects of the scc2-4 mutation on global protein synthesis, 35S-incorporation was examined. A mild reduction in protein synthesis in the scc2-4 mutant was observed (S8B Fig). Consistently, the mutant failed to grow on plates with a sublethal concentration of cycloheximide, an inhibitor of protein translation (S8C Fig).
We hypothesized that a downregulation of box H/ACA snoRNAs, as observed in the scc2-4 mutant, might phenocopy a CBF5 mutant. Mutations in CBF5 show limited effects on translational efficiency, but significant effects on translational fidelity [26]. We decided to examine translational fidelity in the scc2-4 mutant. We first tested the ability of the mutant to translate IRES-dependent mRNAs. IRES elements are used by both viral and endogenous genes for cap-independent translation initiation [27]. The Cricket Paralysis virus (CrPV) IRES has been shown to be active in both yeast and mammalian cells [26, 33] and able to initiate translation by directly recruiting ribosomes to the initiation sites without initiation factors. Defects in ribosome biogenesis can impair cap-independent translation initiation directed by CrPV IGR IRES elements [26, 33] and in cellular RNAs that utilize the IRES mechanism [27]. We monitored translation initiation from the CrPV IGR IRES elements using a dual luciferase reporter which is able to measure translation in vivo in yeast cells (Fig 8). While cap-dependent translation was very weakly reduced in the scc2-4 mutant, consistent with the results of 35S-methionine incorporation (S8B Fig); IRES usage was reduced by more than 40% in the scc2-4 mutant when compared to WT (Fig 8A), implying that Scc2 promotes cap-independent translation.

Faithfully maintaining the translational reading frame is one of the important functions of ribosomes. Viral mRNA signals that can induce ribosomes to shift frame by one base in either -1 or +1 direction have been tremendously useful for examining translational fidelity [26, 34]. We examined frameshifting in the scc2-4 mutant. Dual luciferase reporters able to detect -1 frameshifting (L-A) or +1 frameshifting (Ty1) were employed. The scc2-4 mutant had an approximately 3-fold increase in L-A mediated -1 frameshifting compared to WT (3.4% to 1.1%; p = 0.006) (Fig 8B). However, no change in +1 frameshifting was observed. We further examined the efficiency of stop codon recognition using dual luciferase reporters (Fig 8C) in the scc2-4 mutant. The scc2-4 mutant was able to recognize stop codons as efficiently as WT.
Since paf1Δ mutants also have defects in snoRNA expression, we examined whether a paf1Δ mutant had defects in translational fidelity. Translation initiation was examined by transforming the CrPV IGR IRES dual luciferase construct in a paf1Δ mutant. Similar to the scc2-4 mutant, use of the IRES sequence to initiate translation was severely compromised in the paf1Δ mutant (S9A Fig). We also observed a significant increase in -1 frameshifting in the paf1Δ mutant when compared to the WT strain (S9B Fig). However, +1 frameshifting was unaffected. Overall the paf1Δ and scc2-4 mutants show deficits in translational fidelity similar to those caused by mutation of CBF5 [26].
Changes in ribosome-tRNA interactions can affect translational fidelity and therefore influence the sensitivity of yeast to translational inhibitors. Translational inhibitors that specifically bind to ribosomes serve as useful tools for examining changes in ribosome function. Anisomycin, which inhibits -1 frameshifting by interfering with binding of aa-tRNA to the A-site, was used to probe for defects in this region of the ribosome [35]. Similarly, paromomycin, which promotes translational misreading, and increases programmed -1 ribosomal frameshifting in a mof2-1 strain [36], was used to probe interactions at the decoding center on the small subunit involving the aa-tRNA [37]. While the scc2-4 mutant was hypersensitive to paromomycin (Fig 8D), as might be expected given the tendency for -1 frameshifting, inhibition of -1 frameshifting with anisomycin [35] did not rescue the growth defect observed in the mutant (S10 Fig). The failure of anisomycin to rescue the growth of the scc2-4 mutant probably reflects the fact that Scc2 contributes to growth in multiple ways. Overall, our results are consistent with Scc2 normally promoting the production of rRNA and translational fidelity.
Discussion
In this report, we characterized the biological processes controlled by Scc2, and show that Scc2 promotes the normal expression of hundreds of genes. With the notable exception of the centromeric region, Scc2 binding at the promoter regions of ribosomal protein genes, snoDNAs, and tDNAs was reduced in the scc2-4 mutant. At the snoDNAs in particular, lower levels of Scc2 recruitment correlated with lower levels of RNA; and at these genes, Scc2 recruitment was partially dependent on the Paf1 transcription complex. We show that lower levels of H/ACA snoRNAs correlate with reduced pseudouridylation in the scc2-4 mutant and decreased translational fidelity. Therefore, Scc2 promotes a gene expression program that supports translational fidelity.
Scc2 coordinates events during RNA transcription and modification
Studies in human, fly, and yeast genomes show that Scc2 binds to the promoter regions of active genes [5, 11, 14, 38]. However, how Scc2 contributes to the expression of active genes is not well understood. From ChIP-seq analysis, we observed that Scc2 not only binds to the promoter regions of genes, but also in the gene body of snoRNA and tRNA genes (Fig 2). Binding of Scc2 to both promoter and genes bodies (in particular the snoDNAs) suggest that its role in regulating transcription might not be restricted to initiation. In support of this hypothesis, we observed that Paf1, a protein required for transcription elongation by RNA Pol II [29], is significantly reduced in the scc2-4 mutant. Studies in budding yeast have shown that Paf1 is recruited to the promoter and coding regions of genes and aids in the processing of snoRNA transcripts [30]. The observation that Scc2 and Paf1 recruitment depend on each other at snoDNAs suggests the hypothesis that Paf1 and Scc2 together promote production of snoRNAs. This hypothesis is further supported by the observation that paf1Δ mutants have translational fidelity defects similar to the scc2-4 mutant. Paf1 levels are reduced in the scc2-4 mutant and there is a global reduction in ChIP signal, suggesting this could contribute to differential gene expression. The translation of at least one subunit of the Paf1 complex could be compromised in the scc2-4 mutant, and this could then impact the stability of the other subunits [39]. Alternatively, the mutation in Scc2 could directly impact the stability of the Paf1 complex, for instance, at shared binding sites. The cooperation of Paf1 and Scc2 could have wider implications in humans where the Paf1 complex is overexpressed in a wide range of cancers including prostate, breast, renal, and gastric cancers [40, 41].
Different groups of Scc2 target genes show different responses to the loss of binding. This could be due in part to other factors that operate at those gene groups. For instance, Scc2 binding to snoDNAs appears to depend on Paf1, but this is not the case at other regions of the genome such rDNA and RP genes. Instead, RP genes are expressed at higher levels with reduced Scc2 binding. The elevation may be due to competition between cohesin binding and RNA Pol II binding at these promoters. As the scc2-4 mutant decreases cohesin loading, it may allow RNA Pol II more access (Patrick Grant, in prep).
Mutations that affect cohesin result in differential gene expression and translation in budding yeast
The scc2-4 mutant has translation defects. Some phenotypes are similar to what we previously reported in the acetyltransferase mutant, eco1-W216G [22], such as defects in nucleolar morphology, ribosome distribution, and rRNA production. In both cases these phenotypes could result from reduced cohesin and condensin at the rDNA. Therefore cohesin may be important for nucleolar structure and function [23]. However, a closer look reveals significant differences in both gene expression and translational processes in the two mutants. For example, only the scc2-4 mutation is associated with reduced levels of box H/ACA snoRNAs and poor translational fidelity as well as elevated levels of ribosomal protein gene messages. The acetyltransferase mutation has reduced levels of ribosomal protein gene messages, increased levels of the stress induced transcription factor Gcn4, and a pronounced effect on translational efficiency. Reduced translational efficiency is observed in cells derived from RBS patients and in zebrafish models for CdLS, and these cohesinopathies models are partially rescued by stimulating translation [16, 42]. We speculate that the differences in gene expression between scc2 and eco1 mutants might be based in the molecular mechanisms by which they influence gene expression. While Scc2 may maintain nucleosome free regions and promote cohesin and condensin loading, Eco1 helps to maintain a high level of stable acetylated cohesin. Loss of function mutations in these two genes may have different effects on gene expression. This is consistent with mutations in these two genes causing distinct human syndromes.
Cohesinopathies and ribosomopathies have similar characteristics
Ribosomopathies are diseases caused by mutations that affect ribosome biogenesis. One such disease is dyskeratosis congenita (DKC). Reduced pseudouridylation of RNA is part of the etiology of DKC, a ribosomopathy characterized by bone marrow failure, skin abnormalities and increased susceptibility to cancer [27, 43]. Mutations in a pseudouridine synthase that modifies ribosomal RNAs and other RNAs (DKC1/CBF5) cause some cases of DKC. Studies from X-DC patient lymphoblasts and fibroblasts show downregulation of Box H/ACA snoRNAs [44], a characteristic feature in scc2-4 mutant. Interestingly, the scc2-4 mutant mimics cbf5 mutants which have an impaired ability to translate IRES containing genes. Our results suggest that the scc2-4 mutation reduces rRNA production as well as modification, possibly in part by affecting the production of H/ACA snoRNAs, which are important for pseudouridylation, resulting in reduced translational fidelity. Reduced levels of some box C/D RNAs could also affect translation due to impaired methylation of rRNA. We previously reported that human Roberts syndrome (RBS) cells and zebrafish models of RBS have defects in ribosome biogenesis and protein synthesis [22, 42], suggesting that RBS is at least in part a ribosomopathy. Zebrafish models for CdLS also show reduced translation [16]. Our findings suggest a shared feature of cohesinopathies and ribosomopathies is defective translation. Translation might therefore serve as a therapeutic target for CdLS.
Materials and Methods
Yeast strains
All strains were derived from BY4741 (MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0) (S1 Table). The scc2-4 mutations was originally isolated by the Nasmyth laboratory [1]. We constructed scc2-4 mutant strains in the laboratory by PCR amplifying the mutation fused to a drug resistance marker from yeast genomic DNA, transforming the PCR product into the desired strain background, and isolating temperature sensitive colonies. The presence of the mutation was confirmed by sequencing.
Measurement of RNA synthesis in vivo
Ribosomal RNA production was examined as previously described with some modifications [45]. Briefly, duplicate cultures of WT and scc2-4 mutant strains were grown to mid-log phase (OD600 = 0.3) in SD-ura medium supplemented with 6.7 ng/μl uracil. 3H-uridine (5 μCi) was added to 500 μL of each culture and incubated at 30°C for 5 min with aeration. Samples were treated with 2.5 mL 10% trichloroacetic acid (TCA) and 2.5 mg/ml of uridine. After filtration through nitrocellulose, each membrane was washed with 5% TCA, dried, and counted in a Beckman LS 6500 multipurpose scintillation counter.
Metabolic labeling-protein
WT and scc2-4 strains were grown till mid-log phase YPD+CSM medium. Cells were pelleted, washed in PBS and resuspended in a similar volume of pre-warmed SD-met medium. Aliquot for zero time point was taken. Samples were then supplemented with 27.5 μCi 35S-methionine and 1mg/ml unlabeled-methionine. Samples were withdrawn at 15 min intervals for 2 hr. Amount of incorporated 35S-methionine in proteins was measured by adapting Kang and Hershey approach [46]. Briefly, cells were lysed in 1.8 N NaOH buffer containing 0.2 M β-mercaptoethanol. Proteins were precipitated with hot 10% trichloroacetic acid and precipitates washed twice in acetone. The precipitates were dissolved in 1% SDS and boiled for 10 min. Aliquots of samples were counted for 35S-methionine incorporation using a scintillation counter.
rRNA processing
WT and scc2-4 mutants strains were grown in SD-methionine medium to mid-log phase (OD600 = 0.3) Cells were pulse-labeled for 2 min with 250 μCi/ml 3H-methylmethionine and chased with 5 mM cold methionine. Samples were removed at 0, 2, 5 and 15 min intervals and flash-frozen in liquid nitrogen. RNA was extracted from cells, run on 1% formaldehyde agarose gel, transferred to a Hybond-N+ nylon membrane (GE Healthcare), and detected by autoradiography.
RNA sequencing
WT and scc2-4 mutant strains were grown in triplicate to mid-log phase (~OD600 = 0.8) in YPD+CSM medium. Cells were pelleted and RNA was extracted. RNA integrity was checked following isolation using an Agilent Technologies 2100 Bioanalyzer and a 1.2% formaldehyde agarose gel. Samples were depleted of ribosomal RNA using the Epicentre Ribo-zero Gold kit (human/mouse/rat, cat# RZG1224) according to manufacturer’s protocol. Libraries for sequencing were prepared using the Illumina Truseq RNA library preparation kit (cat# RS-930-2001) according to the manufacturer’s protocol. Libraries were pooled and run on an agarose gel, size selected from 200-400bp (with adapters), and run on the Hi-seq. Reads were mapped to saccCer2 using tophat to generate bam alignment files. Gene coordinates from ensembl biomart were then iterated over to count the number of reads mapping to each feature. These counts were then used to generate gene expression coefficients and statistics using the DESeq package in R. The GO term enrichment analysis was carried out using the hypergeometric test as implemented in the GeneAnswers package from Bioconductor. For the metagene analysis, reads were extended to 150 base pairs before calculating coverage. Coverage was adjusted to reads per million. For a group of genes, RPM coverage was extracted for 600 bp on either side of the TSS and the values were averaged per base pair for the group.
Splicing analysis
The generated fastq files from RNA sequencing were aligned using tophat [47] against the S. cerevisiae genome assembly EF4 and gene annotation from the Ensembl release 69 (Oct. 2012) with the default options. Between 17.4M and 26.0M reads were aligned with 82.2% to 85.1% uniquely mapped reads per library. Splicing efficiency (expressed as Percent Spliced Out or PSO) was measured by computing the mean nucleotide coverage over every spliced junctions divided by the mean nucleotide coverage for reads falling within the exonic splicing unit (i.e. counting all the reads overlapping within the two exons flanking the splice junctions and dividing by the length of the two exons). Significant changes in PSO were computed by performing ANOVA for each splicing unit. The computed probabilities of the difference between the means were adjusted for multiple testing using the Benjamini and Hochberg method.
ChIP
Genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) was performed as previously described [48]. Briefly, yeast strains were cultured in duplicate in YPD to mid-log phase (OD600 = 0.8). Strains were crosslinked for either 15 min or 2 hrs (for Scc2 ChIP) with 1% formaldehyde at room temperature with occasional swirling. Crosslinnking was quenched with 2.5M Glycine. Cultures were pelleted, washed with cold PBS, and resuspended in PBS and placed at 4°C overnight. Cells were spheroplasted with 2.5 mg/ml zymolyase. Spheroplasts were washed and sonicated in SDS lysis buffer (1 mM PMSF, 0.μg/ml pepstatin A, 0.6 μg/ml leupeptin) to between 300–1000 bp in length. Chromatin extracts were diluted in immunoprecipitation (IP) buffer, debris was pelleted, and the supernatant was decanted into conical tubes. This chromatin solution was aliquoted for IPs. Chromatin extracts were immunoprecipitated overnight with antibody at 4°C followed by the addition of IgG beads. Beads were washed in several steps, and DNA was recovered in 1% SDS/0.1 M NaHCO3 elution buffer. Crosslinking was reversed by incubation at 65°C overnight, followed by protease treatment, phenol chloroform extraction, and ethanol precipitation of the recovered DNA. IPs were performed in duplicate for each sample. Sequence libraries were constructed and validated. Reads were mapped to saccCer2 using tophat to generate bam alignment files. Antibodies used for ChIP experiments are as follows; Myc antibody from Cell signaling (Cat #2276), HA antibody (12CA5) from Roche and RNA Pol II CTD4H8 antibody from Millipore (4 ul per 1mg protein IP).
Microscopy and cytometry
Accumulation of ribosomal protein GFP fusion proteins in strains was measured with spinning disc confocal microscopy (Zeiss). The GFP intensity in WT and scc2-4 mutant strains was quantified as previously described [22]. Briefly, peak GFP fluorescence intensity in each cell was calculated by measuring the pulse height in the cell using a B1 detector (525/50 emission). For each sample approximately 10,000 cells were measured. To quantify the distance the empirical distribution function of two samples, the Kolmogorov-Smirnov (KS) statistic was used. This statistic enables us to calculate the distance between two biological replicates (same genotype) and between samples with different genotypes since the distribution of fluorescence intensity among GFP positive cells is non-Gaussian. Using KS-distance enables us to determine whether the distance between WT and mutant samples is significantly greater than between replicates of the same genotype.
Luciferase assays
The IRES and frameshifting assays were performed as previously described [33, 49]. Briefly, yeast strains were transformed with the indicated reporter plasmids. To examine IRES activity, overnight cultures of strains harboring CrPV IGR IRES and mutant cc-gg constructs were subcultured in SD-leu medium to mid-log phase (approx. OD600 = 0.5 to 1.0). Cells were lysed with 100 μl 1X passive lysis buffer (PLB) for 2 min. A dual luciferase assay kit (Promega) was used to measure luminescence according to the manufacturer’s protocol. IRES activity was measured using Firefly/Renilla ratio normalized to the Firefly/Renilla ratio of the wild type strain.
The frameshifting and readthrough assays were conducted with dual luciferase reporters [33]. Translational fidelity assays employed the dual-luciferase reporter system using pJD375 (the 0-frame control), pJD377 (containing the Ty1 +1 PRF signal), pJD376 (containing the yeast L-A virus -1 PRF signal). Luciferase activities were determined as previously described [50, 51]. To measure frameshifting, firefly/renilla luciferase ratio generated from the 0-frame control reporter (pJD375) was divided into that from the frameshifting signal-containing constructs (pJD377 and pJD376) and multiplied by 100% to obtain the frameshifting efficiencies for each construct. For the readthrough assay, overnight cultures of strains transformed with dual luciferase construct were subcultured and grown to mid-log phase, pelleted, lysed with 1X passive lysis buffer and dual luciferase assay performed in triplicates according to manufacturer’s protocol (Promega). Percentage readthrough was then calculated as previously described [33]. Readthrough occurred at a stop codon if firefly luciferase was translated following the Renilla luciferase ORF. Values obtained from firefly luciferase were normalized to Renilla luciferase activity as an internal control. The value obtained was then divided by the luciferase activity normalized to Renilla luciferase from a reporter with no stop codon and theoretically could be 100% readthrough for each reporter. Thus the percentage readthrough is expressed as Firefly/Renilla luciferase activity ratio divided by the firefly/Renilla luciferase activity ratio of the sense codon reporter and multiplied by 100.
Antibiotic sensitivity
Growth and temperature sensitivity assays were performed using 10-fold serial dilutions of cells spotted onto the indicated medium and incubated at 30°C for 2–3 days. Maximal growth rates and sensitivity to translational inhibitors were determined using the TECAN machine in YPD medium containing 5 μg/mL anisomycin, or 800 μg/mL paromomycin.
Western blotting
Western blot experiments were conducted using trichloroacetic acid (TCA) precipitation as previously described [52, 53]. Briefly overnight cultures were subcultured in YPD medium until mid-log phase (OD600 = 0.6). Cell were pelleted and washed 2 times in 20% TCA. Cells were resuspended in residual TCA and glass beads added. Cells were vortexed for 4 min at 3000 g and spun for 5 min at 14000 rpm. To each sample, 100 μl sample loading buffer was added and vortexed. Tris-HCl pH 8.8 or more was added and vortexed until the color changed to purple blue. Samples were boiled at 95°C for 5 min and loaded onto an SDS-PAGE gel.
Pseudouridylation assay
Pseudouridylation assay was performed as previously described with a few modifications [54]. The experimental procedure is summarized below.
Reaction with CMC
Overnight cultures of yeast strains were subcultured and grown to mid-log phase (OD600 = 0.6). Cells were pelleted and RNA isolated from strains. RNA was dried and 8 μg RNA sample were treated with 30 μl of 0.17 M N-Cyclohexyl-N’-(2-morpholinoethyl)carbodiimide methyl-p-toluenesulfonate (CMC) in 50 mM Bicine, pH 8.3, 4 mM EDTA, and 7 M urea at 37°C for 20 min. The reaction was stopped with 100 μl of 0.3 M Sodium acetate (NaOAc) and 0.1 mM EDTA, pH 5.6 (buffer A) and 700ul 100% ethanol. Pellets were recovered, washed with 70% ethanol and dissolved in 100 μl buffer A, and reprecipitated with 300 μl 100% ethanol. Pellets were dried, and dissolved in 40 μl 50 mM Na2CO3, pH 10.4 and incubated at 37°C for 2 hrs. RNA was then precipitated with 100 μl buffer A and 700 μl of ethanol. Pellets were washed twice with 70% ethanol, dried and dissolved in 40 μl water.
Reverse transcription and gel analysis
RNA was reversed transcribed using 5’-end labeled primers. Hybridization was performed in a mixture containing 8 μg RNA, 4 * 105 cpm/pmol 5’-labeled primer, and hybridization buffer (50 mM Tris-HCl, pH 8.5, 20 mM KCl) in a total volume of 5 μl. The mixture was incubated at 70°C for 3 min, transferred to a 37°C for 5 min, and then chilled on ice for 2 min or longer. The extension reaction was performed by adding 5 μl of a mixture containing 100 mM Tris-HCl, pH 8.5, 20 mM MgCl2, 20 mM DTT, 5 nM each of dATP, dGTP, dCTP, and dTTP, and 1.6 units of reverse transcriptase to the hybridization mixture and incubated at 37°C for 30 min. The extension reaction was stopped by adding 2 μl of a solution containing 0.5 mg/ml RNase A and 0.1 M EDTA, pH 6.5 and incubated at 37°C for 45 min. The sample mixture was then precipitated with 3 volumes of ethanol, dried and dissolved in 15 μl 75% Formamide, 1.4 mM Tris-HCl, pH 7.4, 1.4 mM EDTA, and 0.08% each of xylene cyanol FF and bromophenol blue. Samples were run on 8% polyacrylamide-7 M urea gel and detected with phosphoImager screens. Oligonucleotides for monitoring pseudouridylation were designed as previously described [55].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997 Oct 3;91(1):35–45. 9335333
2. Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell. 2003 Mar 21;112(6):765–77. 12654244
3. Haering CH, Lowe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Molecular cell. 2002 Apr;9(4):773–88. 11983169
4. Strunnikov AV, Larionov VL, Koshland D. SMC1: an essential yeast gene encoding a putative head-rod-tail protein is required for nuclear division and defines a new ubiquitous protein family. The Journal of cell biology. 1993 Dec;123(6 Pt 2):1635–48. 8276886
5. D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, et al. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes & development. 2008 Aug 15;22(16):2215–27.
6. Dowen JM, Bilodeau S, Orlando DA, Hubner MR, Abraham BJ, Spector DL, et al. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem cell reports. 2013;1(5):371–8. doi: 10.1016/j.stemcr.2013.09.002 24286025
7. Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Shevchenko A, et al. Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Molecular cell. 2000 Feb;5(2):243–54. 10882066
8. Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Molecular cell. 2004 Dec 22;16(6):991–1002. 15610741
9. Rollins RA, Korom M, Aulner N, Martens A, Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Molecular and cellular biology. 2004 Apr;24(8):3100–11. 15060134
10. Horsfield JA, Anagnostou SH, Hu JK, Cho KH, Geisler R, Lieschke G, et al. Cohesin-dependent regulation of Runx genes. Development. 2007 Jul;134(14):2639–49. 17567667
11. Misulovin Z, Schwartz YB, Li XY, Kahn TG, Gause M, MacArthur S, et al. Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma. 2008 Feb;117(1):89–102. 17965872
12. Lopez-Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. The Scc2-Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nature genetics. 2014 Oct;46(10):1147–51. doi: 10.1038/ng.3080 25173104
13. Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Human mutation. 2013 Dec;34(12):1589–96. doi: 10.1002/humu.22430 24038889
14. Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS biology. 2009 May 5;7(5):e1000119. doi: 10.1371/journal.pbio.1000119 19468298
15. Kaur M, DeScipio C, McCallum J, Yaeger D, Devoto M, Jackson LG, et al. Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. American journal of medical genetics Part A. 2005 Sep 15;138(1):27–31. 16100726
16. Xu B, Sowa N, Cardenas ME, Gerton JL. l-leucine partially rescues translational and developmental defects associated with zebrafish models of Cornelia de Lange syndrome. Human molecular genetics. 2014 Nov 6.
17. Lindgren E, Hagg S, Giordano F, Bjorkegren J, Strom L. Inactivation of the Budding Yeast Cohesin Loader Scc2 alters Gene Expression both Globally and in Response to a Single DNA Double Strand Break. Cell cycle. 2014 Sep 19 : 0.
18. Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, et al. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell. 2014 Sep 25;159(1):148–62. doi: 10.1016/j.cell.2014.08.028 25219674
19. Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014 Sep 5.
20. Ofengand J. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS letters. 2002 Mar 6;514(1):17–25. 11904174
21. Hu B, Itoh T, Mishra A, Katoh Y, Chan KL, Upcher W, et al. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Current biology: CB. 2011 Jan 11;21(1):12–24. doi: 10.1016/j.cub.2010.12.004 21185190
22. Bose T, Lee KK, Lu S, Xu B, Harris B, Slaughter B, et al. Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS genetics. 2012;8(6):e1002749. doi: 10.1371/journal.pgen.1002749 22719263
23. Harris B, Bose T, Lee KK, Wang F, Lu S, Ross RT, et al. Cohesion promotes nucleolar structure and function. Molecular biology of the cell. 2014 Feb;25(3):337–46. doi: 10.1091/mbc.E13-07-0377 24307683
24. Zhang Y, Sikes ML, Beyer AL, Schneider DA. The Paf1 complex is required for efficient transcription elongation by RNA polymerase I. Proceedings of the National Academy of Sciences of the United States of America. 2009 Feb 17;106(7):2153–8. doi: 10.1073/pnas.0812939106 19164765
25. Reichow SL, Hamma T, Ferre-D'Amare AR, Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic acids research. 2007;35(5):1452–64. 17284456
26. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Molecular cell. 2011 Nov 18;44(4):660–6. doi: 10.1016/j.molcel.2011.09.017 22099312
27. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006 May 12;312(5775):902–6. 16690864
28. Preti M, Ribeyre C, Pascali C, Bosio MC, Cortelazzi B, Rougemont J, et al. The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Molecular cell. 2010 May 28;38(4):614–20. doi: 10.1016/j.molcel.2010.04.016 20513435
29. Sheldon KE, Mauger DM, Arndt KM. A Requirement for the Saccharomyces cerevisiae Paf1 complex in snoRNA 3' end formation. Molecular cell. 2005 Oct 28;20(2):225–36. 16246725
30. Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, et al. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. The EMBO journal. 2003 Apr 15;22(8):1846–56. 12682017
31. Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, et al. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Molecular and cellular biology. 2002 Oct;22(20):6979–92. 12242279
32. Tomson BN, Crisucci EM, Heisler LE, Gebbia M, Nislow C, Arndt KM. Effects of the Paf1 complex and histone modifications on snoRNA 3'-end formation reveal broad and locus-specific regulation. Molecular and cellular biology. 2013 Jan;33(1):170–82. doi: 10.1128/MCB.01233-12 23109428
33. Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes & development. 2009 Dec 1;23(23):2753–64.
34. Dinman JD, Kinzy TG. Translational misreading: mutations in translation elongation factor 1alpha differentially affect programmed ribosomal frameshifting and drug sensitivity. Rna. 1997 Aug;3(8):870–81. 9257646
35. Dinman JD, Ruiz-Echevarria MJ, Czaplinski K, Peltz SW. Peptidyl-transferase inhibitors have antiviral properties by altering programmed -1 ribosomal frameshifting efficiencies: development of model systems. Proceedings of the National Academy of Sciences of the United States of America. 1997 Jun 24;94(13):6606–11. 9192612
36. Cui Y, Dinman JD, Kinzy TG, Peltz SW. The Mof2/Sui1 protein is a general monitor of translational accuracy. Molecular and cellular biology. 1998 Mar;18(3):1506–16. 9488467
37. Vicens Q, Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure. 2001 Aug;9(8):647–58. 11587639
38. Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, et al. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS genetics. 2014 Feb;10(2):e1004153. doi: 10.1371/journal.pgen.1004153 24550742
39. Mueller CL, Porter SE, Hoffman MG, Jaehning JA. The Paf1 complex has functions independent of actively transcribing RNA polymerase II. Molecular cell. 2004 May 21;14(4):447–56. 15149594
40. Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochimica et biophysica acta. 2010 May-Jun;1799(5–6):379–88. doi: 10.1016/j.bbagrm.2010.01.001 20060942
41. Chaudhary K, Deb S, Moniaux N, Ponnusamy MP, Batra SK. Human RNA polymerase II-associated factor complex: dysregulation in cancer. Oncogene. 2007 Nov 29;26(54):7499–507. 17599057
42. Xu B, Lee KK, Zhang L, Gerton JL. Stimulation of mTORC1 with L-leucine rescues defects associated with Roberts syndrome. PLoS genetics. 2013;9(10):e1003857. doi: 10.1371/journal.pgen.1003857 24098154
43. Dokal I. Dyskeratosis congenita in all its forms. British journal of haematology. 2000 Sep;110(4):768–79. 11054058
44. Bellodi C, McMahon M, Contreras A, Juliano D, Kopmar N, Nakamura T, et al. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell reports. 2013 May 30;3(5):1493–502. doi: 10.1016/j.celrep.2013.04.030 23707062
45. Snyder M, Huang XY, Zhang JJ. The minichromosome maintenance proteins 2–7 (MCM2-7) are necessary for RNA polymerase II (Pol II)-mediated transcription. The Journal of biological chemistry. 2009 May 15;284(20):13466–72. doi: 10.1074/jbc.M809471200 19318354
46. Kang HA, Hershey JW. Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. The Journal of biological chemistry. 1994 Feb 11;269(6):3934–40. 8307948
47. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25 19261174
48. Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, et al. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS biology. 2004 Sep;2(9):E259. 15309048
49. Deniz N, Lenarcic EM, Landry DM, Thompson SR. Translation initiation factors are not required for Dicistroviridae IRES function in vivo. Rna. 2009 May;15(5):932–46. doi: 10.1261/rna.1315109 19299549
50. Harger JW, Dinman JD. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. Rna. 2003 Aug;9(8):1019–24. 12869712
51. Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF, Atkins JF. A dual-luciferase reporter system for studying recoding signals. Rna. 1998 Apr;4(4):479–86. 9630253
52. Kim DH, Koepp DM. Hect E3 ubiquitin ligase Tom1 controls Dia2 degradation during the cell cycle. Molecular biology of the cell. 2012 Nov;23(21):4203–11. doi: 10.1091/mbc.E12-07-0548 22933573
53. Kim DH, Zhang W, Koepp DM. The Hect domain E3 ligase Tom1 and the F-box protein Dia2 control Cdc6 degradation in G1 phase. The Journal of biological chemistry. 2012 Dec 28;287(53):44212–20. doi: 10.1074/jbc.M112.401778 23129771
54. Bakin A, Ofengand J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry. 1993 Sep 21;32(37):9754–62. 8373778
55. Ganot P, Bortolin ML, Kiss T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell. 1997 May 30;89(5):799–809. 9182768
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 7
Nejčtenější v tomto čísle
- Functional Constraint Profiling of a Viral Protein Reveals Discordance of Evolutionary Conservation and Functionality
- Reversible Oxidation of a Conserved Methionine in the Nuclear Export Sequence Determines Subcellular Distribution and Activity of the Fungal Nitrate Regulator NirA
- Modeling Implicates in Nephropathy: Evidence for Dominant Negative Effects and Epistasis under Anemic Stress
- Nutritional Control of DNA Replication Initiation through the Proteolysis and Regulated Translation of DnaA