
YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
The YAP1/Hippo signaling pathway is a key regulator of organ size and tissue homeostasis, and its dysregulation is linked to cancer development. Elevated activity of YAP1, a transcriptional coactivator and well-established oncogene has been reported to occur in human cancers. Comprehensive identification of YAP1 regulated genes and its mode of action will be of high importance to uncover YAP1 biology that could be exploited for a therapeutic intervention. To this end, we performed genome-wide analyses to identify YAP1 occupied sites in cancer cell lines representing different YAP1/Hippo pathway tumor etiologies and in non-transformed fibroblasts. Our data demonstrate that YAP1 activity is mediated predominantly via TEAD transcription factors supporting the importance of TEADs as main mediators of YAP1-coactivator activity. We further show that YAP1 and TEAD1 exert their transcriptional control via binding to enhancers, leading to characteristic chromatin changes and distal activation of genes. By linking enhancers to genes, we provide a list of novel YAP1 target genes in an oncogenic setting that we show can readily be exploited in tumor classification and provides a foundation for further investigations.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005465
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005465
Summary
The YAP1/Hippo signaling pathway is a key regulator of organ size and tissue homeostasis, and its dysregulation is linked to cancer development. Elevated activity of YAP1, a transcriptional coactivator and well-established oncogene has been reported to occur in human cancers. Comprehensive identification of YAP1 regulated genes and its mode of action will be of high importance to uncover YAP1 biology that could be exploited for a therapeutic intervention. To this end, we performed genome-wide analyses to identify YAP1 occupied sites in cancer cell lines representing different YAP1/Hippo pathway tumor etiologies and in non-transformed fibroblasts. Our data demonstrate that YAP1 activity is mediated predominantly via TEAD transcription factors supporting the importance of TEADs as main mediators of YAP1-coactivator activity. We further show that YAP1 and TEAD1 exert their transcriptional control via binding to enhancers, leading to characteristic chromatin changes and distal activation of genes. By linking enhancers to genes, we provide a list of novel YAP1 target genes in an oncogenic setting that we show can readily be exploited in tumor classification and provides a foundation for further investigations.
Introduction
YAP1 (Yes-associated protein 1) is a major transcriptional effector of the evolutionary and functionally conserved Hippo pathway, which is a crucial regulator of organ size, proliferation but also tumor growth [1–3]. Activation of the Hippo pathway leads to phosphorylation and inactivation of the transcriptional co-activator YAP1 by cytoplasmic retention or enhanced degradation [4–8].
YAP1 has a potent growth promoting activity and the YAP1/Hippo pathway has been tightly linked to cancer [8–11]. Loss of Hippo signaling by mutations or down-regulation of core pathway components is associated with cancer development, while YAP1 is reported as a potent oncogene that can promote tumorigenesis in a wide range of tissues [2, 12, 13]. Elevated expression or activity of YAP1 occurs through multiple mechanisms. YAP1 gene amplification and mutations in upstream pathway regulators, such as NF2, have been described in various human tumors [2, 14–20].
YAP1 lacks an intrinsic DNA-binding domain and is thought to exert its co-activator function through binding to promoter sequences via interaction with transcription factors (TF), such as TEAD1/-2/-3/-4, Smads, Runx1/-2, p73, ErbB4, Pax3, AP-1, or TBX5 [12, 21]. Among these the TEAD TF family members play a dominant role as primary mediators of YAP1-dependent gene regulation and YAP1 growth-promoting activity [22–28]. Although the tumor-promoting function of YAP1 and TEAD by controlling a remarkable range of cellular processes is undisputed [1, 13, 27], the comprehensive ensemble of direct downstream target genes and the underlying mechanisms of target gene regulation remain poorly understood.
In the past decade, gene expression studies have identified several YAP1-responsive genes [22, 29–31]. In contrast, the number of validated direct target genes remains small. Besides validating YAP1 binding to proximal promoter regions of individual genes using ChIP-qPCR [22, 29, 31–38], a ChIP-on-chip approach using a microarray consisting of promoter regions has been conducted to identify direct YAP1-target genes in MCF10A mammary epithelial cells [22]. While focusing on YAP1-binding to promoter proximal regions a substantial set of functional YAP1 genomic binding sites might have been missed given the importance of distal regulatory elements in establishing a precise pattern of gene expression [39–43].
Here, we comprehensively mapped YAP1 chromatin binding genome-wide, independent of gene location, using ChIP-seq in two human cancer cell lines from different lineages with elevated YAP1 activity (SF268 and NCI-H2052) as well as in non-transformed cells (IMR90) enabling an unbiased identification of YAP1 binding sites and their dependence on cellular context. We demonstrate that YAP1 chromatin recruitment is primarily mediated by binding of TEAD1 to single as well as double TEAD motifs with 3bp spacer at distal enhancers. Aside from presenting a global view of YAP1 and TEAD1 binding in a cancer context, our study also provides novel mechanistic insights into YAP1 transcriptional co-activation of TEAD TFs. We show that YAP1-dependent enhancer activation entails characteristic chromatin changes at lysine 27 of histone H3 and activation of associated genes. Finally we identify a set of YAP1 targets genes by expression profiling following YAP1 knockdown representing a gene signature that can predict YAP1 activity in tumor samples.
Results
Genome-wide YAP1 chromatin-binding in YAP1-amplified cancer cells
To gain insight into YAP1 genomic recruitment in a YAP1-relevant cancer context, we used SF268 glioblastoma cells, previously demonstrated to have elevated YAP1 activity due to a 13-fold genomic amplification of the YAP1 locus [44]. Accordingly, YAP1 mRNA and protein levels are increased in SF268 cells as compared to LN229 glioblastoma cells that do not harbor any genetic aberrations of YAP1/Hippo pathway components (Fig 1A and S1 Fig). As a consequence, YAP1 transcriptional activity appears significantly elevated, as suggested by an increased expression of known YAP1 target genes ANKRD1, CYR61, and NPPB but not of unrelated genes FAM171A1 and HAX1 (Fig 1A).

To identify YAP1 binding sites genome-wide we performed chromatin immunoprecipitation with a YAP1-specific antibody followed by high-throughput sequencing (ChIP-seq). The chosen antibody proved to be highly specific and sensitive as measured by western blot analysis as well as immunoprecipitation (S2 Fig). We observed high reproducibility between two independent biological ChIP-seq replicates with a Pearson correlation coefficient (PCC) of 0.95 (Fig 1B). We identified 2,498 binding sites enriched over matching input using the ChIP-seq peak-finder peakzilla [45] (S1 and S2 Tables). To further benchmark our approach we have analyzed the dataset for the presence of peak regions in the most commonly described YAP1 target genes. As anticipated, peaks were identified in the vicinity of published YAP1 target genes, such as CTGF [22], CYR61 [6], NPPB [32], CCND1 [31], AXL [36], DKK1 [33], ITGB2 [22], WWC1 [35], and ANKRD1 (Fig 1C and S3 Fig). Although ANKRD1 expression is commonly used to monitor YAP1 transcriptional activity, to our knowledge, it has not formerly been proven as a direct YAP1 target gene. Our data proofs direct YAP1 binding to the promoter of ANKRD1 (Fig 1C and 1D). ChIP-qPCR validation for several randomly selected loci confirmed YAP1 occupancy at those sites (Fig 1D), further supporting the specificity of binding and overall reliability of the dataset.
The TEAD consensus motif is enriched in YAP1 binding sites
YAP1 does not contain a DNA-binding domain and thus, relies on interactions with other TFs for recruitment to chromatin. To investigate which TFs mediate binding in SF268 cells we searched the YAP1 peak regions for motifs de novo using MEME [46]. This identified CATTCC, the known consensus motif for TEAD, as the predominant hit (Fig 2A). When allowing for 1 base pair (bp) mismatches to the TEAD consensus motif (S3 Table) we observed that more than 86% of all YAP1 peak regions contained at least one TEAD binding site. This represents a 2.3-fold enrichment over random control regions (hypergeometric P < 10−288) and provides evidence that TEADs are the predominant co-factors facilitating YAP1 association with chromatin in YAP1-amplified glioblastoma cancer cells.

To ask whether additional TFs might recruit YAP1, we searched YAP1 peak regions for enrichment of other known TF motifs. Besides the TEAD consensus motif, we identified only the AP-1/JDP2 motif TGACTCA to be significantly enriched (Fig 2B and S4 and S5 Tables). AP-1 is a heterodimeric protein complex composed of c-Fos and c-Jun, both highly expressed in SF268 cells (S5 Table). Cooperative binding of AP-1 with other TFs has been previously reported as a mechanism of context specific gene regulation [47]. Therefore YAP1/TEAD might act cooperatively with AP-1 in a stimulation-dependent manner or dependent on the pathway genetic context of the analyzed cell type to regulate context-specific gene expression programs. In support of this, c-Fos has recently been described to regulate YAP1 transcriptional activity in the context of KRAS-driven cancers [30] and AP-1/TEAD were found to act as regulators of the invasive gene network in melanoma [48]. When allowing for 1bp mismatches we identified TGACTCA motifs in 60% of YAP1 peak regions that do not contain a TEAD motif but observed the motif as well in 45% of peak regions with a TEAD binding motif. Furthermore we observe that peak regions containing a TEAD binding motif have significantly higher YAP1 ChIP occupancy (as defined by the peakzilla peak score), while the presence of an AP-1 motif does not significantly increase YAP1 occupancy (Fig 2C). Our genome-wide binding data therefore do not provide convincing evidence that AP-1 might serve as an alternative factor for the recruitment of YAP1 to chromatin. However we cannot exclude that AP-1 might serve as a co-factor for YAP1/TEAD under specific experimental conditions.
Taken together, our genome-wide binding data support the notion that TEADs account for the vast majority of YAP1 binding to chromatin.
A double TEAD motif with a 3bp spacer is enriched and functional in YAP1 binding sites
We noted that 52% of peak regions contained more than one TEAD binding motif (Fig 2D) with two consecutive sites (double motif) being particularly prevalent. Binding of TEADs and other TFs to double motifs has been recently shown in vitro using high-throughput SELEX [49]. Indeed, we found a specific enrichment of double motifs oriented in the same direction separated by a 3bp spacer (18%) as compared to other spacing or random control regions (hypergeometric P <10−145 vs. control and P < 10−40 vs. other spacer lengths) (Fig 2E). This is consistent with a cooperative mechanism of TEAD1 binding to DNA that has previously been suggested based on structural analyses [50] and in vitro binding experiments [51, 52].
We observed that peak regions are significantly conserved as compared to random control regions especially at their peak summit (Fig 2F and 2G). Further, in contrast to peaks without a TEAD motif, peaks with single or double motifs had significantly higher ChIP occupancies (Fig 2H).
To directly investigate the functionality of the TEAD double motif we utilized a luciferase reporter gene assay. Double motifs from two independent peak regions (CATTCC-NNN-CATTCC) were cloned upstream of a luciferase reporter. Both constructs caused an increase in luciferase reporter expression as compared to control regions. Importantly, mutations in either one or both of the double motif sites reduced reporter gene expression to the levels of control regions (Fig 2I, red line), indicating that both sites of the double motif are required to enhance transcription.
We conclude that, at a subset of binding sites, TEAD binds homotypic clusters of motifs as previously shown for other human TFs [53].
YAP1 binding sites are co-occupied by TEAD1 genome-wide
The four different TEAD proteins display distinct expression patterns in cultured cell lines even though they have been suggested to be functionally redundant [54]. To establish which TEADs are essential for YAP1-mediated transcriptional activity in SF268 cells, we assessed the expression of the TEAD-dependent MCAT-luciferase reporter upon siRNA-mediated depletion of individual TEADs. This revealed that the depletion of TEAD1 had a potent effect on reporter gene activity, while knockdown of TEAD4 had only marginal effects (Fig 3A).

As TEAD1 appears to be the primary transcriptionally active TEAD family member in SF268 cells, we next mapped its genome-wide binding profile by ChIP-seq (S4 Fig). This led to the identification of 2,652 TEAD1 binding sites based on two independent, but highly reproducible biological replicates and matching input (S1 Table). We first noted a high similarity between TEAD1 and YAP1 ChIP samples, which is reflected in a high positive correlation (PCC = 0.71) (Fig 3B) and a remarkable overlap of 90% with YAP1 peaks regions (Fig 3C and S3 Fig). siRNA-mediated depletion of TEADs strongly reduced TEAD1 binding to all tested loci, thereby confirming the specific binding of TEAD1 to the identified peak regions (Fig 3D and S5 Fig). Reduction of TEADs also reduced YAP1 levels at all tested sites (Fig 3E). This further argues that YAP1 association with chromatin is mainly mediated via TEAD TFs and specifically by TEAD1 in the tested glioblastoma setting. Reciprocally, we observed that the majority of TEAD1 peaks overlap with YAP1 peaks arguing that all TEAD1 binding sites recruit YAP1.
YAP1 and TEAD1 bind and activate distal enhancers
Previous studies focused primarily on the association of YAP1 with proximal promoters [22, 31, 33, 34, 36, 38, 55–57]. It is therefore not surprising that the majority of target genes described up to now contain a YAP1/TEAD1 peak in their promoter region. However, less than 4% of the YAP1/TEAD1 peaks identified in our study are actually located within 2Kb of a gene TSS, and only 15% are located in the 5’UTR of known genes (Fig 4A and S6 Fig). Thus, the majority of YAP1/TEAD1 binding sites reside distal to gene TSSs (Fig 4B, top) providing evidence that YAP1 acts at distal enhancers, which account for a large fraction of regulatory regions [58].
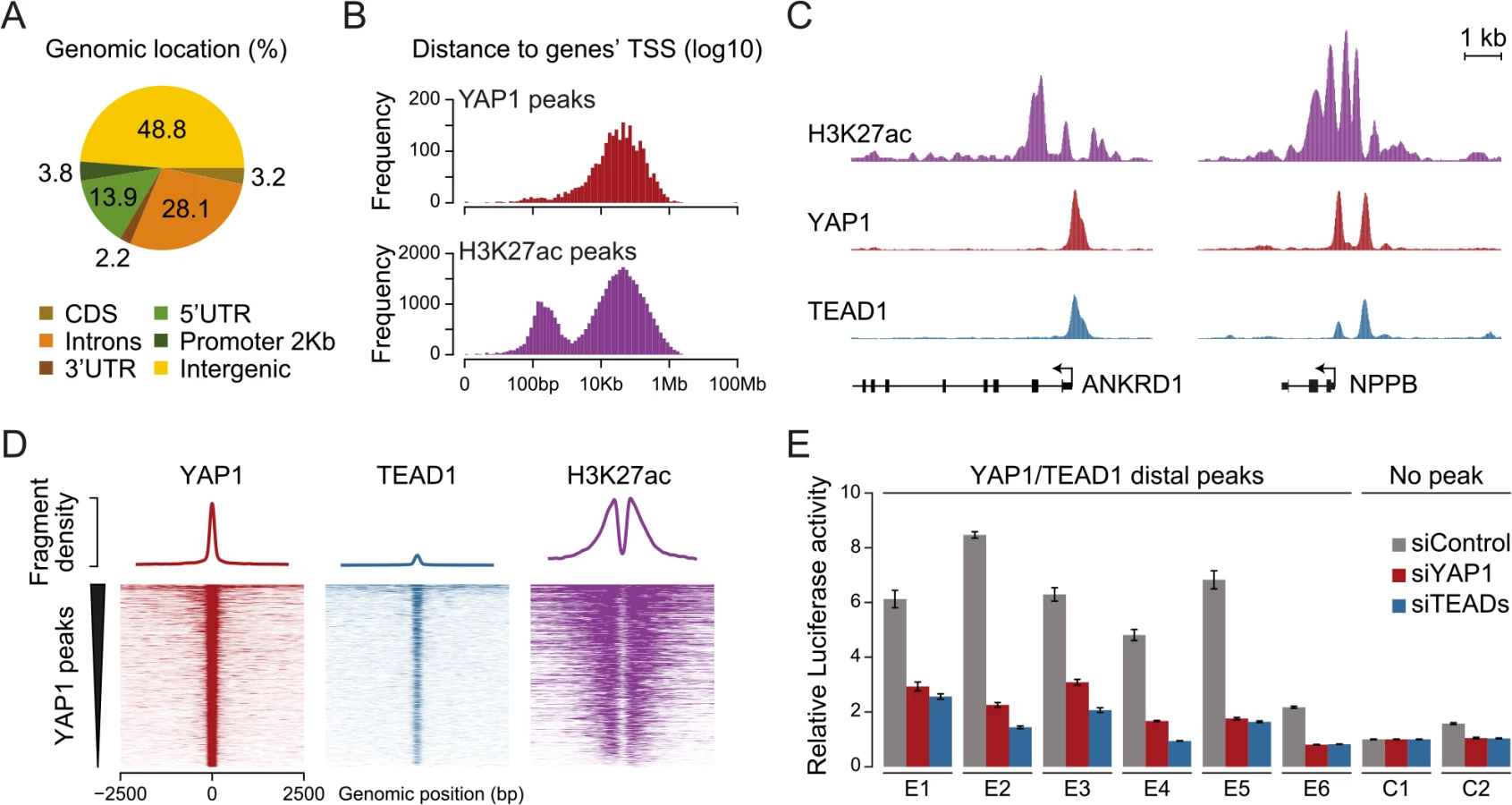
To evaluate whether these distal binding sites occur indeed within functional regions such as enhancers, we took advantage of the fact that acetylation at lysine 27 of histone H3 (H3K27ac) can serve as a signature mark of active enhancers [59, 60]. We performed ChIP-seq in SF268 cells using an H3K27ac-specific antibody in two independent biological replicates and matched input (S1 Table). This identified 38,331 H3K27ac positive regions both proximal and distal to gene TSSs (Fig 4B, bottom). Intersecting this dataset with YAP1 and TEAD1 reveals that 95% of the YAP1/TEAD1 peaks overlap with H3K27ac in particular on nucleosomes flanking YAP1/TEAD1 peaks (Fig 4C and 4D and S3 Fig). Thus, most of YAP1/TEAD1 binding appear to occur within active enhancers and likely represent functional binding events.
To test this hypothesis, we inserted several YAP1/TEAD1 occupied putative enhancer regions into reporter plasmids. In this experiment indeed five out of six tested elements were able to activate the transcription of a luciferase reporter. Notably, siRNA-mediated depletion of YAP1 or TEADs blunted their enhancer activity demonstrating their necessity for proper enhancer function (Fig 4E). These results provide experimental evidence that YAP1/TEAD1 bind primarily at active distal regulatory regions, contributing to enhancer activity.
YAP1/TEAD1 regulate the H3K27ac enhancer chromatin mark
To gain further mechanistic insight into YAP1/TEAD1 transcriptional regulation, we assessed the impact of YAP1 inactivation on TEAD1 chromatin recruitment, target gene expression, and the H3K27ac enhancer chromatin mark. More specifically we took advantage of contact inhibition as a physiological impetus to control YAP1 activity [7]. Although SF268 cells overexpress YAP1, they are nevertheless fully responsive to contact inhibition. When cultivated at high density, YAP1 translocates to the cytoplasm and is degraded as reflected by decreased protein levels (Fig 5A and 5B). This results in reduced target gene expression (Fig 5C) and coincides with diminished YAP1 recruitment (Fig 5D). Interestingly, inactivation of YAP1 also leads to a reduction of TEAD1 expression (Fig 5C), which resulted in reduced cellular TEAD1 protein levels (Fig 5B) and subsequently diminished chromatin occupancy (Fig 5E).

Importantly, YAP1 nuclear depletion also decreases H3K27ac at YAP1/TEAD1 peaks (Fig 5F). This observation appears highly specific since global H3K27ac levels were not affected and regions not bound by YAP1/TEAD1 showed no reduction (Fig 5B and 5F). To gain further mechanistic insight into how YAP1 affects H3K27ac we performed ChIP-qPCR for p300, the major histone acetyltransferase that has been linked to enhancers [61, 62]. This reveals that p300 indeed binds to YAP1 positive H3K27 acetylated sites (Fig 5G). Next we asked if p300 recruitment to these sites is YAP1-dependent by testing p300 occupancy upon YAP1 inactivation under high cell density conditions. This revealed reduced p300 levels mirroring the reduction in H3K27 acetylation. To independently test the link between YAP1 activity and enhancer chromatin, we furthermore depleted YAP1 using siRNA, which similarly led to reduced chromatin binding (S7 Fig). In agreement with YAP1 inactivation by high cell density, siRNA-mediated depletion of YAP1 resulted in diminished H3K27ac levels, p300 occupancy, and reduced TEAD1 expression and chromatin occupancy probably through disruption of a positive feedback loop (S3 and S7 Figs). Together, these data confirm the link between YAP1 chromatin binding and transcriptional activation of target genes and establish a requirement for YAP1 for proper chromatin structure at enhancers.
YAP1 binds similar sites in YAP1-activated cancer cells
Next, we asked if the observed YAP1 binding to distal enhancers is specific for cellular situations with extensive YAP1 amplification such as in SF268 cells. Towards this goal we investigated YAP1 binding in NCI-H2052 malignant mesothelioma cells, a cell line of different lineage and with a different mechanism of YAP1 activation (NF2 mutation, LATS2 deletion) [63]. ChIP-seq analysis of YAP1 in two independent biological replicates and matching input identified 16,470 binding sites (S1 Table). This larger number of peak regions is due to many weak peaks that were not detected in SF268 cells. However, YAP1 binding is well conserved between SF268 and NCI-H2052 cells particularly at strong peaks. This is evident in a global positive correlation (PCC = 0.32) but also at the level of individual loci (Fig 6A and 6B). Indeed 82% of the YAP1 peaks identified in SF268 overlap with peaks in NCI-H2052 cells. As expected, we also identified 1,142 SF268 - and 2,510 NCI-H2052-specific YAP1 binding sites using stringent thresholds but only 48% and 36%, respectively, were assigned to genes not targeted by shared peaks (S8 Fig) suggesting a common function for YAP1 in cancer cells. We also found that YAP1 binds mainly to distal regulatory regions in NCI-H2052 cells (S8 Fig) and that occupied sites were also enriched in TEAD single and double motifs as well as AP-1 motifs (S8 Fig). Similarly to SF268 cells, genetic knockdown of TEADs resulted in reduced YAP1 chromatin binding to NCI-H2052-specific and shared loci with SF268 cells, supporting that TEADs are the main mediators of YAP1 binding also in NCI-H2052 cells (S9 Fig).

Cell-type and context-dependent binding of TFs involves chromatin architecture and epigenetic modifications which are often altered during tumor development [64]. Thus, YAP1 binding in cancer cells might differ from non-transformed cells. To investigate whether YAP1 also binds primarily at TEAD-mediated enhancers in non-transformed cells, we investigated its binding profile in non-transformed lung fibroblast cells (IMR90) as primary cells for which many genomic datasets exist [65, 66]. ChIP-seq analysis of YAP1 in two independent biological replicates and matching input identified 1,111 binding sites (S1 Table). Notably, we found no significant global correlation (PCC = 0.002) of YAP1 binding profiles between SF268 and IMR90 (Fig 6B and 6C). Indeed only 42% of YAP1 peaks in SF268 overlapped peaks in IMR90. This difference in binding also holds true at the gene level (S8 Fig). Despite these differences binding nevertheless takes place predominantly at distal regulatory regions (S8 Fig). Furthermore, cell type-specific binding generates cell type-specific presence of the H3K27ac mark suggesting that those sites are functional (Fig 6D). Finally, YAP1 binding sites in IMR90 are also predominantly enriched in TEAD motifs. Importantly, depletion of TEADs using siRNAs resulted in reduced YAP1 occupancy at cell type-specific and shared loci confirming the general observations made in the cancer cell lines (S9 Fig). Different from the cancer cell lines the consensus motif for forkhead TFs (FOX) is significantly enriched as a secondary motif at IMR90-specific sites (S8D Fig). This might indicate that FOX factors act as cell type-specific contributors to YAP1/TEAD transcriptional regulation. This is compatible with a recent publication that shows a functional interaction of YAP1 and FoxO1 in cardiomyocytes [67]. This however remains challenging to test experimentally due to the fact that more than 20 FOX TFs are expressed in these particular cells that are all predicted to bind to this consensus motif. While it remains to be determined if FOX TFs contribute to cell type-specific TEAD binding our data clearly reveal that also YAP1 binding in non-transformed cells is mainly mediated by TEAD (S8 Fig).
Taken together, these findings indicate that YAP1 binding to enhancers, as well as the presence of double TEAD motifs with a 3bp spacer, are general features of YAP1-mediated transcription in YAP1-activated cancer as well as non-transformed cells even though targeted enhancers can largely differ.
A novel set of YAP1/TEAD1 target genes
The high occurrences of YAP1 binding sites that we identify at distal enhancers suggest that the number of direct YAP1 target genes is much larger than previously anticipated based on studies that focused on promoter regions. While it is undisputed that enhancers are highly relevant for gene activation it remains challenging to correctly assign their target genes due to the fact that enhancers can regulate genes over long distances [68]. Nevertheless, assigning enhancers to the gene in their nearest vicinity is a useful approximation that is correct in the majority of cases [62, 69]. Based on this observation we assigned each YAP1/TEAD1 peak to its nearest gene TSS, yielding 1,738 genes in SF268 cells (S2 Table). In agreement with the notion that YAP1 mainly functions as a transcriptional co-activator this gene set was expressed at a significantly higher level as compared to a random control set (Fig 7A) indicating that the peak-to-gene assignment is overall accurate. Distal YAP1/TEAD1 peaks (over 2Kb away from gene TSS) were assigned to 1580 genes, 52 of which also had a proximal peak. Importantly, the 1528 genes with only distal YAP1/TEAD1 peaks were also more highly expressed than a random control set (Fig 7A). To further, determine the accuracy of the peak to gene assignment we directly tested the expression of 19 randomly selected genes upon YAP1 or TEAD siRNA-mediated depletion (S10 Fig). Gene expression levels were affected in all tested cases arguing for a direct link between binding events and target gene expression.

Gene Ontology (GO) analysis of this gene set showed enrichment for previously reported YAP1 functions such as regulation of cell migration (hypergeometric P < 10−17), extracellular matrix organization (P < 10−15), actin cytoskeleton organization (P < 10−07), and regulation of epithelial cell proliferation (P < 10−6) (Fig 7B and S6 Table). In agreement with recent studies demonstrating a complex interaction network between the YAP1/Hippo and others signaling pathways such as WNT, BMP, TGF-β and PI3K-mTOR [70], our analysis reveals an enrichment of terms associated with signaling (Fig 7B and 7C and S6 Table). We noted that this set of genes contains a number of core components and downstream targets of diverse signaling pathways (S11 Fig). In addition, a number of YAP1/Hippo pathway components including WWC1, LATS, NF2, and AMOT are bound by YAP1/TEAD1. This suggests an extensive feedback mechanism in vertebrates and confirms previous reports in Drosophila [71, 72].
Gene Signature Analysis predicts YAP1 levels in primary tumor samples
Next we set out to determine which genes are transcriptionally activated by YAP1 and performed RNA-seq profiling following YAP1 siRNA-mediated depletion in SF268 cells (Fig 7D and S12 Fig). This identified 219 and 360 genes that were down - or up-regulated by at least 2-fold, respectively, upon YAP1 knockdown compared to control siRNA-treated cells. Among the down-regulated genes, 70 (32%) contained a YAP1/TEAD1 peak assignment in SF268 cells (Fig 7D).
To evaluate the physiological relevance of these YAP1-activated target genes, we sought to predict YAP1 expression in tumor samples using expression data for 528 primary glioblastoma and 279 head and neck squamous cell tumor samples [73]. For each indication, we labeled the samples as YAP1 “high” or “low” expression and divided the datasets into training and test datasets (2/3 and 1/3 of the samples respectively) over 1000 randomized iterations. Using a naïve Bayes classifier this allowed to predict YAP1 expression level with high accuracy (an area under the receiver operating characteristics curve (AUC) = 0.83 for glioblastoma samples and AUC = 0.78 for head and neck squamous tumor samples) (S13 Fig). Feature selection allowed reducing the full YAP1 gene signature to ten genes without losing prediction performance. Hierarchical clustering of the samples shows consistent patterns of expression depending on YAP1 “high” or “low” expression (Fig 7D). This result supports the use of the acquired gene signature to identify YAP1-activated cancers.
Discussion
By providing a comprehensive account of YAP1 genomic binding and its impact on transcription this study establishes that transcriptional regulation of YAP1 target genes is predominantly mediated by TEAD binding to distal enhancers. In addition to demonstrating this mode of regulation we show that this activity entails the establishment of chromatin marks typical to enhancers linking YAP1 activity to H3K27ac. The identified distal regions enabled us to largely expand the set of YAP1 target genes, which we foresee to be a valuable source for functional studies and which we show to have predictive power to identify YAP1-activated cancers.
Due to lack of a DNA-binding domain, YAP1 requires TFs for genomic recruitment. TEAD family members are considered the main TFs for YAP1-mediated regulation of gene expression [22, 23, 25]. In support of a dominant function of TEADs in cancer cells, overexpression of an artificial TEAD2-VP16 construct in NIH3T3 cells was reported to mimic the effects of YAP1 overexpression at the transcriptional level and lead to cell transformation [23]. Furthermore, mutations in YAP1 that prevent binding to TEAD were shown to abolish YAP1-induced transcription and cell transformation in NIH3T3 and MCF10A cells [22].
Here, we comprehensively mapped YAP1 chromatin binding genome-wide in two different cancer cell lines and in non-transformed cells, enabling an unbiased assessment of the sequence features that direct YAP1-mediated regulation. Our genome-wide map of TEAD1 binding sites revealed that the vast majority of YAP1 binding sites were co-occupied by TEAD1 confirming the dominant role of TEAD factors in the control of YAP1 transcriptional activity. To the best of our knowledge this is the first study demonstrating a genome-wide co-occupancy of both factors in cancer cells. Our data extends the results from a previous ChIP-on-chip study that used a promoter specific microarray and demonstrated a comparable overlap of >80% for YAP1 and TEAD1 binding around start sites in MCF10A mammary epithelial cells [22].
Despite the major role of TEADs to mediate YAP1 co-activator activity, additional TFs are described to interact with YAP1 (reviewed in [12]). Our data, however, do not provide evidence for the importance of additional TFs in targeting YAP1 chromatin binding. We demonstrate that this finding is not just limited to a cellular situation where YAP1 is amplified since we observe a similar predominant enrichment of TEAD motifs in YAP1 peak regions in a YAP1-activated NCI-H2052 cancer cell line as well as in non-transformed IMR90 cells.
TEAD1 also referred to as TEF-1, for transcription enhancer factor 1, was first cloned in HeLa cells as an activator of the simian virus 40 (SV40) “enhancer”, which is a short 72bp sequence element that is a component of the viral early promoter [54, 74, 75]. So far, however, binding and function at endogenous elements that by the current definition of an enhancer act distal to promoters had not been investigated.
Our genome-wide analysis of YAP1/TEAD1 binding indicates that the vast majority of endogenous sites, in cancer and non-cancer cells (SF268, NCI-H2052 and IMR90), are actually located within distal regulatory regions representing enhancer elements. This mimics the typical distribution of sequence-specific TFs and is in line with the concept that distal TF and co-activator binding are key determinants of enhancer activity and in turn cell-type specific gene expression patterns [39, 68, 76]. Recent efforts in mapping enhancers in different tissues revealed that the human genome contains up to several hundred thousand distal regulatory regions, most of which are cell-type specific [77]. Their misregulation can be highly disease relevant since mutations in these regions have extensively been associated with disease susceptibility [78].
Distal binding has not been reported for Yki (Yorki) the Drosophila homolog of YAP1 and it remains open if this reflects a functional difference or the organization of the smaller and gene denser fly genome [57, 79]. Similarly, enhancer binding of Yap1 has not been reported in mouse embryonic stem cells [55]. However, when reanalyzing an available list of Yap1 peak regions from Lin et al., we observed that a large fraction of these Yap1 binding sites are located in regions distal from promoters. In support of our data two recent reports demonstrated that YAP1/TEAD regulate transcription by binding to distal enhancers [67, 80]. Together this argues that YAP1 distal binding is a general feature of YAP1/TEAD-driven transcription activation also in non-transformed cells and is not an acquired feature of cancer cells.
Distinct chromatin modifications are associated with various aspects of gene expression. In particular H3K27ac was found to be an effective means to determine enhancer activity [59, 81]. Our data show that the vast majority of YAP1 binding sites overlap H3K27ac positive regions and that cell-type specific YAP1 sites match cell type specific H3K27ac regions. Interestingly we show that, in SF268 cells, YAP1 chromatin association is a prerequisite for the deposition of H3K27ac supporting the fact that YAP1 binding sites represent functional enhancers.
Interestingly, genome-wide binding analysis in Drosophila revealed a correlation between Yki chromatin binding and trimethylation of H3K4 (H3K4me3) [57]. Consistent with this finding, nuclear receptor coactivator 6 (Ncoa6), a subunit of the Trithorax-related H3K4 methyltransferase complex, has been identified as a Yki binding protein that is required for transcriptional regulation [79]. Importantly, besides the H3K4 methyltransferases, the mammalian Ncoa6 has been reported to enhance the activity of TFs by interacting with histone acetyltransferases CBP/p300 [82]. Whether this NCOA6 function possibly facilitates YAP1-dependent acetylation of H3K27 (H3K27ac) and which additional cofactors are recruited to trigger transcriptional activity warrants further investigations.
The determination of genes targeted by specific enhancers remains a challenge. We observed that only a minority of genes nearest to binding sites was transcriptionally affected by depletion of YAP1. Notably however, only 10–25% of TF binding events in higher eukaryotes contribute to the expression of the closest proximal gene in any given cell type. This is likely to be an underestimate given the nature of enhancers and the complexity of transcription regulatory networks [76]. Besides the uncertainty of assigning binding sites to target genes, enhancers function in a modular manner such that they contribute additively and redundantly to the expression of their target genes [76, 83]. Therefore YAP1/TEAD contribution to transcriptional activity might not be apparent at many target genes.
In addition to shedding light on basic principles of YAP1 transcriptional regulation, the identification of distal regulation as the primary means of YAP1 transcriptional control enabled us to identify an extended list of target genes based on both YAP1 chromatin binding and gene expression changes. This novel YAP1 signature from YAP1-amplified glioblastoma cells should have predictive potential for the identification of YAP1-dependent tumors.
Materials and Methods
Cell culture and transfections
SF268 cells (NCI DCTD tumor/cell line repository) were maintained in RPMI 1640 Medium, GlutaMAX supplement, 25 mM HEPES, 10% (v/v) fetal calf serum and 1 mM sodium pyruvate. NCI-H2052 cells (ATCC) were maintained in RPMI, 10% (v/v) fetal calf serum, 1% (v/v) non-essential amino acids, 1 mM sodium pyruvate. IMR90 cells (ATCC) were maintained in EMEM (Sigma M 4655) supplemented with 1% non-essential amino acids (NEAA) and 10% FBS. All media and supplements were from Life Technologies. To obtain low density (LD) or high density (HD) cultures, SF268 cells were plated at 10.000 cells/cm2 or 100.000 cells/cm2, respectively and harvested 48h or 96 hours after seeding. Transient transfections of SF268, IMR90 and NCI-H2052 cells with siRNA (final concentration: 25 nM) were performed using Lipofectamine RNAiMAX (Life Technologies). Transient transfection of SF268 cells with plasmid DNA was performed using Cell Avalanche Transfection Reagent (EZ Biosystems). Cells were harvested at 48 hours or 72 hours post-transfection.
Antibodies
The following antibodies were used for western blot, immunoprecipitation and chromatin immunoprecipitation (ChIP): anti-YAP1 [EP1674Y] (ab52771), anti-KAT3B/p300 (ab14984), and anti-H3 (ab1791) from Abcam; anti-TEAD1 (610922) from BD Transduction Laboratories; anti-TEAD4 (ARP33426_P050) from Aviva Biosystems; anti-β-Actin (A2066) from Sigma-Aldrich; anti-H3K27ac (AM 39133) from Active Motif. YAP1 and TEAD1 antibodies for ChIP-seq were characterized in western blot, immunoprecipitation and ChIP-qPCR (S2 and S4 Figs).
Chromatin immunoprecipitation (ChIP)
ChIP was essentially carried out as previously described [84], with slight modifications. Chromatin was sonicated for 14 minutes using a Covaris E210 (Settings: 5% duty cycle, intensity 4). 60μg of chromatin were incubated over night at 4°C with 5μg of the corresponding antibody and for 2 hours with preblocked (tRNA, BSA) Dynabeads protein G. DNA was purified using the Minielute PCR purification kit (Qiagen).
ChIP-qPCR
Quantitative PCR was performed using Maxima SYBR Green / ROX qPCR Master Mix (Thermo Scientific) and the ViiA 7 Real-Time PCR System (Life Technologies) and 1/80th of the ChIP sample or 0.01% of input chromatin per PCR, respectively. Amplifications were performed in triplicate, and mean values were expressed as percentage input. Standard deviation was calculated from the triplicates, and error bars are indicated accordingly. Primers are listed in Table 1 and Table 2.


ChIP-seq
YAP1, TEAD1 and H3K27ac ChIPs from SF268 cells and YAP1 ChIPs from NCI-H2052 and IMR90 cells were subjected to high-throughput sequencing on a 356R Illumina HiSeq 2500 sequencer using standard NEB library preparation kits and protocols.
ChIP-seq data processing
Additional ChIP-seq dataset for H3K27ac in IMR90 cells was obtained from the Gene Expression Omnibus under the accession number GSM469967. We mapped the ChIP-seq sequencing reads (single-end, 50bp) to the human reference genome (hg19 only chromosomes 1 to 22, X, Y and M) using bowtie [85] version 1.0.0 with parameters-v 3-m 1—best—strata. We extended the reads to 150bp (average estimated fragment length) and calculated for each genomic position the read density normalized to one million reads in the library to generate wiggle files. Genome screenshots were taken using the UCSC genome browser [86].
Peak calling and overlap
We identified peaks in YAP1 and TEAD1 ChIP samples compared to the corresponding input samples using peakzilla [45] and in H3K27ac ChIP using MACS [87] version 1.4.2. The strategy used to define and overlap peak regions is described in [88] and S1 Table. We defined control peak regions by shuffling the peaks randomly within the same chromosome. We calculated the Pearson correlation coefficient (PCC) and plotted scatterplots between two samples using the mean fragment density of each peak region from all samples. Differentially bound regions were identified with the R package DESeq [89] using an adjusted p-value threshold of 10−5 and a 2-fold enrichment with enrichment in the reference sample below 100 normalized reads per kilobase.
Motif enrichment analysis
We searched for motif de novo using MEME [46] within 31bp around peak summits and for occurrences of the known motifs from Jaspar [90], and [49] using MAST [91] (from the MEME suite programs version 4.1.1) with a P-value of 10−3 in an area of 151bp (average genomic fragment length) around each peak summit.
Functional analyses
We assigned each peak to its closest gene transcriptional start site (TSS) using the reference transcriptome (GRCh37.71). For each gene ontology biological processes [92] and WikiPathways [93], we calculated the enrichment and associated hypergeometric P-values of genes in each class compared to all genes. We calculated the conservation rate of regions using the PhastCons 46 way placental mammals [94].
RT-qPCR
RNA was isolated using RNeasy Mini Kit (Qiagen) and cDNA synthesis was performed using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems). cDNA was subjected to quantitative PCR (qPCR) analysis in triplicates with gene-specific primers (see Table 3) using Maxima SYBR Green / ROX qPCR Master Mix (Thermo Scientific) and the ViiA 7 Real-Time PCR System (Life Technologies).

RNA-seq
Total RNA of three biological replicates was extracted from SF268 cells 48h after transfection with two individual siRNAs targeting YAP1 and unspecific control siRNAs using the Total RNA purification kit from Norgen Biotek. RNAseq libraries were prepared using the Illumina TruSeq RNA Sample Prep kit v2 and sequenced using the Illumina HiSeq2500 platform (76-bp paired-end reads).
RNA-seq data processing
Additional RNA-seq datasets for SF268 and LN229 cells were obtained from the Cancer Genomics Hub (https://browser.cghub.ucsc.edu). We mapped the RNA-seq sequencing reads (paired-end, 100bp) to the human reference transcriptome (GRCh37.71) using tophat [95] version 1.3.1 with parameter—no-novel-juncs. We calculated genes FPKMs (fragments per kilobase of transcript per million mapped reads) using cufflinks [96] version 2.0.2 with parameter-G using the reference transcriptome (GRCh37.71). Differentially expressed genes in YAP1 knockdown were identified with the R package DESeq [89]. Genes either down - or up-regulated were selected using an adjusted p-value threshold of 10−5 in all four pairwise comparisons of YAP1 and control siRNA treated samples and an at least 2-fold enrichment in one comparison and at least 1.2-fold in the other three.
Luciferase reporter assays
GeneArt Strings DNA fragments encompassing approximately 200bp of six distal enhancers bound by YAP1/TEAD1 (see Table 4) and two negative regions carrying BglII restriction sites were cloned into pGL3 promoter vector (E1761, Promega) upstream of the luciferase gene with SV40 minimal promoter. For two regions mutations were introduced in either one or both motif sites of the double TEAD motif with 3bp spacer (see Table 5). One day prior transfection SF268 cells were plated on 384-well plates (1800 cells/well). Cells were co-transfected with 28.5 ng of the respective reporter constructs and 1.5 ng pRenilla.


For luciferase assays in YAP1 and TEADs-depleted cells, SF268 cells were transfected with the indicated siRNAs (see Table 6) at the day of seeding (1800 cells/well) in 384-well plates. The next day, the medium was changed and cells were transfected with DNA (pGL3 reporter constructs and pRenilla). Firefly and Renilla luminescence signals were measured at 24 hours after DNA transfection using Dual-Glo luciferase assay system (Promega). Firefly luminescence signals were normalized according to their corresponding Renilla signals resulting in relative luciferase activity. Each sample was transfected in triplicate, and each experiment was repeated independently at least three times.

SF268 cells stably expressing the MCAT-Luc YAP1/TEAD responsive reporter [44] were transfected with siRNAs targeting YAP1, TEAD1, TEAD2, TEAD3, and TEAD4 with 8 siRNAs per gene (see Table 7). At 72 hours after transfection medium was aspirated and cells were incubated with fresh medium containing 1.4μM resazurin (SIGMA; MO, USA) for 2 hours before measuring fluorescence (Ex: 540 nm, Em: 590 nm) as a read-out for cell viability. Subsequently the cells were lysed in fresh medium containing 1 : 10 (v/v) Steady-Glo luciferase assay reagent (Promega; WI, USA). Luciferase measurements were taken according to the manufacturer's protocol. Fold change in MCAT-Luc reporter activity was calculated by normalizing luminescence signal to resazurin and to negative control siRNA. Each experiment was carried out in triplicate.

Protein isolation and western blot analysis
SF268 cells were lysed in FT lysis buffer (20 mM Tris / HCl at pH 7.8, 600 mM NaCl, 20% glycerol, proteinase inhibitor), and proteins including histones were extracted by repeated freeze-thaw cycles followed by Benzonase (Novagen) treatment. Lysates were separated using Novex NuPAGE SDS-PAGE gel system transferred to Immobilon-P membranes (Millipore) and subjected to immunoblotting.
Immunofluorescence
Cells were fixed with 4% PFA (Paraformaldehyde 20% solution, EM grade #15713-S) for 15 minutes at room temperature. Subsequently cells were washed 1x PBS and permeabilized in PBS / 0.1% Triton X-100 at room temperature for 10 minutes. Cells were rinsed in PBS and incubated with anti-YAP1 diluted 1 : 300 in PBS / 1.5% BSA over-night at 4°C. Cells were washed with PBS and incubated with the secondary antibody, anti-rabbit Alexa647 (1 : 1000, Life Technologies) and Hoechst (1 : 10.000) for DNA staining for 2 hours at room temperature. After washing with PBS, staining was analyzed by fluorescence microscopy (Operetta, Perkin Elmer), 20x objective.
Expression analysis in tumor samples
We used as gene signature 70 genes that were 2-fold down-regulated in YAP1 depleted SF268 cells and had a YAP1/TEAD1 binding peak in their vicinity. Expression data were collected from cBioPortal [73, 97] for 528 primary glioblastoma and 279 Head and neck squamous cell tumor samples generated by the TCGA Research Network. We used either all or the top and bottom 10% of samples according to the ranksum for YAP1 expression and copy number. Over 1000 iterations we randomly divided the datasets into training and test subsets (2/3 and 1/3 respectively) and used a naïve Bayes predictor from the Bioconductor package e1071 to predict the YAP1 expression level (“high” or “low”). Prediction accuracy was measured using recall statistics and receiver-operating-characteristic (ROC) curves and performance statistics were generated using the ROCR package [98].
Accession numbers
Raw and processed ChIP-seq data are deposited in the Gene Expression Omnibus (GEO) under the accession number GSE61852. The raw RNA-seq reads are available in the NCBI Short Read Archive under the accession number SRP056665.
Supporting Information
Zdroje
1. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011 20951342
2. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–57. doi: 10.1038/nrc3458 23467301
3. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13(8):877–83. doi: 10.1038/ncb2303 21808241
4. Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. 2014;127(Pt 4):709–17. doi: 10.1242/jcs.140103 24532814
5. Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283(9):5496–509. 18158288
6. Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68(8):2789–94. doi: 10.1158/0008-5472.CAN-07-6205 18413746
7. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61. 17974916
8. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122(3):421–34. 16096061
9. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17(23):2054–60. 17980593
10. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–33. 17889654
11. Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell. 2014;26(2):273–87. doi: 10.1016/j.ccr.2014.05.029 25087979
12. Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014;15(6):642–56. doi: 10.15252/embr.201438638 24825474
13. Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24(9):862–74. doi: 10.1101/gad.1909210 20439427
14. Lorenzetto E, Brenca M, Boeri M, Verri C, Piccinin E, Gasparini P, et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget. 2014;5(9):2608–21. 24810989
15. Hu X, Xin Y, Xiao Y, Zhao J. Overexpression of YAP1 is Correlated with Progression, Metastasis and Poor Prognosis in Patients with Gastric Carcinoma. Pathol Oncol Res. 2014.
16. Liu JY, Li YH, Lin HX, Liao YJ, Mai SJ, Liu ZW, et al. Overexpression of YAP 1 contributes to progressive features and poor prognosis of human urothelial carcinoma of the bladder. BMC Cancer. 2013;13 : 349. doi: 10.1186/1471-2407-13-349 23870412
17. Song M, Cheong JH, Kim H, Noh SH. Nuclear expression of Yes-associated protein 1 correlates with poor prognosis in intestinal type gastric cancer. Anticancer Res. 2012;32(9):3827–34. 22993325
18. Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010;101(5):1279–85. doi: 10.1111/j.1349-7006.2010.01511.x 20219076
19. Wang Y, Xie C, Li Q, Xu K, Wang E. Clinical and prognostic significance of Yes-associated protein in colorectal cancer. Tumour Biol. 2013;34(4):2169–74. doi: 10.1007/s13277-013-0751-x 23558963
20. Xia Y, Chang T, Wang Y, Liu Y, Li W, Li M, et al. YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PLoS One. 2014;9(3):e91770. doi: 10.1371/journal.pone.0091770 24622501
21. Kodaka M, Hata Y. The mammalian Hippo pathway: regulation and function of YAP1 and TAZ. Cell Mol Life Sci. 2014.
22. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–71. doi: 10.1101/gad.1664408 18579750
23. Ota M, Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008;135(24):4059–69. doi: 10.1242/dev.027151 19004856
24. Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14(3):388–98. doi: 10.1016/j.devcel.2008.01.007 18258486
25. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15(10):1229–41. 11358867
26. Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell. 2008;14(3):377–87. doi: 10.1016/j.devcel.2008.01.006 18258485
27. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109(37):E2441–50. doi: 10.1073/pnas.1212021109 22891335
28. Tian W, Yu J, Tomchick DR, Pan D, Luo X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc Natl Acad Sci U S A. 2010;107(16):7293–8. doi: 10.1073/pnas.1000293107 20368466
29. Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, et al. Switch enhancers interpret TGF-beta and Hippo signaling to control cell fate in human embryonic stem cells. Cell reports. 2013;5(6):1611–24. doi: 10.1016/j.celrep.2013.11.021 24332857
30. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell. 2014.
31. Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31(49):5117–22. doi: 10.1038/onc.2012.5 22286761
32. Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288(6):3977–88. doi: 10.1074/jbc.M112.436311 23275380
33. Seo E, Basu-Roy U, Gunaratne PH, Coarfa C, Lim DS, Basilico C, et al. SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage. Cell reports. 2013;3(6):2075–87. doi: 10.1016/j.celrep.2013.05.029 23791527
34. Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11(12):1444–50. doi: 10.1038/ncb1993 19935651
35. Xiao L, Chen Y, Ji M, Dong J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem. 2011;286(10):7788–96. doi: 10.1074/jbc.M110.173468 21233212
36. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30(10):1229–40. doi: 10.1038/onc.2010.504 21076472
37. Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y, et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013;73(12):3615–24. doi: 10.1158/0008-5472.CAN-12-3793 23576552
38. Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, et al. Hippo coactivator YAP1 upregulates SOX9 and endows stem-like properties to esophageal cancer cells. Cancer Res. 2014.
39. Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144(3):327–39. doi: 10.1016/j.cell.2011.01.024 21295696
40. Sakabe NJ, Savic D, Nobrega MA. Transcriptional enhancers in development and disease. Genome Biol. 2012;13(1):238. doi: 10.1186/gb-2012-13-1-238 22269347
41. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. doi: 10.1016/j.cell.2013.09.053 24119843
42. Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336(6082):736–9. doi: 10.1126/science.1217277 22499810
43. Atchison ML. Enhancers: mechanisms of action and cell specificity. Annu Rev Cell Biol. 1988;4 : 127–53. 2848550
44. Michaloglou C, Lehmann W, Martin T, Delaunay C, Hueber A, Barys L, et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS One. 2013;8(4):e61916. doi: 10.1371/journal.pone.0061916 23613971
45. Bardet AF, Steinmann J, Bafna S, Knoblich JA, Zeitlinger J, Stark A. Identification of transcription factor binding sites from ChIP-seq data at high resolution. Bioinformatics. 2013;29(21):2705–13. doi: 10.1093/bioinformatics/btt470 23980024
46. Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):W369–73. 16845028
47. Bottomly D, Kyler SL, McWeeney SK, Yochum GS. Identification of {beta}-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010;38(17):5735–45. doi: 10.1093/nar/gkq363 20460455
48. Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nature communications. 2015;6 : 6683. doi: 10.1038/ncomms7683 25865119
49. Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152(1–2):327–39. doi: 10.1016/j.cell.2012.12.009 23332764
50. Anbanandam A, Albarado DC, Nguyen CT, Halder G, Gao X, Veeraraghavan S. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc Natl Acad Sci U S A. 2006;103(46):17225–30. 17085591
51. Jiang SW, Desai D, Khan S, Eberhardt NL. Cooperative binding of TEF-1 to repeated GGAATG-related consensus elements with restricted spatial separation and orientation. DNA Cell Biol. 2000;19(8):507–14. 10975468
52. Halder G, Carroll SB. Binding of the Vestigial co-factor switches the DNA-target selectivity of the Scalloped selector protein. Development. 2001;128(17):3295–305. 11546746
53. Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I. Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res. 2010;20(5):565–77. doi: 10.1101/gr.104471.109 20363979
54. Jacquemin P, Hwang JJ, Martial JA, Dolle P, Davidson I. A novel family of developmentally regulated mammalian transcription factors containing the TEA/ATTS DNA binding domain. J Biol Chem. 1996;271(36):21775–85. 8702974
55. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010;24(11):1106–18. doi: 10.1101/gad.1903310 20516196
56. Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A. 2011;108(6):2270–5. doi: 10.1073/pnas.1019603108 21262812
57. Oh H, Slattery M, Ma L, Crofts A, White KP, Mann RS, et al. Genome-wide association of Yorkie with chromatin and chromatin-remodeling complexes. Cell reports. 2013;3(2):309–18. doi: 10.1016/j.celrep.2013.01.008 23395637
58. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232 22955617
59. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–6. doi: 10.1073/pnas.1016071107 21106759
60. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–83. doi: 10.1038/nature09692 21160473
61. Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–8. 17277777
62. Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–8. doi: 10.1038/nature07730 19212405
63. Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011;71(3):873–83. doi: 10.1158/0008-5472.CAN-10-2164 21245096
64. Dowell RD. Transcription factor binding variation in the evolution of gene regulation. Trends Genet. 2010;26(11):468–75. doi: 10.1016/j.tig.2010.08.005 20864205
65. Nichols WW, Murphy DG, Cristofalo VJ, Toji LH, Greene AE, Dwight SA. Characterization of a new human diploid cell strain, IMR-90. Science. 1977;196(4285):60–3. 841339
66. Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell stem cell. 2010;6(5):479–91. doi: 10.1016/j.stem.2010.03.018 20452322
67. Morikawa Y, Zhang M, Heallen T, Leach J, Tao G, Xiao Y, et al. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Science signaling. 2015;8(375):ra41. doi: 10.1126/scisignal.2005781 25943351
68. Schwarzer W, Spitz F. The architecture of gene expression: integrating dispersed cis-regulatory modules into coherent regulatory domains. Curr Opin Genet Dev. 2014;27 : 74–82. doi: 10.1016/j.gde.2014.03.014 24907448
69. Sikora-Wohlfeld W, Ackermann M, Christodoulou EG, Singaravelu K, Beyer A. Assessing computational methods for transcription factor target gene identification based on ChIP-seq data. PLoS Comput Biol. 2013;9(11):e1003342. doi: 10.1371/journal.pcbi.1003342 24278002
70. Mauviel A, Nallet-Staub F, Varelas X. Integrating developmental signals: a Hippo in the (path)way. Oncogene. 2012;31(14):1743–56. doi: 10.1038/onc.2011.363 21874053
71. Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev Cell. 2010;18(2):300–8. doi: 10.1016/j.devcel.2009.12.011 20159599
72. Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8(1):27–36. 16341207
73. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088 23550210
74. Davidson I, Xiao JH, Rosales R, Staub A, Chambon P. The HeLa cell protein TEF-1 binds specifically and cooperatively to two SV40 enhancer motifs of unrelated sequence. Cell. 1988;54(7):931–42. 2843293
75. Xiao JH, Davidson I, Matthes H, Garnier JM, Chambon P. Cloning, expression, and transcriptional properties of the human enhancer factor TEF-1. Cell. 1991;65(4):551–68. 1851669
76. Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nature reviews Genetics. 2012;13(9):613–26. doi: 10.1038/nrg3207 22868264
77. Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–61. doi: 10.1038/nature12787 24670763
78. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794 22955828
79. Qing Y, Yin F, Wang W, Zheng Y, Guo P, Schozer F, et al. The Hippo effector Yorkie activates transcription by interacting with a histone methyltransferase complex through Ncoa6. eLife. 2014;3.
80. Cebola I, Rodriguez-Segui SA, Cho CH, Bessa J, Rovira M, Luengo M, et al. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat Cell Biol. 2015;17(5):615–26. doi: 10.1038/ncb3160 25915126
81. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–12. doi: 10.1038/nature07829 19295514
82. Mahajan MA, Samuels HH. Nuclear receptor coactivator/coregulator NCoA6(NRC) is a pleiotropic coregulator involved in transcription, cell survival, growth and development. Nuclear receptor signaling. 2008;6:e002. doi: 10.1621/nrs.06002 18301782
83. Biggin MD. Animal transcription networks as highly connected, quantitative continua. Dev Cell. 2011;21(4):611–26. doi: 10.1016/j.devcel.2011.09.008 22014521
84. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–66. 17334365
85. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25 19261174
86. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. 12045153
87. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137 18798982
88. Bardet AF, He Q, Zeitlinger J, Stark A. A computational pipeline for comparative ChIP-seq analyses. Nat Protoc. 2012;7(1):45–61.
89. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106 20979621
90. Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32(Database issue):D91–4. 14681366
91. Bailey TL, Gribskov M. Methods and statistics for combining motif match scores. J Comput Biol. 1998;5(2):211–21. 9672829
92. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. 10802651
93. Kelder T, van Iersel MP, Hanspers K, Kutmon M, Conklin BR, Evelo CT, et al. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2012;40(Database issue):D1301–7. doi: 10.1093/nar/gkr1074 22096230
94. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50. 16024819
95. Trapnell C, Salzberg SL. How to map billions of short reads onto genomes. Nat Biotechnol. 2009;27(5):455–7. doi: 10.1038/nbt0509-455 19430453
96. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5. doi: 10.1038/nbt.1621 20436464
97. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095 22588877
98. Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–1. 16096348
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in