
HIV gp41 Engages gC1qR on CD4+ T Cells to Induce the Expression of an NK Ligand through the PIP3/H2O2 Pathway
CD4+ T cell loss is central to HIV pathogenesis. In the initial weeks post-infection, the great majority of dying cells are uninfected CD4+ T cells. We previously showed that the 3S motif of HIV-1 gp41 induces surface expression of NKp44L, a cellular ligand for an activating NK receptor, on uninfected bystander CD4+ T cells, rendering them susceptible to autologous NK killing. However, the mechanism of the 3S mediated NKp44L surface expression on CD4+ T cells remains unknown. Here, using immunoprecipitation, ELISA and blocking antibodies, we demonstrate that the 3S motif of HIV-1 gp41 binds to gC1qR on CD4+ T cells. We also show that the 3S peptide and two endogenous gC1qR ligands, C1q and HK, each trigger the translocation of pre-existing NKp44L molecules through a signaling cascade that involves sequential activation of PI3K, NADPH oxidase and p190 RhoGAP, and TC10 inactivation. The involvement of PI3K and NADPH oxidase derives from 2D PAGE experiments and the use of PIP3 and H2O2 as well as small molecule inhibitors to respectively induce and inhibit NKp44L surface expression. Using plasmid encoding wild type or mutated form of p190 RhoGAP, we show that 3S mediated NKp44L surface expression on CD4+ T cells is dependent on p190 RhoGAP. Finally, the role of TC10 in NKp44L surface induction was demonstrated by measuring Rho protein activity following 3S stimulation and using RNA interference. Thus, our results identify gC1qR as a new receptor of HIV-gp41 and demonstrate the signaling cascade it triggers. These findings identify potential mechanisms that new therapeutic strategies could use to prevent the CD4+ T cell depletion during HIV infection and provide further evidence of a detrimental role played by NK cells in CD4+ T cell depletion during HIV-1 infection.
Published in the journal:
. PLoS Pathog 6(7): e32767. doi:10.1371/journal.ppat.1000975
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000975
Summary
CD4+ T cell loss is central to HIV pathogenesis. In the initial weeks post-infection, the great majority of dying cells are uninfected CD4+ T cells. We previously showed that the 3S motif of HIV-1 gp41 induces surface expression of NKp44L, a cellular ligand for an activating NK receptor, on uninfected bystander CD4+ T cells, rendering them susceptible to autologous NK killing. However, the mechanism of the 3S mediated NKp44L surface expression on CD4+ T cells remains unknown. Here, using immunoprecipitation, ELISA and blocking antibodies, we demonstrate that the 3S motif of HIV-1 gp41 binds to gC1qR on CD4+ T cells. We also show that the 3S peptide and two endogenous gC1qR ligands, C1q and HK, each trigger the translocation of pre-existing NKp44L molecules through a signaling cascade that involves sequential activation of PI3K, NADPH oxidase and p190 RhoGAP, and TC10 inactivation. The involvement of PI3K and NADPH oxidase derives from 2D PAGE experiments and the use of PIP3 and H2O2 as well as small molecule inhibitors to respectively induce and inhibit NKp44L surface expression. Using plasmid encoding wild type or mutated form of p190 RhoGAP, we show that 3S mediated NKp44L surface expression on CD4+ T cells is dependent on p190 RhoGAP. Finally, the role of TC10 in NKp44L surface induction was demonstrated by measuring Rho protein activity following 3S stimulation and using RNA interference. Thus, our results identify gC1qR as a new receptor of HIV-gp41 and demonstrate the signaling cascade it triggers. These findings identify potential mechanisms that new therapeutic strategies could use to prevent the CD4+ T cell depletion during HIV infection and provide further evidence of a detrimental role played by NK cells in CD4+ T cell depletion during HIV-1 infection.
Introduction
CD4 cell depletion and the resulting immune dysfunction are hallmarks of HIV-1 infection. Although this phenomenon appears to be the principal component of HIV disease, its underlying mechanisms remain controversial. The CD4+ T cell loss observed during the chronic phase of infection was initially thought to result from the death of productively infected CD4+ T cells, due to the cytopathic effects of viral replication and cytotoxic CD8-mediated killing [1], [2]. Several studies have demonstrated, however, that most dying cells are uninfected and thus suggest that the death of uninfected bystander CD4+ T cells plays a major role in CD4 depletion [1], [3]. Since then, numerous studies have strengthened this hypothesis. Several HIV-1 proteins, including gp120, Tat, and Nef, activate a variety of disparate pathways to initiate apoptosis in uninfected cells [4]. An alternative proposal is that the high level of immune activation in HIV-infected individuals may cause the killing of uninfected CD4+ T cells, by activation-induced cell death (AICD) through Fas [5]. The importance of CD4+ T cell preservation in this disease is highlighted by findings in African green monkeys infected with SIVagm: despite high viral replication, their CD4+ T cell counts remain normal, and they do not develop AIDS [6].
Increasing evidence suggests that NK cells are involved in CD4+ T cell depletion. We have shown that after HIV infection a significant fraction of the CD4-cell subset expresses NKp44L, an activator ligand of the natural cytotoxicity receptor NKp44 [7]. NKp44L expression renders CD4+ T cells sensitive to autologous NK lysis. This expression is strongly correlated with both the decline in the CD4 cell count and the increase in viral load [8]. The role of NKp44L in CD4+ T cell depletion was confirmed in vivo in an SHIV-infected macaque model [9]. Ward et al. [10] demonstrated that the ligand for NKp44 is expressed only on uninfected bystander CD4+ T cells. More recently, we showed that Nef, one of the main effector proteins involved in HIV immune evasion, mediates intracellular retention of NKp44L in HIV-infected CD4+ T cells [11]. These results suggest that HIV-1 has acquired the ability to use NK cells to disarm the host immune system by selectively triggering the death of uninfected CD4+ T cells.
We have previously showed that a specific motif of HIV-gp41, which is called 3S and is highly conserved in HIV-1 isolates, plays a pivotal role in NKp44L expression [8]. HIV-gp41 protein is a subunit of the Env complex protein and is essential for HIV entry into target cells [12]. The interaction of gp120 with the target cell surface induces conformational changes in the gp120-gp41 complex that expose the gp41 molecules and appear to promote fusion between the viral and cellular membranes [13]. During this process, several gp41 motifs become accessible to the surface, including the 3S motif located between residues 618–623 [8], [14]. Studies in a macaque model have confirmed the important role of this 3S motif in HIV-1 pathogenicity and showed that immunization with the 3S motif prevents NKp44L expression and CD4+ T cell depletion following SHIV infection [15].
Interestingly, it has been reported that the HIV-gp41 motif located between residues 601–620 binds to the complement component 1q (C1q) and then activates the classic complement cascade in the absence of antibody [16]. In addition, the gp41 epitope between residues 615–635, which includes the 3S motif, interacts with complement factor H, a negative regulator of the complement cascade that can induce virus opsonization [17], [18]. Its simultaneous triggering of the complement cascade pathway and binding to one of its negative regulators may make HIV-1 gp41 crucial for viral spread and for T cell susceptibility to HIV infection [17], [19].
In this study, we sought to identify the receptor of the HIV-gp41 3S motif on CD4+ T cells and examined the signaling pathway leading to NKp44L surface expression. We discovered that the 3S motif binds to gC1qR, a receptor for the globular domain of C1q, on CD4+ T cells. This interaction activates successively the PI3K, the NADPH oxidase, and the p190A RhoGAP proteins. Activation of this signaling cascade induces the translocation of pre-existing NKp44L molecules from the cytoplasm to the plasma membrane of CD4+ T cells via a TC10-dependent mechanism.
Results
The 3S motif of HIV-gp41 stimulates ROS production by NADPH oxidase
We investigated the signaling cascade triggered by the 3S peptide to induce NKp44L cell-surface expression on CD4+ T cells. To identify the proteins involved in this cascade, we subjected total protein extracts of U2 cells to a standard two-dimensional polyacrylamide gel electrophoresis (2D PAGE), in the presence or absence of 3S peptide. Proteins differentially expressed by more than twofold were identified by mass spectrometry (MALDI-TOF). Table 1 summarizes in detail the analytic data for each identified spot. The up-regulation of peroxiredoxin and the presence of oxidized forms of GADPH, characterized by their low isoelectric points, indicated the presence of hydrogen peroxide (H202) in cells incubated with the 3S peptide [20].

To confirm the production of H202 during the 3S peptide-mediated signaling, the intracellular concentration of H202 was measured with DCFH-DA in purified CD4+ T cells incubated with or without the 3S peptide. Phorbol 12-myristate 13 acetate (PMA), which induces H202 production, was used as a positive control [21]. As Figure 1A shows, incubation with 20 µg/ml 3S peptide induced an increase in the intracellular H202 concentration, similar to the level observed following PMA incubation.

To confirm the role played by H202 in NKp44L surface expression, we incubated CD4+ T cells with H202 and detected similar levels of NKp44L surface expression on the CD4+ T cells treated with 100 µM H202 and with 5 µg/ml 3S peptide (Figure 1B). Next, assessment of the effect of N-acetyl L cysteine (NAC), an antioxidant, on NKp44L surface expression showed that 10 mM NAC completely abolished this surface expression on purified CD4+ T cells treated with 5 µg/ml 3S peptide (Figure 1C). To confirm a previous report that H202 synthesis in lymphocytes depends on NADPH oxidase activation [20], we treated cells with diphenyleneiodonium chloride (DPI), an NADPH oxidase inhibitor, and observed a drastic decrease in 3S-mediated NKp44L cell-surface expression (Figure 1D).
Together, these data indicate that NADPH oxidase may play a key role in the signaling pathway induced by the 3S peptide that leads to NKp44L surface expression and showed that H202 is sufficient to induce this expression.
The 3S motif of HIV-gp41 activates p190 RhoGAP
Next, we sought to identify the molecules involved downstream of H202 production in 3S-mediated signal transduction. Nimnual et al. [22] described the redox-dependent activation of p190 RhoGAP: H202 produced by NADPH oxidase inhibits the low molecular weight protein tyrosine phosphatase (LMW-PTP), which in turn inhibits p190 RhoGAP A (p190A) activity by dephosphorylating it. To determine whether p190A functions as a downstream effector of H202 in NKp44L surface expression, we used purified CD4+ T cells transfected with plasmid containing either wild-type p190 RhoGAP A (p190 WT) or its dominant negative form (p190 DN). Remarkably, transient transfection with p190 WT was sufficient to induce NKp44L cell-surface expression, while overexpression of p190 DN had no effect (Figure 2A).

As previously reported, p190A stimulates the GTPase activity of some Rho GTPases, including RhoA and its isoforms RhoB-C as well as TC10, and therefore promotes their GDP-bound form [23], [24]. Accordingly, we used a commercial ELISA-based assay to investigate RhoA activity in the lysate of purified CD4+ T cells in the presence of the 3S peptide. As expected, after 3S peptide treatment, RhoA activity decreased transiently by twofold, before returning to its initial level (Figure 2B).
NKp44L surface expression is TC10-dependent
TC10/RhoQ is a member of the Rho GTPase family and, like other members of this family, it cycles between an inactive GDP-bound state and an active GTP-bound form. Interestingly, TC10 is mainly located on vesicles that are visible throughout the cytoplasm [25]. To determine whether TC10 plays a role in 3S-mediated NKp44L surface expression, TC10 was knocked down by RNA interference (RNAi) in purified CD4+ T cells before 3S peptide stimulation. The down-modulation of TC10 in the presence of specific siRNAs was confirmed by western blot (Figure 2C). As Figure 2D shows, two specific TC10 siRNAs strongly reduced NKp44L cell-surface expression after incubation with the 3S peptide, while control siRNA had no effect. Interestingly, the level of NKp44L down-regulation by specific TC10 siRNAs correlated with their ability to down-modulate TC10. This result indicates that TC10 is required for NKp44L cell-surface expression and probably plays a key role in the fusion of vesicles containing NKp44L.
The 3S motif of HIV-gp41 induces NKp44L translocation to the cell surface
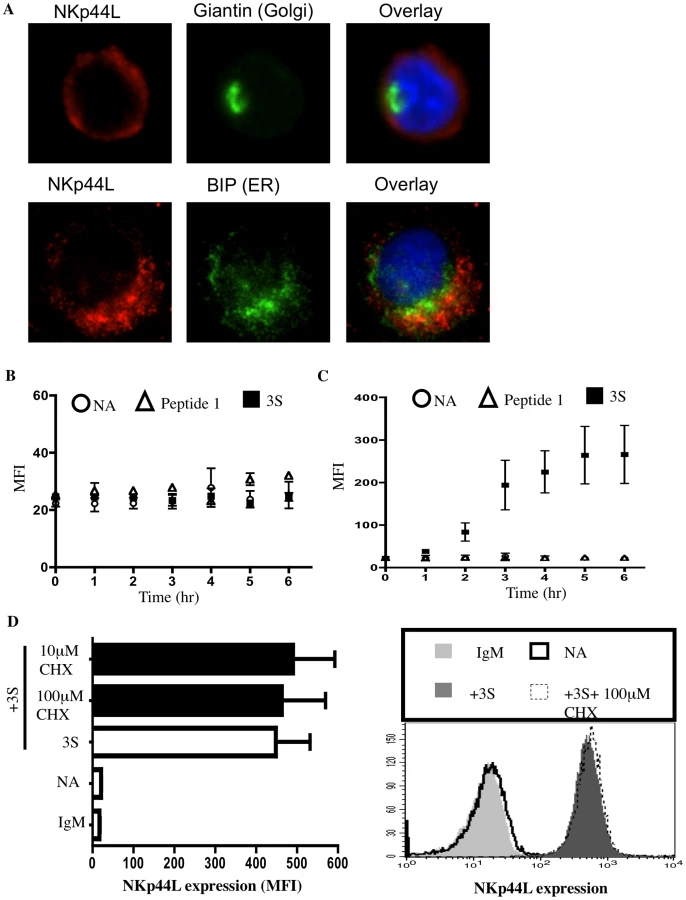
Next, we investigated whether the 3S peptide induces de novo synthesis of NKp44L as well as its translocation to the plasma membrane. High intracellular NKp44L expression was detected by confocal microscopy in purified CD4+ T cells in the absence of 3S-peptide stimulation. NKp44L molecules mainly colocalised with the ER marker, GRP78 BiP, within unstimulated CD4+ T cells (Figure 3A). Similar results were obtained using the Hela cell line (Supplementary Figure S1). The absence of NKp44L on the surface of CD4+ T cells in the absence of 3S-peptide is therefore not due to the absence of NKp44L in the cytoplasm of unstimulated cells but to the inability of these cells to translocate NKp44L to their cell surface. Interestingly, the total level of NKp44L expression remained similar in purified CD4+ T cells, without peptide stimulation or treated with the 3S peptide or control peptide (Figure 3B). Concomitantly, at the cell surface, NKp44L steadily and time-dependently increased in purified CD4+ T cells after 3S peptide incubation. NKp44L was absent from the surface of untreated CD4+ T cells and CD4+ T treated with control peptides (Figure 3C). Moreover, cycloheximide treatment did not prevent NKp44L cell-surface induction on primary CD4+ T cells by the 3S peptide (Figure 3D). This findings indicate that protein neosynthesis might not be necessary in this signaling pathway and suggests that the 3S peptide directly induces the translocation of pre-existing NKp44L molecules from the cytoplasm to the surface of CD4+ T cells.
The 3S motif of HIV-gp41 triggers Class I PI3K activation
We examined the signaling mediators upstream of NADPH oxidase activation in further detail. For cytokine signaling, PI3K indirectly activates Rac, one of the regulatory subunits of the NADPH oxidase that leads to H2O2 production [26]. The role of PI3K in NKp44L translocation was therefore investigated. Purified CD4+ T cells were incubated with 7 µM of PIP3, PIP2, or carrier (neomycin) alone. As shown in Figure 4A, PIP3, but neither PIP2 nor carrier alone, was sufficient to induce NKp44L surface expression. Furthermore, similar levels of NKp44L were observed on the surface of CD4+ T cells after treatment with either PIP3 or 3S peptide (Figure 4A). Of note, PIP3 did not induce other tested ligands of activating NK receptor, including ULBP1-3, MIC-A/B, CD112 or CD48 (Supplementary Figure S2). To confirm the role of PIP3 production in cell-surface expression of NKp44L, CD4+ T cells were pre-incubated, separately, in the presence of two PI3K inhibitors, wortmannin and Ly294,002. These two inhibitors strongly decreased 3S peptide-mediated cell-surface expression of NKp44L, in a dose-dependent manner (Figure 4B). The inability of these two PI3K inhibitors to inhibit H202-mediated NKp44L surface expression demonstrates that NADPH activation occurs downstream of PI3K activation (Figure 4C).

gC1qR is the receptor of the 3S motif of HIV-1 gp41
Finally, we searched for the receptor of the 3S motif of HIV-1 gp41 at the surface of CD4+ T cells. Interestingly, a gp41 epitope, closed to the 3S motif and located in residues 601–620, has been reported to bind to C1q and to activate the complement pathway [16]. In addition, the gp41 epitope located between residues 615–635, which include the 3S motif, binds to soluble factor H, a negative regulator of the complement cascade [18]. We searched the databases for a pleiotropic membrane receptor that is linked to the complement cascade and that activates PI3K and/or NADPH oxidase. Using the above criteria, we identified gC1qR, an atypical receptor of the complement [27], as a potential receptor for the 3S motif of HIV gp41.
To determine if gC1qR is the membrane partner of the 3S motif, immunoprecipitation was performed with magnetic beads covalently linked to the 3S peptide. Beads coupled to BSA were used as a negative control. Both sets of beads were then separately incubated with Jurkat CD4+ T cells, in the presence of 3,3′–Dithiobis (sulfosuccinimidylpropionate) (DTTSSP), a chemical cross-linker, to link the 3S peptide covalently with interacting proteins. Cells were then lysed, and the interacting proteins eluted from the beads. These proteins were analyzed by both SDS PAGE and Western blotting (with a cocktail of two anti-gC1qR mAbs, 60.11 and 74.5.2). As Figure 5A shows, gC1qR was detected solely in the presence of 3S peptide-coated beads. The specific interaction between the 3S peptide and the gC1qR was confirmed by ELISA. Addition of increasing concentrations of biotin-labeled 3S peptide showed strong and specific binding between gC1qR and the 3S peptide (Figure 5B).

To confirm the role of gC1qR as receptor of the 3S motif of HIV-1 gp41, CD4+ T were pretreated with anti-gC1qR mAb (74.5.2 or 60.11) or with the isotype-matched IgG and then stained with FITC-labeled 3S peptide. As Figure 5C shows, the presence of anti-gC1qR mAbs significantly inhibited the binding of FITC-labeled 3S peptide to CD4+ T cells. Similar results were observed using biotin-labeled 3S (Supplementary Figure S3A). Next, the ability of anti-gC1qR mAb to inhibit 3S peptide-mediated induction of cell-surface NKp44L expression was tested in purified CD4+ T cells. Cells were pretreated with anti-gC1qR mAb (74.5.2) and then incubated with the 3S peptide. As shown in Figure 5D, NKp44L cell-surface expression was strongly inhibited in the presence of anti-gC1qR mAb. Of note, another clone of anti-gC1qR mAb (60.11) was also able to decrease 3S mediated inductions of cell-surface expression of NKp44L but to a lower extend to the 74.5.2 clone (Supplementary Figure S3B). Similarly, anti-gC1qR mAb also inhibited NKp44L surface expression on cells incubated with wild type HIV (NL4.3) for 4hr (Figure 5E).
After demonstrating that the 3S peptide is a new viral ligand of gC1qR, we sought to determine if some endogenous ligands, such as C1q and high molecular weight kininogen (HK), both of which bind to gC1qR, also induce cell-surface expression of NKp44L. Purified CD4+ T cells were incubated for 4 h in the presence or absence of C1q, HK, or 3S peptide, and then stained for NKp44L surface expression. As Figure 5F shows, C1q, HK, and the 3S peptide all induced high levels of NKp44L cell-surface expression.
Discussion
In this report, we demonstrated that the gC1qR protein is the receptor for the 3S motif of HIV-1 gp41 and that their interaction induces NKp44L cell-surface expression on CD4+ T cells. A ubiquitous and highly anionic cellular 33-kDa protein, gC1qR was initially identified and characterized as a receptor for the globular heads of C1q (gC1q) [28]. Although it is located mainly in the cytoplasm, particularly in mitochondria, its presence on the surface of diverse human cells has been demonstrated unequivocally, both here and previously [29]. Indeed, we observed here that the 3S peptide interacts with gC1qR expressed on the cell surface of CD4+ T cells. Although gC1qR does not have a transmembrane domain, it is thought to transduce signals from the cell surface through interactions with other membrane-bound proteins. For example, the lateral association of gC1qR with β1-integrin is required for C1q-mediated cell adhesion and spreading [30]. Importantly, we showed that two major endogenous ligands of gC1qR, C1q and HK, also induce cell surface expression of NKp44L on CD4+ T cells.
Induction of NKp44L surface expression could be relevant for additional bacterial and viral infections. Indeed, gC1qR has been reported to interact with and facilitate entry of numerous microbial and parasitic pathogens, including for example Listeria monocytogenes, Staphylococcus aureus as well as Plasmodium falciparum [31], [32], [33]. More importantly, it is also utilized by some viruses, including HCV, rubella virus, HSV and HTNV [27], [34]. In addition, gC1qR is expressed on several immune cells including macrophages, dendritic and endothelial cells, B and T lymphocytes [27]. The pathogens listed earlier may induce NKp44L surface expression on cells of the immune system mentioned above. Here, we showed that gC1qR is also a receptor for the 3S motif of the HIV-1 gp41 protein. However, in vitro the presence of anti-gC1qR mAbs (74,5,2 or 60,11) had no or little impact on the level of infection of purified CD4+ T cells by both CCR5 - and CXCR4-tropic HIV strains (supplementary Figure S3C). This strongly suggests that these anti-gC1qR mAbs lack the potential to neutralize HIV-1 infection and that gC1qR is not required for HIV-1 infection.
Virus binding to cell-surface receptors may trigger signaling cascades in the host cell. Our examination of the signaling cascade triggered by the 3S motif revealed that PI3K activation plays a critical role in the 3S-mediated signaling that leads to NKp44L translocation to the cell surface. We note that Braun et al previously reported that the binding of internalin B of Listeria monocytogenes to gC1qR activates PI3K [31]. In contrast to previous reports of PI3K-mediated activation of Akt [35], Akt phosphorylation was not detected here following 3S peptide stimulation (data not shown). We traced the downstream signaling mediators after 3S-induced PI3K activation. Activated PI3K produced PIP3, which activated NADPH oxidase, probably by promoting GTP-bound Rac. Indeed, one of the Rho GTPase Rac functions that was first characterized in phagocytes was the regulation of the activity of the NADPH oxidase complex, which produces reactive oxygen species (ROS) including H2O2 [36], [37]. We demonstrated that H2O2, like the 3S peptide, was able to induce a high level of NKp44L surface expression on CD4+ T cells. Remarkably, H2O2 can also induce surface expression of some ligands for another NK activating receptor, NKG2D, on airway epithelial cells; these ligands include MICA/B and ULBP1-4. In addition, H2O2 did not modify the intracellular levels of any of these ligands [38]. Similarly, the 3S peptide did not seem to affect intracellular levels of NKp44L. This strongly suggests that H2O2 is a crucial mediator in the translocation of activating NK ligands to the surface of stressed cells that are flagged for destruction by NK-mediated lysis. This hypothesis is supported by previous reports that H2O2 also reduces HLA-A, -B, and -C molecules on epithelial cells, probably favoring NK-mediated lysis [38]. Of note, H2O2 production by the 3S peptide should inactivate PTEN, which would increase PIP3 concentration and therefore H2O2 production. This suggests a positive feedback loop to amplify NKp44L cell-surface translocation. Similar signal amplification has previously been described for EGF and PDGF stimulations [39].
We also demonstrated that in vitro the antioxidant NAC prevented 3S-mediated NKp44L surface expression. Antioxidants have previously been used in clinical trials for treatment of HIV-infected individuals, but with contradictory results. Some have failed to detect any significant beneficial effect on the CD4 cell count of antioxidant supplementation added to HAART treatment, but all showed increased T-lymphocyte proliferation responses [40]. In contrast, other in vivo studies have shown that antioxidant supplementation of HIV-infected individuals under HAART improved their CD4+ cell count compared with placebo-treated patients [41]. The discrepancies between these results may be due to the type and dose of antioxidant used, as well as to characteristics of the patients.
In the present study, we also showed that stimulation by the 3S peptide activates the p190 RhoGAP-A protein. A previous report showed that Rac-dependent ROS production leads to down-regulation of RhoA through oxidative inactivation of low-molecular-weight protein tyrosine phosphatase and the subsequent activation of p190 RhoGAP-A [22]. In line with that study, our data demonstrated that 3S peptide stimulation promotes the inactive (GDP-bound) form of RhoA. It probably inactivates other Rho family GTPases, notably RhoB, RhoC and TC10, which have all been shown to be inactivated by p190 RhoGAP-A [23], [24]. We note that TC10, a Rho family GTPase mainly localized on vesicular structures, plays a significant role in the exocytosis of GLUT4 and other proteins [42]. More recently, Kawase et al. [24] suggested that TC10 may be implicated in three separate steps of exocytosis: loading cargo into secretory vesicles, tethering vesicles to the plasma membrane, and triggering their fusion. Here, we showed that TC10 was necessary for the 3S peptide to induce the translocation of NKp44L to the cell surface. Of note, 3S peptide inactivation of the above Rho family GTPases might, as well as inducing NKp44L surface expression, affect additional signaling pathways and lead to surface expression of other membranes proteins. Indeed, the picture that is emerging today shows that Rho family GTPases transduce signals from pattern recognition receptors and from receptors of antigens, chemokines, and cytokines, as well as adhesion molecules and they function as focal points for crosstalk between different signaling pathways [43].
We propose the following model for the translocation of NKp44L to the plasma membrane after stimulation by the 3S peptide (Figure 6): 1) The 3S motif of HIV-gp41 interacts with gC1qR, its specific receptor; 2) this interaction activates PI3K; 3) activated PI3K promotes the activation of the NADPH-oxidase; 4) activated NADPH-oxidase promotes p190 RhoGAP-A activity; and 5) activated p190 RhoGAP-A then induces GTP hydrolysis by Rho-A and TC10, which are implicated in exocytosis. This signaling cascade is similar to that previously described for insulin-induced GLUT4 translocation. GLUT4 surface expression is also dependent on H2O2 production [44], as well as PI3K [45], and TC10 activation [42].

As we showed previously, the induction of NKp44L surface expression on uninfected bystander cells by the 3S motif renders them highly sensitive to NK lysis, a phenomenon that may lead to the CD4 depletion observed during HIV-1 infection [8], [11]. Interfering with NKp44L expression on the surface of CD4+ T cells should therefore be beneficial for HIV-1 infected individuals, by preventing CD4 depletion. Blocking the interaction between the 3S motif and its receptor with gC1qR antagonists should inhibit NKp44L surface expression on CD4+ T cells. Moreover, we note that HCV is another pathogen that employs gC1qR to subvert the immune response by inhibiting T cell activation and proliferation [46]. Before gC1qR antagonists can be used to treat HCV or HIV-1 infected individuals, suitable animal models are needed to investigate the in vivo effect of gC1qR inhibition in both HCV and HIV-1 infection. Alternatively, HIV-1 infected individuals might be vaccinated with the 3S peptide. A macaque model showed the efficacy of therapeutic immunization with the 3S motif of HIV gp41 against CD4 depletion [15].
Materials and Methods
CD4+ T cell purification
Leukocytes from whole-blood samples from healthy donors were obtained by buffy coat centrifugation from the hospital blood bank (Hôpital Pitié-Salpêtrière, Paris, France), after approval by the local ethics committee “Etablissement Français du sang”. Written consent from each individual was not required, as blood samples were obtained from healthy anonymous donors. CD4+ T cells were purified with CD4 magnetic microbeads (Miltenyi). Flow cytometric analysis demonstrated a purity of >95% CD4+ T cells. Purified CD4+ T cells were incubated for 3 days with 1 µg/ml PHA-L (Murex) in RPMI-1640 medium supplemented with 10% FCS and then cultured with 100 IU/ml Proleukin 2 (Chiron). The human CD4+ T lymphoma Jurkat cell line and the human promonocytic U2 cell line, were both grown in RPMI-1640 medium containing 10% FCS at 37°C and 5% CO2. The U2 cell line, which is derived from the promonocytic U937 cell line, was used as it is sensitive to 3S-mediated NKp44L surface induction.
Reagents
Anti-NKp44L mAb (IgM; #7.1) was previously described [8]. Purified unconjugated and Biotin - or FITC-conjugated 3S peptide from HIV-1 gp41 (NH2-PWNASWSNKSLDDIW-COOH) as well as scramble peptide (NH2-WNWDSKILSDPAWNS-COOH) were purchased from Covalabs (Villeurbanne, France). Complement component C1q (C1q), dibenziodolium chloride (DPI), LY 294,002 hydrochloride, wortmannin, hydrogen peroxide 30% solution, and imidazole were all purchased from SIGMA. Native human high molecular weight kininogen (HK) was purchased from AbD Serotec, PIP3 and PIP2 from TEBU Biosciences, anti-gC1qR (74.5.2 and 60.11), anti-TC10 (T304) and anti GRP78 BiP mAbs from Abcam.
2D PAGE and protein identification
For each sample, approximately 2×107 U2 cells were lysed with IEF buffer (7 M urea, 2 M thiourea, 1% CHAPS, 10% isopropanol, 10% isobutanol, 100 mM DTT, 0.5% SB 3–10, 0.2% Bio-Lyte, and 5% triton ×100). Proteins were separated by 2D PAGE. Briefly, protein lysates were mixed with IEF buffer supplemented with 1% IPG pH 3–7 buffer (GE Healthcare), 12 µL/ml Destreak reagent (GE Healthcare) plus bromophenol blue. Samples were added to 18 cm 3–10 NL strips (GE Healthcare) and treated as previously described [47]. SDS-Page was performed on 10% polyacrylamide, and gels were either stained with Imperial Protein Stain (Pierce) or with the PlusOne Silver Staining Kit (GE Healthcare), according to the manufacturer's instructions. Image Master 2D 6.0 software (GE Healthcare) was used to identify proteins significantly modulated from three separate set of experiments. Imperial protein dye was removed from gel pieces with 25 mM ammonium bicarbonate (AMBIC) in 50% ethanol. Gel pieces were dried in acetonitrile (ACN) and then digested with trypsin (PROMEGA). Peptides were extracted twice in TFA 5%, ACN 60%. The combined extracts were dried and resuspended in 0.1% TFA, 50% ACN. Samples were then analyzed by Maldi-ToF (Autoflex by Bruker).
Measurement of the intracellular H2O2 concentration
Purified CD4+ T cells were pre-incubated with 10 µM DCFH-DA (2′7′ –dichlorofluorescein diacetate) in PBS (without Ca 2+ et Mg2+) and then incubated in the presence or absence of 20 µg/ml 3S peptide or with 0.2 µg/ml PMA as the positive control. Fluorescence readings were performed every 3 min with a microplate reader (Flexstation 3, Molecular Devices).
p190 RhoGAP A activity
Purified CD4+ T cells were transfected by p190 RhoGAP A wild type (pWT) or RhoGAP A dominant negative (pDN) plasmids, a generous gift from Dr K. Burridge (University of North Carolina, USA), with the Human T cell nucleofector kit (AMAXA) according to the manufacturer's instructions. p190RhoGAP-GFP and p190RhoGAPR1283A-GFP plasmids have been previously described [48]. Transfection efficiency was verified by flow cytometry using GFP fluorescence.
Rho activity assay
Purified CD4+ T cells were incubated in the presence or absence of 5µg/ml 3S peptide for various lengths of time, washed with ice-cold PBS, and then rapidly lysed. For each sample, Rho activity was measured from 12.5 µg total lysate with the G-LISA luminometry-based assay (Tebu-Bio), according to the manufacturer's instructions.
TC10 knock-down
Purified CD4+ T cells were resuspended at 2×106 cells/ml in Accell siRNA delivery media (Dharmacon) supplemented with 100 unit/ml Proleukin 2. Cells were incubated for 72 h with 1 µM siRNA control or with 1 µM Accell siRNA against TC10 (Dharmacon). Two separate siRNAs against TC10 were used independently (5′-CUACUGACCUUGAUGGGUU 3′; 5′ - GCAUCAGAUAAUGGUGUUA -3′). Samples were then incubated in the presence or absence of 5 µg/ml 3S peptide before NKp44L staining by flow cytometry. Silencing efficiency of the different siRNAs was first monitored by western blot using anti-TC10 mAb.
Fluorescence microscopy
CD4+ T cells were fixed with 4% paraformaldehyde (PFA) before permeabilisation using 0.1% Triton. Samples were then stained using 1ug anti-NKp44L mAb and 0.75 µg Texas Red conjugated anti mouse IgM mAb. For giantin staining, cells were then incubated with 1/1000 diluted anti giantin mAb and 2 µg diluted Alexa 488 conjugated anti Rabbit IgG. For GRP78 BiP staining, cells were then incubated with 1/1000 diluted anti GRP78 BiP and 2 µg diluted Alexa 488 conjugated anti Rabbit IgG. For TC10 staining, cells were then incubated with 1/100 diluted anti TC10 mAb and 2 µg diluted Alexa 488 conjugated anti Rabbit IgG. Following staining, samples were mounted using ProLong Gold (Invitrogen).
Flow cytometry
FACS analysis was performed on purified CD4+ T cells. Isotype-matched immunoglobulin served as negative controls. For NKp44L surface staining, samples were incubated with 2 µg anti NKp44L antibodies for 1 h at 4°C then incubated in 100 µL of 1∶100 diluted PE anti mouse IgM antibodies (Pharmingen) and CD4-APC antibodies (Beckman Coulter). P190 transfected were stained using only APC anti mouse IgM antibodies (Pharmingen). For total NKp44L level, cells were fixed using BD cellFix solution (BD Biosciences) and permeabilised using 0.5% BSA, 0.1% saponine. Cells were stained using 2 µg purified anti-NKp44L antibodies and 1∶100 diluted PE anti mouse IgM antibodies (Pharmingen). At least 10000 events in the population of interest were detected on a FACScalibur (BD Biosciences). Results were analysed with cellQuest software.
Delivery of PIP (phospho-inositol) derivatives
PIP derivatives were delivered as previously described [49]. Briefly, solutions containing 250 µM PIP3 or PIP2 in the presence of 250 µM carrier in RPMI medium without serum were freshly prepared in HEPES buffer and then sonicated. Carrier, PIP3, or PIP2 mixtures were added for 4 h to purified CD4+ T cells at a final concentration of 7 µM.
Production, detection, and immunoprecipitation of gC1qR
Recombinant gC1qR protein was purified from E. coli transformed with the pET28a-5 plasmid encoding for gC1qR, a kind gift from E. Gouin and P. Cossart (Institut Pasteur, Paris, France) [31]. Briefly, transformed E. coli was incubated with 1 mM IPTG for 3 h, and then lysed. The recombinant gC1qR was purified with the TALON dynabeads (Invitrogen), according to the manufacturer's instructions, and eluted in buffer containing 150 mM imidazole. The interaction between the 3S peptide and gC1qR was determined with ELISA after coating with 0.5µg gC1qR, or with BSA as control.
For immunoprecipitation, magnetic beads were coated with 25 µg of 3S peptide, or BSA, as control, per mg of beads, according to the manufacturer's instructions (Ademtech). For each sample, approximately 40×106 cells were mixed with 2 mg of coated beads for 1 h at 4°C, in the presence of 2 mM DTSSP. After incubation, cells were lysed with 1% SDS for 30 min. The magnetic beads were then collected and washed several times with 1 M KCl, followed by 0.1 M NA2CO3 pH11 and by hypotonic buffer (10 mM Hepes, 10 mM KCl, 1.5 M MgCl2) in the presence of protease inhibitor cocktail, as previously described [50]. Purified proteins were eluted, subjected to SDS-PAGE, and then analyzed by Western blot, in the presence of 1.5 µg/ml anti-gC1qR (74.5.2 mAb) and 1 µg/ml anti-gC1qR (60,11 mAb) mAbs. After extensive washing, membranes were analyzed with the Etan Dige Imager.
Accession numbers
For accession numbers, please see Table 2.

Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AlimontiJB
BallTB
FowkeKR
2003 Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol 84 1649 1661
2. StevensonM
2003 HIV-1 pathogenesis. Nat Med 9 853 860
3. FinkelTH
Tudor-WilliamsG
BandaNK
CottonMF
CurielT
1995 Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV - and SIV-infected lymph nodes. Nat Med 1 129 134
4. VarbanovM
EspertL
Biard-PiechaczykM
2006 Mechanisms of CD4 T-cell depletion triggered by HIV-1 viral proteins. AIDS Rev 8 221 236
5. KaplanD
SiegS
1998 Role of the Fas/Fas ligand apoptotic pathway in human immunodeficiency virus type 1 disease. J Virol 72 6279 6282
6. DiopOM
GueyeA
Dias-TavaresM
KornfeldC
FayeA
2000 High levels of viral replication during primary simian immunodeficiency virus SIVagm infection are rapidly and strongly controlled in African green monkeys. J Virol 74 7538 7547
7. MorettaA
BottinoC
VitaleM
PendeD
CantoniC
2001 Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 19 197 223
8. VieillardV
StromingerJL
DebreP
2005 NK cytotoxicity against CD4+ T cells during HIV-1 infection: a gp41 peptide induces the expression of an NKp44 ligand. Proc Natl Acad Sci U S A 102 10981 10986
9. VieillardV
HabibRE
BrochardP
DelacheB
BovendoHF
2008 CCR5 or CXCR4 use influences the relationship between CD4 cell depletion, NKp44L expression and NK cytotoxicity in SHIV-infected macaques. AIDS 22 185 192
10. WardJ
BonaparteM
SacksJ
GutermanJ
FogliM
2007 HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 110 1207 1214
11. Fausther-BovendoH
Sol-FoulonN
CandottiD
AgutH
SchwartzO
2009 HIV escape from natural killer cytotoxicity: nef inhibits NKp44L expression on CD4+ T cells. AIDS 23 1077 1087
12. WyattR
SodroskiJ
1998 The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280 1884 1888
13. WeissenhornW
DessenA
HarrisonSC
SkehelJJ
WileyDC
1997 Atomic structure of the ectodomain from HIV-1 gp41. Nature 387 426 430
14. SattentauQJ
MooreJP
1991 Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med 174 407 415
15. VieillardV
Le GrandR
DaussetJ
DebreP
2008 A vaccine strategy against AIDS: an HIV gp41 peptide immunization prevents NKp44L expression and CD4+ T cell depletion in SHIV-infected macaques. Proc Natl Acad Sci U S A 105 2100 2104
16. EbenbichlerCF
ThielensNM
VornhagenR
MarschangP
ArlaudGJ
1991 Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med 174 1417 1424
17. StoiberH
BankiZ
WilflingsederD
DierichMP
2008 Complement-HIV interactions during all steps of viral pathogenesis. Vaccine 26 3046 3054
18. StoiberH
SchneiderR
JanatovaJ
DierichMP
1995 Human complement proteins C3b, C4b, factor H and properdin react with specific sites in gp120 and gp41, the envelope proteins of HIV-1. Immunobiology 193 98 113
19. BankiZ
StoiberH
DierichMP
2005 HIV and human complement: inefficient virolysis and effective adherence. Immunol Lett 97 209 214
20. RheeSG
ChaeHZ
KimK
2005 Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 38 1543 1552
21. BassDA
ParceJW
DechateletLR
SzejdaP
SeedsMC
1983 Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 130 1910 1917
22. NimnualAS
TaylorLJ
Bar-SagiD
2003 Redox-dependent downregulation of Rho by Rac. Nat Cell Biol 5 236 241
23. RidleyAJ
SelfAJ
KasmiF
PatersonHF
HallA
1993 rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. EMBO J 12 5151 5160
24. KawaseK
NakamuraT
TakayaA
AokiK
NamikawaK
2006 GTP hydrolysis by the Rho family GTPase TC10 promotes exocytic vesicle fusion. Dev Cell 11 411 421
25. MichaelsonD
SillettiJ
MurphyG
D'EustachioP
RushM
2001 Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol 152 111 126
26. WelchHC
CoadwellWJ
StephensLR
HawkinsPT
2003 Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett 546 93 97
27. GhebrehiwetB
PeerschkeEI
2004 cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol 41 173 183
28. GhebrehiwetB
LimBL
KumarR
FengX
PeerschkeEI
2001 gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol Rev 180 65 77
29. EggletonP
GhebrehiwetB
SastryKN
CoburnJP
ZanerKS
1995 Identification of a gC1q-binding protein (gC1q-R) on the surface of human neutrophils. Subcellular localization and binding properties in comparison with the cC1q-R. J Clin Invest 95 1569 1578
30. FengX
TonnesenMG
PeerschkeEI
GhebrehiwetB
2002 Cooperation of C1q receptors and integrins in C1q-mediated endothelial cell adhesion and spreading. J Immunol 168 2441 2448
31. BraunL
GhebrehiwetB
CossartP
2000 gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. EMBO J 19 1458 1466
32. NguyenT
GhebrehiwetB
PeerschkeEI
2000 Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun 68 2061 2068
33. BiswasAK
HafizA
BanerjeeB
KimKS
DattaK
2007 Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathog 3 1271 1280
34. ChoiY
KwonYC
KimSI
ParkJM
LeeKH
2008 A hantavirus causing hemorrhagic fever with renal syndrome requires gC1qR/p32 for efficient cell binding and infection. Virology 381 178 183
35. ManningBD
CantleyLC
2007 AKT/PKB signaling: navigating downstream. Cell 129 1261 1274
36. AboA
PickE
HallA
TottyN
TeahanCG
1991 Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353 668 670
37. KnausUG
HeyworthPG
EvansT
CurnutteJT
BokochGM
1991 Regulation of phagocyte oxygen radical production by the GTP-binding protein Rac 2. Science 254 1512 1515
38. BorchersMT
HarrisNL
WesselkamperSC
VitucciM
CosmanD
2006 NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 291 L222 231
39. KwonJ
LeeSR
YangKS
AhnY
KimYJ
2004 Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A 101 16419 16424
40. JariwallaRJ
LalezariJ
CenkoD
MansourSE
KumarA
2008 Restoration of blood total glutathione status and lymphocyte function following alpha-lipoic acid supplementation in patients with HIV infection. J Altern Complement Med 14 139 146
41. KaiserJD
CampaAM
OndercinJP
LeoungGS
PlessRF
2006 Micronutrient supplementation increases CD4 count in HIV-infected individuals on highly active antiretroviral therapy: a prospective, double-blinded, placebo-controlled trial. J Acquir Immune Defic Syndr 42 523 528
42. ChiangSH
BaumannCA
KanzakiM
ThurmondDC
WatsonRT
2001 Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410 944 948
43. TybulewiczVL
HendersonRB
2009 Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol 9 630 644
44. MahadevK
WuX
ZilberingA
ZhuL
LawrenceJT
2001 Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem 276 48662 48669
45. RudermanNB
KapellerR
WhiteMF
CantleyLC
1990 Activation of phosphatidylinositol 3-kinase by insulin. Proc Natl Acad Sci U S A 87 1411 1415
46. KittlesenDJ
Chianese-BullockKA
YaoZQ
BracialeTJ
HahnYS
2000 Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest 106 1239 1249
47. TanakaT
KuramitsuY
FujimotoM
NaitoS
OkaM
2008 Downregulation of two isoforms of ubiquitin carboxyl-terminal hydrolase isozyme L1 correlates with high metastatic potentials of human SN12C renal cell carcinoma cell clones. Electrophoresis 29 2651 2659
48. BarberisD
CasazzaA
SordellaR
CorsoS
ArtigianiS
2005 p190 Rho-GTPase activating protein associates with plexins and it is required for semaphorin signalling. J Cell Sci 118 4689 4700
49. SweeneyG
GargRR
CeddiaRB
LiD
IshikiM
2004 Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes incorporation of glucose transporter 4 into the plasma membrane of muscle and fat cells without increasing glucose uptake. J Biol Chem 279 32233 32242
50. ZhaoY
ZhangW
KhoY
2004 Proteomic analysis of integral plasma membrane proteins. Anal Chem 76 1817 1823
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- RNA Virus Replication Complexes
- Functional Genetic Diversity among Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival
- Extreme CD8 T Cell Requirements for Anti-Malarial Liver-Stage Immunity following Immunization with Radiation Attenuated Sporozoites
- A Systems Immunology Approach to Plasmacytoid Dendritic Cell Function in Cytopathic Virus Infections