
Trans-Reactivation: A New Epigenetic Phenomenon Underlying Transcriptional Reactivation of Silenced Genes
We discovered a new epigenetic phenomenon we called trans-reactivation. We found that genes, unable to produce a functional coding transcript, but with the potential of transcribing other RNA’s within their gene body, strongly reactivate the transcription of a wildtype copy of the same gene silenced by heterochomatin. This new epigenetic phenomenon is heritable, relies on the presence of diffusible RNAs able to carry and transfer epigenetic information and is affected by mutations in genes involved in Post-Transcriptional Gene Silencing. Our data strongly suggest that homologous non-coding RNA can reactivate the expression of genes silenced by heterochromatin, thus defining a new unpredicted level of gene expression control in the context of heterochromatic genes.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005444
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005444
Summary
We discovered a new epigenetic phenomenon we called trans-reactivation. We found that genes, unable to produce a functional coding transcript, but with the potential of transcribing other RNA’s within their gene body, strongly reactivate the transcription of a wildtype copy of the same gene silenced by heterochomatin. This new epigenetic phenomenon is heritable, relies on the presence of diffusible RNAs able to carry and transfer epigenetic information and is affected by mutations in genes involved in Post-Transcriptional Gene Silencing. Our data strongly suggest that homologous non-coding RNA can reactivate the expression of genes silenced by heterochromatin, thus defining a new unpredicted level of gene expression control in the context of heterochromatic genes.
Introduction
In recent years it has become increasingly evident that the expression of eukaryotic genomes is far more complex than it had been previously explored. An emerging body of evidence, coming from next generation sequencing approaches, is showing that the genomes of all studied eukaryotes are almost entirely transcribed, generating an enormous number of non-coding RNAs (ncRNAs) [1]. The ENCODE project showed that at least 90% of analyzed eukaryote genome is transcribed in different cell types, indicating that there is a huge reservoir of RNA molecules with potentially unexplored biological function [2,3,4]. These RNAs are remarkably different in their number, size, subcellular localization, and mechanisms of action, and many are essential to finely control gene expression as well as genomic plasticity [5].
It is becoming increasingly clear that some ncRNAs are part of epigenetic regulatory networks with striking evolutionary conservation [6,7]. For example, dynamic changes in chromatin function are frequently transacted by nuclear RNA signaling pathways. Although the evolutionarily conservation and precise molecular mechanisms are poorly understood, a differential recruitment of a hierarchy of chromatin modifying complexes to specific loci by RNAs sets precise transcriptional states leading to differentiation [8,9,10,11,12]. Moreover, the unusual epigenetic phenomena of paramutation [13,14,15], trans-induction [16] and transvection [17,18] observed in a variety of higher eukaryotes involve the activity of ncRNAs that ‘rewrite’ the transcriptional state of an allele, in processes that apparently escape classic Mendel’s laws of genetic inheritance. These epigenetic phenomena are clear examples of ncRNA-directed regulatory processes that transfer epigenetic information both across cells, between tissues and across generations, though the mechanisms underlying these phenomena still remain elusive.
In order to unveil the role played by cellular RNA pools produced by homologous genomic loci in changing the transcriptional state of a silenced gene, we used classic Position Effect Variegation (PEV) assays in the model system D. melanogaster [19,20] and tested the effect of non-functional alleles of the white gene in the presence of a functional copy of white, silenced by heterochromatin (wm4h). Surprisingly, we found that several non-functional white alleles, unable to produce the main wildtype white coding transcript but with the potential of transcribing other RNA’s from the white locus, trans-reactivate the variegating wm4h line thus increasing eye pigmentation. Strikingly, the presence of non-functional white alleles cause an increase in the wm4h gene transcript as well as an opening in the chromatin structure at the wm4h locus. Remarkably, this new epigenetic phenomenon is heritable, relies on the presence of diffusible homologous RNA’s, and is affected by mutations in genes involved in post-transcriptional gene silencing. Overall, our data strongly indicate that trans-reactivation is a new epigenetic phenomenon that positively control gene expression in the context of heterochromatin through homologous RNA molecules.
Results
Alleles of white can suppress wm4h eye color variegation
The white (w) gene encodes an ABC transporter essential for the red pigment transportation in the compound Drosophila eye. The In(1)wm4h X chromosome inversion places the fully functional wildtype euchromatic w gene adjacent to a region of pericentromeric heterochromatin, creating the variegated white-mottled 4 (wm4h) allele that is characterized by the random inactivation of w by the heterochromatin spreading from the inversion chromosome breakpoint (Fig 1A, upper panel). This cell autonomous transcriptional inactivation once established is clonally inherited, and it is responsible for an eye with a variegated expression of the red pigment, constituting an example of the genetic phenomenon known as Position Effect Variegation (PEV) [20].
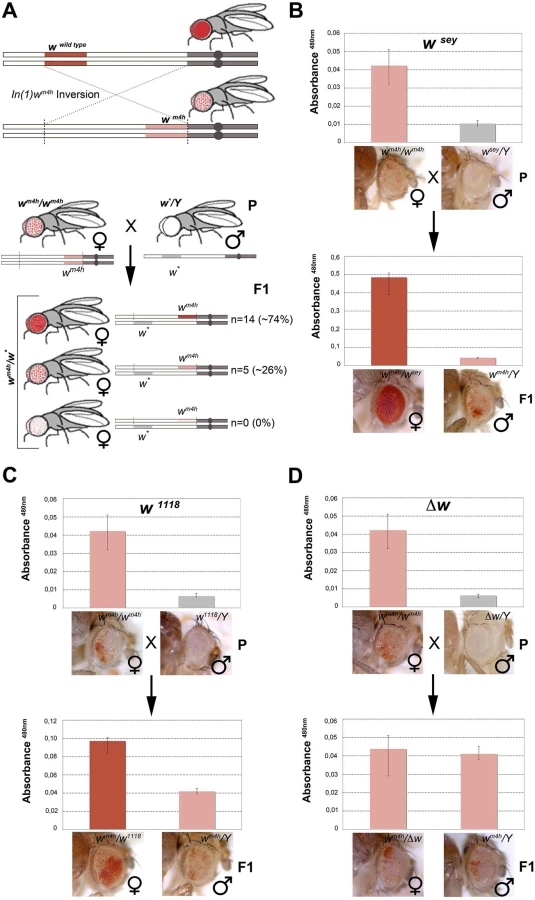
Genetic screens have shown that a large number of mutations alter PEV phenotypes, resulting in the isolation of enhancers E(var)s or suppressors Su(var)s of variegating phenotypes. The vast majority of these modifiers were originally isolated in Drosophila as dominant mutations that suppressed or enhanced eye color variegation caused by the wm4h allele [21]. The molecular characterization of those mutants have shown that the products of E(var)’s and Su(var)’s are structural components of chromatin, or enzymes that covalently modify chromatin proteins [19]. However, to date the effect exerted by non-functional w alleles on the wm4h chromatin-dependent variegation is unknown. Therefore, we took advantage of the classic wm4h PEV assay to analyze if non-functional alleles of the w gene were able to modify eye color variegation when in trans-heterozygosis with the wm4h allele (Figs 1A, lower panel, and S1A and S1C).
We screened 19 loss-of-function or hypomorphic w alleles (w*) for genetic interaction with wm4h, and measured eye red pigment to quantify the strength of the genetic interaction [22]. The pilot screen resulted in the isolation of 14 suppressors (74% of total w* alleles screened) able to increase wm4h eye color pigmentation (Fig 1A and S1 Table). Among the tested hypomorphic alleles that behaved as robust suppressors of wm4h variegation, the wsey allele interacted more strongly (Figs 1B and S2). Moreover, loss-of-function alleles of w, including the widely used w1118 allele, showed a weak but highly reproducible suppression effect (Figs 1C and S3). Notably, F1 wm4h/Y males derived from the crosses did not show any increase in eye color pigmentation (Fig 1B and 1C), indicating that the genetic locus responsible for the increased eye color pigmentation in the wm4h/w* trans-heterozygous females is carried by the w* bearing X chromosome. Reciprocal crosses using multiple wm4h balanced lines (S1B, S1D, S1E and S1F Fig) and classic recombination mapping (S1G and S1H Fig) confirmed the effect of the w* loci tested in increasing eye color pigmentation of wm4h. Interestingly, deletions spanning the w locus (Δw) did not show any modification of the wm4h variegation (Fig 1D and S1 Table). Moreover, none of the w* alleles tested was able to reduce the red pigmentation of wm4h, while 5 of them (26%) had no effect (Fig 1A, lower panel, and S1 Table). The pilot screen we conducted suggested that the presence of white genomic homologous sequences of non-functional w* alleles could reactivate in trans-heterozygosis the wm4h heterochromatic silenced locus.
The suppression of wm4h variegation by w* alleles is a robust effect, unrelated to classic Su(var) function and independent from chromosome pairing
To further characterize the effect of wsey and w1118 alleles (from now on used as representative examples of w* alleles giving a strong and weak interaction with wm4h, respectively), we first decided to measure the strength of their suppression on the wm4h variegation. Interestingly, in the presence of a strong E(var) mutation encoding for a factor contributing to the opening of chromatin [23], both w1118 and wsey retain their ability to increase eye pigmentation of wm4h, though w1118 with a weaker strength (Figs 2A and S4A). This data indicate that the suppressing w* alleles are able to increase eye pigmentation even when the wm4h allele is in a strongly silenced heterochromatin context.
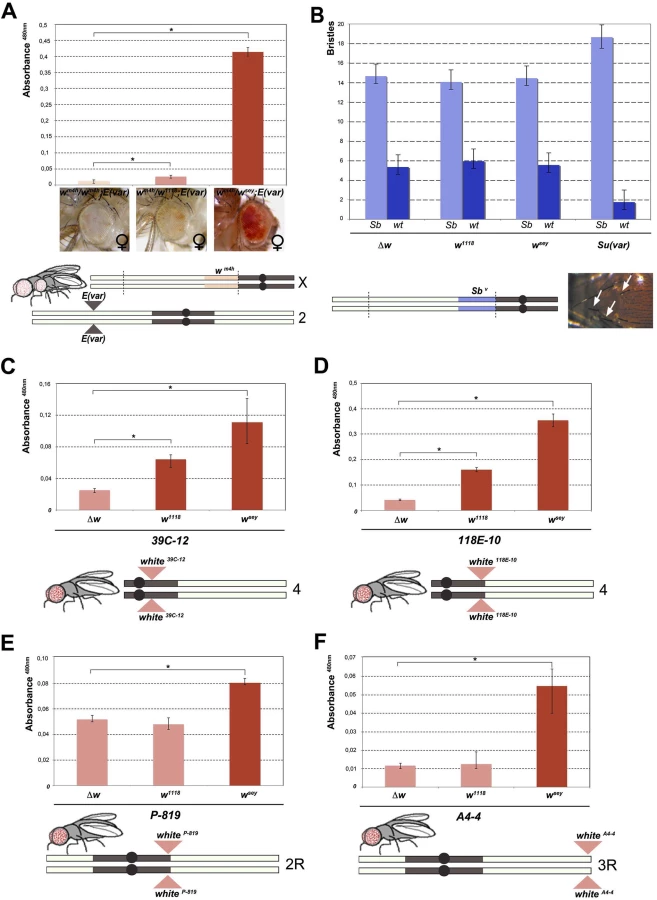
Next we wanted to test the ability of w* alleles to behave as suppressors in other PEV assays, a feature that is shared by classic Su(var)s encoding for factors responsible for the establishment and maintenance of inactive chromatin [24]. We choose the Sbv PEV assay, characterized by the T(2;3)Sbv translocation that juxtaposes the dominant Sb mutation to centric heterochromatin, resulting in mosaic flies with both short (Sb) and normal bristles (Fig 2B). As expected, when the Sbv stock was crossed with a strong Su(var) [25] a significant suppression was observed in the frequency of Sb bristles (Figs 2B, upper graph, and S4B). However, both w1118 and wsey as well as the w locus deletion (Δw) were not able to increase the number of short bristles when crossed with Sbv. These results suggest that the reactivating w* alleles do not likely encode for a factor that, beyond its role in eye pigmentation, it is also responsible for chromatin structure organization. Thus, our data strongly indicate that the mechanism underlying wm4h suppression by w* alleles is very likely unrelated to classic Su(var)’s function.
The suppression effect of w* alleles we observed over wm4h is highly reminiscent of transvection, a well-studied epigenetic phenomenon that results from the interaction between an allele on one chromosome and the corresponding allele on the homologous chromosome. Transvection may lead to both gene activation or repression, and is strongly dependent upon chromosome pairing [18]. However, because of the large In(1)wm4h inversion, the wm4h bearing chromosome is never paired with the w* containing sister chromatid in the suppressed trans-heterozygous females (Fig 1A), thus suggesting that the effect we observed could not be assimilated to classic transvection. However, to exclude intra-chromosomal long distance pairing or X chromosome specific effects, we tested the ability of w* alleles to de-repress variegated wildtype w insertions in pericentric and telomeric autosomal chromosome heterochromatin [26,27,28]. In all lines tested the strong wsey and in some cases also the weak w1118 allele, but not the Δw deletion, consistently increased eye pigmentation of variegated wildtype w pericentric or telomeric autosomal insertions (Figs 2C–2F and S4C). Our data strongly suggest that the increase in eye pigmentation exerted by w* alleles over wm4h is independent from the type of heterochromatin tested, it does not rely on intra-chromosomal pairing and it is chromosome independent.
w* alleles can increase wm4h eye pigmentation by opening chromatin at the wm4h locus and increasing the levels of wild type white coding transcripts
To explore the correlation of increased eye pigmentation in trans-heterozygous interacting females (wm4h/w*) and transcriptional activation of the w gene, we conducted semi-quantitative RT-PCR on total RNA extracted from adult heads. To measure the amount of full-length wildtype white coding mRNA we designed primers against the 3’ of the transcript that are able to amplify the fully transcribed spliced and unspliced mRNA products (Fig 3A). As expected, the RNA extracted from wildtype flies shows a robust amplification of w, while flies carrying the w1118 loss-of-function allele or the Δw deletion do not show detectable levels of white transcripts (Fig 3A, compare lanes 1 with 2 and 7). Moreover, while the wm4h stock produced very low levels of wildtype coding white transcripts (Fig 3A, lane 3), the hypomorphic wsey flies produced only modest levels of unspliced white transcripts (Fig 3A, lane 4). Remarkably, when w1118 or wsey are introduced in trans with wm4h, we observed a highly reproducible and robust amplification of white transcripts (Fig 3, lanes 5 and 6). These data strongly correlate the levels of eye pigmentation, we observed in the trans-heterozygous w*/wm4h adults, with the levels of expressed coding white transcripts.

However, an increased white transcript stability in w*/wm4h trans-heterozygous could in theory also explain the observed high levels of amplification of coding white transcripts. Therefore, we tested the ability of mutants in genes known to be involved in maternal and zygotic fly mRNA stabilization for their ability to modify the levels of eye pigmentation normally scored in w*/wm4h adults. However, none of the mutants tested, including pumilio (pum) and smaug (smg), were able to significantly change eye pigmentation in our PEV assay (S5A Fig). These data strongly suggest that the increased levels of eye pigmentation observed in w*/wm4h trans-heterozygous females, cannot be directly correlated with general changes in white mRNA stability (S5A Fig).
While, for all the interacting loss-of-function w* alleles, we could be sure that any white coding transcript we detect could only be produced from the wildtype wm4h locus, the same conclusion cannot be drawn for the interacting hypomorphic w* alleles. Therefore, we decided to use genomic probes from the white genomic locus to conduct fluorescent in situ hybridization (FISH) analyses on polytene chromosomes, for monitoring the level of accessibility of the interacting w* and wm4h chromatin loci. Polytene chromosomes represent a special structural organization of Drosophila salivary glands, consisting of polyploid interphase nuclei, which originate by repeated rounds of DNA replication without cell division. Drosophila polytene chromosomes have proven to be an invaluable cytogenetic tool to examine chromosome structure. Moreover, DNA FISH probes hybridization is dependent on the level of replication of specific genomic regions on polytene chromosomes, which in turn depends on the overall level of transcriptional activity present on that locus [24].
Exploiting this special feature of polytene chromosomes, we designed genomic probes covering both the w locus and the coding sequences of the hsp70 gene as internal positive control for FISH probe chromatin accessibility. As expected, wm4h homozygous female chromosomes gave a FISH signal for the hsp70 gene but failed to show a detectable band from the wm4h pericentric chromatin region because this heterochromatic locus is under-replicated [24] (See arrowhead and asterisk in Fig 3B). On the other hand, homozygous chromosomes for the w1118 and wsey alleles or wildtype flies gave detectable FISH signals, for both the w and hsp70 loci (see arrowheads in Figs 3C and 3D and S5B). Remarkably, wm4h/w1118 and wm4h/wsey but not wm4h/∆w trans-heterozygous female chromosomes, on top of the expected signals coming from the w and hsp70 loci, gave a highly reproducible FISH band originating from the pericentric wm4h chromatin locus (see arrowhead in the vicinity of the asterisk in Figs 3E and 3F and S5B). This analysis strongly suggests that FISH signals detected from the wm4h locus in trans-heterozygous w*/wm4h chromosomes are the result of increased DNA replication, consistent with an increase in transcription at the wm4h locus, as also found for other classic Su(var)s [24]. In conclusion, our data strongly indicate that the increase in eye pigmentation, observed in the wm4h/w* trans-heterozygous combinations, is strongly correlated with a reopening of wildtype heterochromatic wm4h locus by the presence of homologous genomic sequences present in the interacting loss of function and hypomorphic w* alleles. We called the ability of w* alleles to re-reactivate wild type white at the wm4h locus: trans-reactivation.
trans-reactivation is heritable
The epigenetic interaction between the w* and wm4h alleles, leading to an increase in white transcription as a consequence of chromatin reopening at the silenced wm4h locus, is a phenomenon highly reminiscent of the epigenetic switch occurring in paramutation. In this epigenetic phenomenon, identified in plants, mice, and recently also in flies, the paramutating allele has the ability to change the activity state of its partner allele on the homologous chromosome (usually to a silent state) [14,15,29]. This effect is dependent on homologous sequences present in the two interacting alleles, is heritable and once established, is independent from the presence of the paramutating allele. To investigate if the reactivation of the wm4h locus was heritable through meiosis, we crossed F1 trans-reactivated wm4h/w* females with w*/Y males to look for trans-generational inheritance of trans-reactivation in the F2 progeny (Figs 4A and S6A).
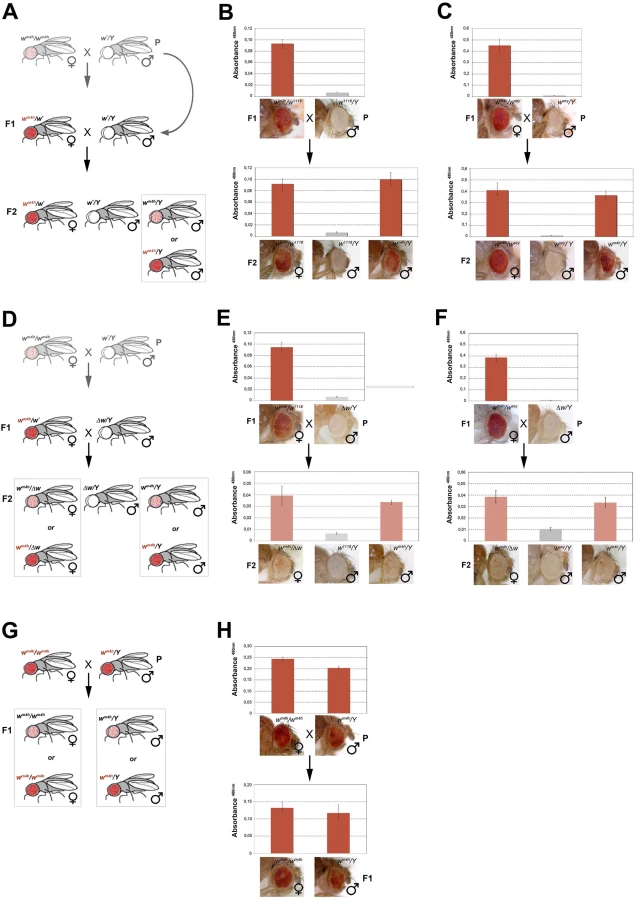
In the F2 progeny we expect to score wm4h/w* females (that should continue to show increased levels of eye pigmentation), w*/Y males (deficient of red eye pigment), and finally wm4h/Y males where the F1 trans-reactivated wm4h allele, inherited from the F1 mothers has segregated from the w* trans-reactivating allele. If F2 wm4h/Y males have normal variegated eyes, the F1 trans-reactivated wm4h allele had lost its trans-reactivating potential during meiosis. On the other hand, if F2 wm4h/Y males showed increased eye pigmentation, this would be a strong indication that the trans-reactivated state of the F1 wm4h allele is transmitted in the germline and inherited in the F2 generation (Fig 4A).
Remarkably, both w1118 and wsey trans-reactivating alleles when segregated away from the F1 activated wm4h allele were able to generate F2 wm4h/Y males with strong eye pigmentation (Fig 4B and 4C). However, when the same F1 trans-heterozygous wm4h/w* females were crossed with Δw /Y males not bearing any homologous genomic w sequence (Figs 4D and S6B), the resulting F2 wm4h/Y males and wm4h/Δw females failed to trans-reactivate showing levels of eye variegation indistinguishable from parental (P) wm4h stocks (Fig 4E and 4F). Remarkably, when wild type w+ carrying flies are tested for their ability to trans-reactivate in F2 wm4h males, we did not observe any trans-reactivation (S6C Fig). However, when we crossed homozygous trans-reactivated wm4h/wm4h female with trans-reactivated wm4h/Y male, where the trans-reactivating w* allele was no longer present in both lines, we could reproducible score only trans-reactivated progeny up to the F5 (Fig 4G and 4H).
Although, trans-reactivation also works in reciprocal crosses where the trans-reactivating w* allele is carried by the mother (S1E and S1F Fig), our data show that wm4h trans-reactivation is dependent upon the presence of non-functional w* homologous genomic sequences present in at least one gamete (the sperm, in this set of experiment). Moreover, since fully functional coding wild type w+ alleles are unable to trans-reactivate, our data also indicate that the w* reactivating alleles likely encode trans-reactivating factors able to maintain the trans-reactivated state of wm4h coming from the other gamete, a feature that does not meet the classic definition of paramutation. Indeed, paramutated alleles maintain their state independently from the presence of the paramutating allele. However, crosses in which both parents have exclusively wm4h trans-reactivated alleles strongly maintain the trans-generational inheritance of trans-reactivated wm4h, making trans-reactivation a new epigenetic phenomenon, distinct from paramutation, causing the trans-generational inheritance of trans-reactivated wm4h.
A diffusible factor carrying genetic information is responsible for trans-reactivation
A long-standing question in the understanding of the mechanisms underlying epigenetic memory inheritance is whether a genetic information could be transferred from one allele to another through physical pairing of homologous chromosomes, or via a diffusible intermediate carrying genetic information. Although, our data indicate that trans-reactivation of w* over wm4h is independent from classic chromosome pairing (Fig 2C–2F), some of the features of w* trans-reactivation are highly reminiscent of the process known as trans-inactivation observed for the brown (bw) locus in flies, and of trans-induction occurring at the globin gene cluster in mammals, two epigenetic phenomena opposite in their final transcriptional outcome, but highly dependent on long-range chromosome physical interaction [16,30].
Indeed, in trans-inactivation when two bw alleles are paired, heterochromatin insertion in one causes both alleles to associate with the heterochromatic centromere, suppressing brown expression [30]. On the other hand, in trans-induction intergenic transcription of the globin cluster specific for the erythroid cells can be induced in non-erythroid cells by the expression of transiently transfected globin genes [16]. Physical interaction of homologous chromatin region of the affected alleles appears to inactivate, in the case of trans-inactivation, or activate, in the case of trans-induction, chromosomal transcription. Indeed, the activating effect of w* alleles over wm4h could be explained by the two homologous inverted loci engaging on a long-range physical interaction. Under this model, the silent wm4h locus could dilute out heterochromatic factors over the non-functional w* homologous genomic sequences, causing a partial de-repression of wm4h, thus explaining the opening of the chromatin locus, as well as the increased white transcription and eye pigmentation.
In order to determine if w* trans-reactivation is mediated by a long range physical interaction between the homologous w* and wm4h loci or instead through diffusible factors produced by the homologous genomic sequences present in the w* alleles, we made use of the unique reproductive features of flies able to generate adults through the process of gynogenesis [31,32]. Gynogenesis is like parthenogenesis in that diploid zygotes inheriting all chromosomes from their mother can develop without a genetic contribution from fathers. However, even though gynogenetic diploid eggs do not require paternal chromosomes, they do require the physical penetration of the sperm into the mature egg and the contribution of paternal diffusible nuclear and cytoplasmic factors in order to initiate zygotic development (Fig 5A). In particular, when crossing the paternal effect lethal ms(3)K81 males with gynogenetic gyn2, gyn3 females, gynogenesis occurs and the only possible progeny resulting from this cross are gyn2, gyn3 females (Fig 5A).

Therefore, we asked what happened to eye color pigmentation in the progeny resulting from wm4h gynogenetic gyn2; gyn3 eggs fertilized by ms(3)K81 males carrying the strong trans-reactivating wsey allele (Fig 5B). If a long range physical interaction between the male-carrying wsey and female-carrying wm4h chromosomes is required for trans-reactivation we should observe no increase in eye pigmentation in the wm4h; gyn2; gyn3 F1 female progeny, because the father chromosomes do not contribute to the gynogenetic zygote (Fig 5B). However, if diffusible factors carrying epigenetic information are present in the ms(3)K81 males encoding the trans-reactivating wsey allele, then diploid wm4h; gyn2; gyn3 F1 female progeny should show trans-reactivated eyes (Fig 5B). Remarkably, F1 wm4h; gyn2; gyn3 females, not carrying any genomic contribution from the father, show strong trans-reactivated eyes when fertilized by ms(3)K81 males carrying the trans-reactivating wsey allele (Figs 5C and S7A). However, no effect on eye pigmentation was observed in the F1 progeny of gynogenetic females crossed with ms(3)K81 males carrying the Δw deletion (Figs 5D and S7B).
The data resulting from the gynogenesis experiment strongly rule out the possibility that trans-reactivation is due to any type of long-range physical genomic interactions between the wm4h and w* alleles. Instead, our data strongly indicates that diffusible factors, present in the gamete carrying the homologous trans-reactivating w* allele, are responsible for the phenomenon of trans-reactivation of wm4h.
Mutations in genes encoding for factors involved in the generation, modification, and processing of ncRNA influence trans-reactivation
The diffusible factors able to mediate trans-reactivation should carry a genetic information coming from the w* allele, epigenetically transfer this information to the wm4h locus, and amplify this information in the zygote in order to exert their effect later in the fully differentiated adult eye tissue. The high diffusibility, self-amplification property, and capacity to epigenetically transfer information make RNA a great candidate for such a trans-reactivating diffusible factor. We therefore looked at the effect of known mutations in genes involved in the generation, modification and processing of a variety of family of small ncRNA in the process of trans-reactivation (S7C and S7D Fig).
With the single exception of spd-E allele, mutations in genes encoding for factors involved in piRNA (armi, piwi, aub, hsp83, and ago2) and siRNA (dcr2 and r2d2) biogenesis enhanced the trans-reactivating effect of w* over wm4h (Fig 6A). On the other hand, mutations in genes encoding factors involved in the production of miRNA (ago1 and dcr1) suppress trans-reactivation, with the exception of ago2 that is also involved in the piRNA pathway (Fig 6A). Recently, a non-coding RNA expressed from a human pseudogene was reported to regulate the corresponding protein-coding mRNA by acting as a decoy for microRNAs [33]. This study raised the questions about the potential ability of non-coding transcripts to act as ‘sponges’ to attract miRNAs, thus boosting the expression of miRNA target genes. If the trans-reactivating w* alleles produced miRNAs sponge RNA’s we should expect that mutations in genes affecting miRNA biogenesis should enhance the effect of the trans-reactivating w* alleles over wm4h. However, our data clearly show that mutations in components of the miRNA processing machinery suppress trans-reactivation (Fig 6A and 6B), making it very unlikely that trans-reactivation could be explained by a miRNAs sponge effect. Remarkably, mutation in dnmt2, an RNA-dependent Methyl Transferase (RdMT) very recently shown to be involved in the phenomenon of paramutation in mice [34,35], has also strong suppressing effect on trans-reactivation (Fig 6A). Overall, our genetic interaction data strongly indicate that altering the biogenesis, the post-translational modification and processing of a variety of small ncRNA families interfere at various levels the efficiency of trans-reactivation (Fig 6B), strongly supporting the involvement of RNA molecules in the onset and maintenance of this new epigenetic phenomenon.

In order to identify unique specific RNA species produced by the w* alleles during the process of trans-reactivation of wm4h we conducted high throughput RNA sequencing analysis of total as well as small RNA enriched fractions from larval Malpighian tubules, an abundant fly larval tissue that express high levels of the w gene. However, the analysis of short RNAs did not result in the identification of specific or enriched RNA species in the trans-reactivated wsey/wm4h line when compared to the wsey or wm4h lines alone (lower panel Fig 6C). On the other hand, the analysis of long RNA species clearly showed that the trans-reactivated wsey/wm4h line overall produces more coding RNA from the w locus than the single wsey or wm4h lines alone (upper panel Fig 6C and 6D), in line with our previous semi quantitative RT-PCR analysis (Fig 3A). In conclusion, our RNA sequencing data did not allow us to identify specific or discrete RNA species present in the wsey trans-reactivating allele that could explain the onset of trans-reactivation.
However, if RNA species produced at the homologous w* alleles could influence in trans the transcription of the silenced wm4h locus, we expect that injection of wm4h eggs with total RNA extracted from w* individuals should result in trans-reactivated adult flies. Remarkably, when we injected wm4h eggs with total RNA extracted from wsey testis (Fig 6E) we obtained adults with trans-reactivated eyes. However, wm4h eggs injected with RNA derived from Δw testis had no effect (Fig 6E), strongly indicating that RNA molecules produced by the homologous wsey allele are responsible for the trans-reactivation of the wm4h locus. In conclusion, we propose that the RNA produced by the w* alleles from the w locus can induce in trans the activation of the silenced wm4h locus through a yet to be defined mechanism in which post transcriptional gene silencing and RNA methylation factors are involved.
Discussion
Role for ncRNA in transcriptional regulation
Over the last decade we have come to appreciate that the eukaryotic chromatin is full of coding and non-coding overlapping transcripts, and that a lot of the genetic information is transacted by coding and non-coding RNAs. Indeed, the vast majority of genomes of all metazoans are transcribed into complex patterns of ncRNAs. Recent observations strongly suggest that ncRNAs contribute to the complex transcriptional networks needed to regulate cell function [36,37]. However, despite a rich variety of mechanisms by which RNA acts by repressing transcription have been identified, fewer studies have explored the role of RNA in the positively regulation of transcription. RNA is an integral component of chromatin [38] and many transcription factors and chromatin modifying enzymes have the capacity to bind RNA or complexes containing RNA or RNA-binding proteins [8]. Moreover, RNA-directed processes help to establish chromatin architecture and epigenetic memory [10]. Indeed, ncRNAs can act locally to regulate the epigenetic state of the nearby chromatin, often recruiting either repressing or activating chromatin remodeling complexes [8]. Given the high degree of sequence and locus specificity information, RNA could recruit chromatin remodelers and modifiers in a sequence-specific manner. Indeed, recent observations are beginning to reveal that RNA molecules can stimulate gene transcription. These RNA activators employ a wide array of mechanisms to up-regulate transcription of target genes, often functioning as DNA-tethered activation domains, as coactivators or modulators of general transcriptional machinery [39].
trans-reactivation: A new level of gene expression control of silenced genes?
In search for a possible role played by cellular RNA pools produced by homologous genomic loci in changing the transcriptional state of a silenced gene, we found that non-functional alleles of the classic fly marker white could trans-reactivate the expression of wild type copy of white silenced by heterochromatin. We called this new epigenetic phenomenon trans-reactivation. This process is heritable over many generations, it relies on the presence of diffusible RNAs, and it is affected by mutations in genes involved in post-transcriptional gene silencing. Our data strongly suggest that the presence of a gene that does not produce a full coding transcript but only spurious transcription could trans-reactivate the expression of a functional copy of the same gene silenced by heterochromatin, defining a new unpredicted level of gene expression control in the context of heterochromatic genes.
The mechanism underlying trans-reactivation
The mechanisms of action and many features of trans-reactivation appear to be different from the previously characterized epigenetic reprogramming events occurring in paramutation, transinduction and transvection. In trans-reactivation genomic regions, transcribing RNAs unable to produce a full coding transcript, can positively influence the transcriptional state of an homologous locus silenced by heterochromatin. However, how can the information present in the non-functional RNA molecules be ‘read’ and telecasted to specific silenced homologous genomic regions? RNA molecules have the potential to pair with other RNA molecule or with DNA. Possibly through their ability to form paired structures, RNA may function as a sensor and/or a messenger of homology. RNA can form duplexes with sequence homology with genomic promoter sequences thus promoting transcription [40]. Indeed, there is a great deal of evidence that enhancers and other promoter regulatory sequences are transcribed in the cell in which they are active, and that enhancer transcription is often needed to activate the transcription on their target gene [41]. Moreover, given that mutations in genes encoding for factors involved in posttranscriptional gene silencing modulate trans-reactivation, it remains to be determined whether some sequence specific small ncRNAs, that we failed to identify, may explain the mechanism of trans-reactivation. Furthermore, we do not know why some non-functional w* null or hypomorphic alleles failed to trans-reactivate their homologous silenced locus. Indeed, differences in trans-reactivation ability between different w* alleles will need future investigation, and may provide key information to understand the mechanism of action of trans-reactivation. However, independently from the mechanistic details of trans-reactivation, we are convinced that we identified a new epigenetic phenomenon of allelic communication that acts through diffusible RNA molecules. Nevertheless, much work need to be done to study the evolutionary conservation and the general ability of trans-reactivating RNAs in resetting the epigenetic and transcriptional state of homologous genetic loci.
Materials and Methods
Drosophila stocks and genetic crosses
Flies were raised and crossed at 20° on K12 Medium (US Biological). Unless otherwise stated, strains were obtained from Bloomington, Szeged, and DGRC Stock Centers. Recombination mapping of the trans-reactivating effect of w* alleles is described in S1 Fig. The white alleles used for the trans-reactivation assays are listed in S1 Table. The w1; gyn-21;gyn-31 and ms(3)K811/TM3, Sb1, Ser1 lines were a generous gift from Dr. Mia Levine. The wm4h; the CyORoi/Sco—wm4h; PrDr/TM3 Sb,Ser—E(var)3−101- lines were donated by Dr. Gunter Reuter. The 39C12 and 118E-10 variegating autosomal lines were a gift from Dr. Sarah Elgin. The cn1 P{PZ}AGO104845/CyO—ry506, r2d21/CyGFP—aubHN2/CyO—y1 w67c23;P{EPgy2}AGO2EY04479/ Cy—Armi/TM3 ser, GFP—ru1 st1 spn-E1 e1 ca1/TM3, Sb1 Ser1—Dcr1/TM6c—Hsp83Scratch/TM3 Sb,Ser and T(2;3)SbV⁄ TM3,Ser lines were obtained from Dr. Maria Pia Bozzetti and Dr. Laura Fanti. The Ago2null, Dcr2null /CyO, piwi06843/CyO, aubQC42/CyO lines derive from Dr. Valerio Orlando Lab and the P-819 and A4-4 lines from Dr. Stephane Ronsseray lab. Dr Frank Lyko provided the Dnmt2Δ99/CyO line.
Pigment and variegation assays
The eye pigment assay was performed in triplicate for each of the three biological replicate we conducted, as previously described [22]. Eyes from representative individuals of the genotype tested were photographed with an Olympus SZ61 stereomicroscope. For the dominant Stubble variegation (SbV) assay ten pairs of major dorsal bristles were analysed in 20 flies. The extent of Sb variegation was represented as the mean of Sb and WT bristles per genotype tested in three biological replicate.
Fluorescence in situ hybridization (FISH)
Cytological preparations and FISH analysis were carried out as previously described [42] The P(CaSpeRA) plasmid containing the genomic sequences of the mini white as well as the hsp70 genes was used as a template for generating the FISH probe. FISH signals were acquired with a Leica DM 4000B microscope (Leica).
Semi-quantitative RT-PCR
Total RNA was purified from 20 heads of adult flies of the desired genotype, using TRIzol reagent following the manufacturer’s (Invitrogen) recommended protocol. First strand cDNA was synthesized using MuLV reverse transcriptase (Applied Biosystems). Semi-Quantitative PCR was conducted using specific primer pairs that amplify the fourth, fifth and sixth exons of the white gene (forward, 5’ - CTATAGGTCATATCTTGTTTTTATTGGCAC-3’) (Reverse 3’ - CGTGGGTGCCCAGTGTCC-5’) and the following parameters: denaturation 2 min at 94°C, followed by 20 cycles of 30” at 94°C, 30” at 53°C, 2’ at 72°C and final extension at 72°C for 3’. The PCR products were analyzed by agarose electrophoresis and the images were acquired and quantified with the ChemiDoc XRS imager (BioRad). The Act5C RNA was amplified as loading control with the primer pairs (forward, 5’-CCTCGTTCTTGGGAATGG-3’, Act5C reverse, 3’ - CGGTGTTGGCATACAGATCCT-5’) using the same PCR cycle parameters.
RNA-sequencing
Total long RNAs were extracted from malpighian tubuli tissues using the miRNeasy Mini Kit, following an RNeasy MinElute Cleanup Kit (Qiagen) step for the enrichment of small RNAs. The 100bp paired-end sequencing was performed at IGA using a HiSeq2000TM machine. Long RNAs were mapped against the Drosophila genome (assembly BDGP Release 5) using TopHat (PUBMED 19289445) with default parameters. Mapped reads were associated with gene annotations (FlyBase 5.12), and transcript levels of annotated genes, expressed as Fragments per Kilobase of exon Per Million fragments mapped (FPKM), were estimated by CuffCompare (PUBMED 20436464). For small RNAs, sequencing adapters were trimmed from sequence reads with custom scripts. Resulting sequences were mapped against the Drosophila genome using Bowtie (PUBMED 19261174) allowing at most two substitutions within the first 20 base pairs.
RNA-injection
wm4h; CyORoi/Sco 3h starved flies were allowed to lay eggs for 50 minutes on apple juice agar plates with yeast. Approximately 100–150 decorionated eggs were injected with 1–2 pl of an injection solution containing 5 mM NaCl, 5% Texas Red conjugated dextran (Molecular Probes), and 1 ng/μl of total RNA extracted and purified from testis of either Δw or wsey Drosophila males.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Amaral PP, Dinger ME, Mercer TR, Mattick JS (2008) The eukaryotic genome as an RNA machine. Science 319 : 1787–1789. doi: 10.1126/science.1155472 18369136
2. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489 : 57–74. doi: 10.1038/nature11247 22955616
3. Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, et al. (2010) Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330 : 1775–1787. doi: 10.1126/science.1196914 21177976
4. Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, et al. (2010) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330 : 1787–1797. doi: 10.1126/science.1198374 21177974
5. Berretta J, Morillon A (2009) Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO reports 10 : 973–982. doi: 10.1038/embor.2009.181 19680288
6. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, et al. (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America 106 : 11667–11672. doi: 10.1073/pnas.0904715106 19571010
7. Nagano T, Fraser P (2011) No-nonsense functions for long noncoding RNAs. Cell 145 : 178–181. doi: 10.1016/j.cell.2011.03.014 21496640
8. Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF (2009) RNA regulation of epigenetic processes. BioEssays: news and reviews in molecular, cellular and developmental biology 31 : 51–59.
9. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 : 281–297. 14744438
10. Bernstein E, Allis CD (2005) RNA meets chromatin. Genes & development 19 : 1635–1655.
11. Grewal SI, Elgin SC (2007) Transcription and RNA interference in the formation of heterochromatin. Nature 447 : 399–406. 17522672
12. White SA, Allshire RC (2008) RNAi-mediated chromatin silencing in fission yeast. Current topics in microbiology and immunology 320 : 157–183. 18268844
13. Brink RA (1973) Paramutation. Annual review of genetics 7 : 129–152. 4361265
14. Chandler VL (2007) Paramutation: from maize to mice. Cell 128 : 641–645. 17320501
15. de Vanssay A, Bouge AL, Boivin A, Hermant C, Teysset L, et al. (2012) Paramutation in Drosophila linked to emergence of a piRNA-producing locus. Nature 490 : 112–115. doi: 10.1038/nature11416 22922650
16. Ashe HL, Monks J, Wijgerde M, Fraser P, Proudfoot NJ (1997) Intergenic transcription and transinduction of the human beta-globin locus. Genes & development 11 : 2494–2509.
17. Gubb D, Ashburner M, Roote J, Davis T (1990) A novel transvection phenomenon affecting the white gene of Drosophila melanogaster. Genetics 126 : 167–176. 2121594
18. Duncan IW (2002) Transvection effects in Drosophila. Annual review of genetics 36 : 521–556. 12429702
19. Girton JR, Johansen KM (2008) Chromatin structure and the regulation of gene expression: the lessons of PEV in Drosophila. Advances in genetics 61 : 1–43. doi: 10.1016/S0065-2660(07)00001-6 18282501
20. Reuter G, Spierer P (1992) Position effect variegation and chromatin proteins. BioEssays: news and reviews in molecular, cellular and developmental biology 14 : 605–612.
21. Schotta G, Ebert A, Dorn R, Reuter G (2003) Position-effect variegation and the genetic dissection of chromatin regulation in Drosophila. Seminars in cell & developmental biology 14 : 67–75.
22. Lloyd VK, Sinclair DA, Grigliatti TA (1997) Competition between different variegating rearrangements for limited heterochromatic factors in Drosophila melanogaster. Genetics 145 : 945–959. 9093849
23. Netter S, Fauvarque MO, Diez del Corral R, Dura JM, Coen D (1998) white+ transgene insertions presenting a dorsal/ventral pattern define a single cluster of homeobox genes that is silenced by the polycomb-group proteins in Drosophila melanogaster. Genetics 149 : 257–275. 9584101
24. Hayashi S, Ruddell A, Sinclair D, Grigliatti T (1990) Chromosomal structure is altered by mutations that suppress or enhance position effect variegation. Chromosoma 99 : 391–400. 2125551
25. Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, et al. (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes & development 18 : 1251–1262.
26. Levis R, Hazelrigg T, Rubin GM (1985) Effects of genomic position on the expression of transduced copies of the white gene of Drosophila. Science 229 : 558–561. 2992080
27. Sun FL, Cuaycong MH, Craig CA, Wallrath LL, Locke J, et al. (2000) The fourth chromosome of Drosophila melanogaster: interspersed euchromatic and heterochromatic domains. Proceedings of the National Academy of Sciences of the United States of America 97 : 5340–5345. 10779561
28. Josse T, Boivin A, Anxolabehere D, Ronsseray S (2002) P element-encoded regulatory products enhance Repeat-Induced Gene Silencing (RIGS) of P-lacZ-white clusters in Drosophila melanogaster. Molecular genetics and genomics: MGG 268 : 311–320. 12436253
29. Chandler VL, Stam M (2004) Chromatin conversations: mechanisms and implications of paramutation. Nature reviews Genetics 5 : 532–544. 15211355
30. Henikoff S (1997) Nuclear organization and gene expression: homologous pairing and long-range interactions. Current opinion in cell biology 9 : 388–395. 9159074
31. Fuyama Y (1986) Genetics of Parthenogenesis in DROSOPHILA MELANOGASTER. II. Characterization of a Gynogenetically Reproducing Strain. Genetics 114 : 495–509. 17246347
32. Fuyama Y (1986) Genetics of Parthenogenesis in DROSOPHILA MELANOGASTER. I. the Modes of Diploidization in the Gynogenesis Induced by a Male-Sterile Mutant, ms(3)K81. Genetics 112 : 237–248. 17246314
33. Ebert MS, Sharp PA (2010) Emerging roles for natural microRNA sponges. Current biology: CB 20: R858–861. doi: 10.1016/j.cub.2010.08.052 20937476
34. Kiani J, Grandjean V, Liebers R, Tuorto F, Ghanbarian H, et al. (2013) RNA-mediated epigenetic heredity requires the cytosine methyltransferase Dnmt2. PLoS genetics 9: e1003498. doi: 10.1371/journal.pgen.1003498 23717211
35. Liebers R, Rassoulzadegan M, Lyko F (2014) Epigenetic Regulation by Heritable RNA. PLoS genetics 10: e1004296. doi: 10.1371/journal.pgen.1004296 24743450
36. Mattick JS (2005) The functional genomics of noncoding RNA. Science 309 : 1527–1528. 16141063
37. Prasanth KV, Spector DL (2007) Eukaryotic regulatory RNAs: an answer to the 'genome complexity' conundrum. Genes & development 21 : 11–42.
38. Rodriguez-Campos A, Azorin F (2007) RNA is an integral component of chromatin that contributes to its structural organization. PloS one 2: e1182. 18000552
39. Ansari AZ (2009) Riboactivators: transcription activation by noncoding RNA. Critical reviews in biochemistry and molecular biology 44 : 50–61. doi: 10.1080/10409230902734044 19280431
40. Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, et al. (2007) Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nature chemical biology 3 : 166–173. 17259978
41. Lempradl A, Ringrose L (2008) How does noncoding transcription regulate Hox genes? BioEssays: news and reviews in molecular, cellular and developmental biology 30 : 110–121.
42. Pimpinelli S, Bonaccorsi S, Fanti L, Gatti M (2010) Fluorescent in situ hybridization (FISH) of mitotic chromosomes from Drosophila larval brain. Cold Spring Harbor protocols 2010: pdb prot5391.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in