
Virus-Infection or 5′ppp-RNA Activates Antiviral Signal through Redistribution of IPS-1 Mediated by MFN1
In virus-infected cells, RIG-I-like receptor (RLR) recognizes cytoplasmic viral RNA and triggers innate immune responses including production of type I and III interferon (IFN) and the subsequent expression of IFN-inducible genes. Interferon-β promoter stimulator 1 (IPS-1, also known as MAVS, VISA and Cardif) is a downstream molecule of RLR and is expressed on the outer membrane of mitochondria. While it is known that the location of IPS-1 is essential to its function, its underlying mechanism is unknown. Our aim in this study was to delineate the function of mitochondria so as to identify more precisely its role in innate immunity. In doing so we discovered that viral infection as well as transfection with 5′ppp-RNA resulted in the redistribution of IPS-1 to form speckle-like aggregates in cells. We further found that Mitofusin 1 (MFN1), a key regulator of mitochondrial fusion and a protein associated with IPS-1 on the outer membrane of mitochondria, positively regulates RLR-mediated innate antiviral responses. Conversely, specific knockdown of MFN1 abrogates both the virus-induced redistribution of IPS-1 and IFN production. Our study suggests that mitochondria participate in the segregation of IPS-1 through their fusion processes.
Published in the journal:
. PLoS Pathog 6(7): e32767. doi:10.1371/journal.ppat.1001012
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001012
Summary
In virus-infected cells, RIG-I-like receptor (RLR) recognizes cytoplasmic viral RNA and triggers innate immune responses including production of type I and III interferon (IFN) and the subsequent expression of IFN-inducible genes. Interferon-β promoter stimulator 1 (IPS-1, also known as MAVS, VISA and Cardif) is a downstream molecule of RLR and is expressed on the outer membrane of mitochondria. While it is known that the location of IPS-1 is essential to its function, its underlying mechanism is unknown. Our aim in this study was to delineate the function of mitochondria so as to identify more precisely its role in innate immunity. In doing so we discovered that viral infection as well as transfection with 5′ppp-RNA resulted in the redistribution of IPS-1 to form speckle-like aggregates in cells. We further found that Mitofusin 1 (MFN1), a key regulator of mitochondrial fusion and a protein associated with IPS-1 on the outer membrane of mitochondria, positively regulates RLR-mediated innate antiviral responses. Conversely, specific knockdown of MFN1 abrogates both the virus-induced redistribution of IPS-1 and IFN production. Our study suggests that mitochondria participate in the segregation of IPS-1 through their fusion processes.
Introduction
Type I and III interferons (IFNs) play central roles in innate immune responses to viral infections [1], [2], [3], [4]. In a variety of tissues, IFN production is triggered by a cytoplasmic sensor, retinoic acid inducible gene I (RIG-I)-like Receptor (RLR), which specifically senses viral RNA and induces antiviral signaling [5], [6]. Once RLR is activated, its signal is relayed through physical interaction to IFN-β promoter stimulator 1 (IPS-1, also known as MAVS, VISA or Cardif) [7], [8], [9], [10]. IPS-1 interacts with multiple signal transducers and protein kinases that activate transcription factors to induce IFN and other cytokine genes [11]. IPS-1 is expressed on the mitochondrial outer membrane and this localization is essential for signaling to occur [9]. However the reason for this underlying mechanism is unknown. Here, we investigated the cellular distribution of IPS-1 in virus-infected cells. We observed that IPS-1 is usually distributed evenly in all mitochondria in uninfected cells, however upon viral infection or the introduction of 5′ppp-RNA, which mimics viral RNA [12], [13], [14], a redistribution of IPS-1 occurred, resulting in a speckle-like pattern on mitochondria. Furthermore, we demonstrated that a mitochondrial GTPase, Mitofusin 1 (MFN1), which regulates mitochondrial fission and fusion [15], plays a critical role in the redistribution of IPS-1, as well as in virus-induced IFN production. Our study highlights the novel mitochondrial regulatory function of specifically sorting IPS-1 and providing a signaling platform for antiviral responses.
Results
Dynamic redistribution of IPS-1 in virus-infected or 5′ppp-RNA-transfected cells
To examine the localization of IPS-1 during viral infections, we generated HeLa cell lines stably expressing FLAG-tagged IPS-1 (IPS-1-HeLa clones, Fig. 1). Although the temporary expression of wild type (wt) IPS-1 results in constitutive signaling [7], [8], [9], [10], the stable cell lines did not exhibit the constitutive activation of downstream target genes. However, upon infection with the Sendai virus (SeV), the cells exhibited increased expression of IFN and chemokine genes (IFNB1, IL29, IL28A, IL28B and CXCL11) and interferon-stimulated genes (DDX58, IFIH1, DHX58, IFIT1-3, and OASL) (Fig. 1B and C). Furthermore, the IPS-1-HeLa clones exhibited diminished susceptibility to Encephalomyocarditis virus (EMCV) replication (1 to 2 log) (Fig. 1D). The low basal activity and elevated signaling after SeV-infection suggest that FLAG-IPS-1 is under a regulatory control similar to that of endogenous IPS-1.

Like endogenous IPS-1, FLAG-tagged IPS-1 is expressed on mitochondria in uninfected cells as shown by co-staining with MitoTracker (Fig. 2A, Mock). However, compared to the even cytoplasmic staining in the mock-infected cells, the staining pattern of IPS-1 became noticeably speckled in SeV-infected cells (Fig. 2A, SeV). Quantification of the fluorescence image revealed that mitochondria heavily stained with MitoTracker but lightly stained with anti-FLAG antibody were produced in SeV-infected cells. This redistribution was also observed with another mitochondrial marker, endoplasmic reticulum-associated amyloid β-peptide-binding protein (ERAB) (Fig. 2B), and different viruses (Newcastle disease virus (NDV), Sindbis virus, EMCV, Influenza virus, and Vesicular stomatitis virus (VSV)) (Fig. 3).


We also examined the distribution of IPS-1 in 5′ppp-RNA-transfected cells. Unlike synthetic single stranded RNA (5′OH-RNA), 5′ppp-RNA is a chemical ligand for RIG-I and is known to mimic viral signaling [12], [13], [14]. Interestingly, as with a viral infection, 5′ppp-RNA induced a redistribution of IPS-1, suggesting that the redistribution was triggered through RIG-I signaling. It is worth noting that EMCV, which selectively activates another RLR, melanoma differentiation-associated gene 5 (MDA5), also caused the redistribution of IPS-1, suggesting that this effect is common to RLRs. We suspected that IPS-1-HeLa cells exhibit enhanced redistribution of IPS-1 due to enhanced signaling (>10 IFN-β mRNA accumulation, Fig. 1B). This led us to analyze the distribution pattern of endogenous IPS-1 in HeLa cells, and we observed that the distribution pattern of endogenous IPS-1 changed in SeV-infected cells, although exclusive staining by mitochondrial marker was not observed (Fig. 4A). Similar to IPS-1-HeLa cells, we observed that hepatocellular carcinoma SKHep1 cells NDV, SeV, Influenza virus, or Sindbis virus infection also induced a speckled staining pattern in endogenous IPS-1 (Fig. 4B), and displayed enhanced IRF-3 dimerization when compared with HeLa cells (our unpublished data). This suggests that the redistribution is not simply an artifact due to the overexpression of FLAG-IPS-1.

Localization of viral nucleocapsid, RIG-I, and IPS-1
In order to activate RLR signaling, we used NDV to infect cells because it is available an anti-nucleocapsid protein (NP) antibody, a probe for the viral RNA-NP complex. NDV infection resulted in foci of NP in the cytoplasm and induced foci of RIG-I to form (Fig. 5A) [16]. RIG-I was evenly distributed in the cytoplasm, however some of the foci co-localized with those of NP (Fig. 5A). A similar formation of foci and co-localization with viral nucleoprotein complex was observed with other viruses (Ko.O. unpublished observations). IPS-1 accumulated on the periphery of the foci of RIG-I (Fig. 5B) and NP (Fig. 5C). We speculate that activated RIG-I recruits IPS-1, because RIG-I and IPS-1 interacted with each other through CARD-CARD interaction [7], [8]. IPS-1 did not co-localize with RIG-I nor NP presumably because mitochondria do not penetrate these foci nor is IPS-1 released from mitochondria. Immunoelectron microscopy using the anti-NP antibody clearly identified the NP foci (Fig. 6A), and anti-FLAG staining (Fig. 6B) showed that mitochondrial IPS-1 accumulated on the periphery of NP foci in NDV infected cells.


A dominant negative mutant of RIG-I does not induce IPS-1 redistribution
To determine if the observed redistribution of IPS-1 is functionally relevant, we used a point mutant of RIG-I (K270A), which normally recognizes ligand RNA but functions as a dominant negative inhibitor (Fig. 7A) [14]. It was observed that NDV infection induced foci of both wt and K270A RIG-I to form (Fig. 7B), however wt but not K270A promoted the speckled staining pattern of IPS-1 (Fig. 7C). The results indicate that the redistribution of IPS-1 is strongly correlated with the activation of antiviral signaling.

Mitofusin 1, but not Mitofusin 2, plays a critical role in RIG-I-induced antiviral signaling
RIG-I was originally identified by screening an expression cDNA library [17]. In addition to the cDNA encoding RIG-I, there were several other candidate cDNA clones which enhance virus-responsive reporter activity. Two of the independent clones encoded a full-length protein, Mitofusin 1 (MFN1). Human MFN1 is composed of 741 amino acids and domains of GTPase and transmembranes (Fig. 8A). MFN1 together with its related protein Mitofusin 2 (MFN2) is expressed on the outer membrane of mitochondria and regulates mitochondrial dynamics [18], [19]. Hyper - and hypo-functioning of either MFN1 or MFN2 result in elongated/aggregated and fragmented mitochondria, respectively. GTPase activity was previously shown to be essential for mitochondrial morphological change, particularly the fragmentation of mitochondria induced by a GTP-binding-deficient mutant of MFN1 (MFN1 T109A) [18].
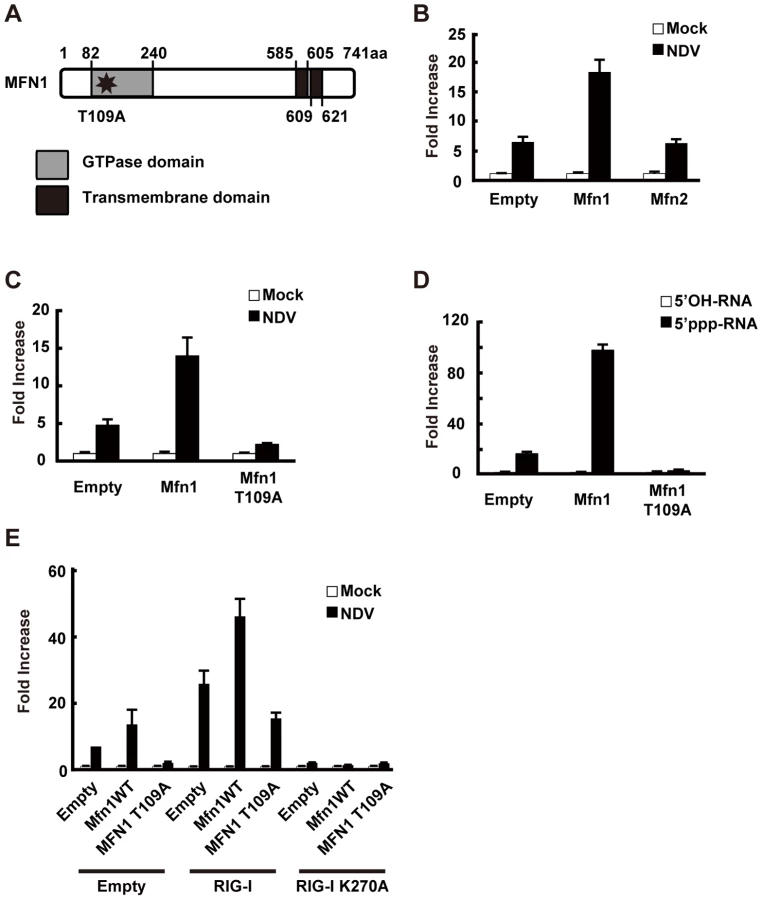
Consistent with the screening results, overexpression of MFN1, but not MFN2, augmented IFN-β promoter activity (Fig. 8B). The GTPase activity is involved in this MFN1 function, since MFN1 T109A significantly inhibited the signaling induced by NDV or 5′ppp-RNA (Fig. 8C and D). It is worth noting that overexpression of MFN1, which results in elongated mitochondria, is not by itself sufficient to deliver the signal. To confirm that the increased signaling observed by MFN1 overexpression was correlated with RIG-I activation, we transfected cells with a combination of RIG-I and MFN1. The RIG-I/MFN1 combination showed enhanced IFN-β promoter activity, but the RIG-I K270A mutant/MFN1 combination failed to do so (Fig. 8E). MEFs derived from mice with disrupted Mfn1 or Mfn2 gene was used to confirm the specific involvement of MFN1 in virus-induced antiviral signaling (Fig. 9A and B). The results indicated that MFN1, but not MFN2, is essential for the signal transduction mediated by RIG-I.

We examined other regulatory proteins for mitochondrial fission/fusion mechanism. Optic atrophy protein 1 (OPA1) is expressed on, and implicated in the fusion of the mitochondrial inner membrane [20]. Three independent siRNA targeting OPA1, down-regulated OPA1 expression (Fig. 9C) and partially (up to 50%) blocked NDV-induced signaling (Fig. 9D). However, the knockdown of dynamin-related protein 1 (DRP1) (Fig. 9E), which regulates mitochondrial fission [21] resulting in elongated mitochondria, did not have a significant effect (Fig. 9F). To explore the site where MFN1 is active, we temporarily overexpressed the dominant active RIG-I (RIG-I CARD) [17] or IPS-1 in wt and Mfn-knockout MEFs. Unlike the signal generated by the overexpression of IPS-1, the signal generated by overexpression of the RIG-I tandem caspase recruitment domain (CARD) clearly required MFN1. MFN1 however, is dispensable if IPS-1 is overexpressed (Fig. 9G). Again, MFN2 exhibited little influence on the signaling triggered by either stimulus. These results indicate that MFN1, but not MFN2, is essential for signal transduction mediated by RIG-I and IPS-1.
Physical interaction between IPS-1 and MFN1
To explore the molecular mechanism of how IPS-1 is regulated by MFN1, co-immunoprecipitation was performed using cells stably expressing IPS-1. FLAG-IPS-1 was precipitated by anti-FLAG and the associated proteins were analyzed by immunoblotting (Fig. 10). Both MFN1 and MFN2 constitutively associated with IPS-1 in the cells, but an unrelated mitochondrial outer membrane protein, BCL-XL, did not associate with IPS-1. Furthermore, OPA1 and DRP1 did not co-immunoprecipitate with FLAG-IPS-1. These data suggest that IPS-1 selectively associates with MFN1 and MFN2.

Knockdown of MFN1 inhibits the redistribution of IPS-1 induced by viral infection
Next, we examined what effect the knockdown of MFN1 would have on the virus-induced redistribution of IPS-1. Three independent siRNA efficiently knocked down MFN1 expression (Fig. 11A) resulting in a strong inhibition of the NDV-induced IFN-β gene expression in HeLa cells and IPS-1-HeLa cells (Fig. 11B and 11C). This once again suggests that IPS-1-HeLa cells tend to behave like normal cells. Upon NDV infection, IPS-I displayed a speckled staining pattern in control cells, but not in the MFN1-knockdown cells (Fig. 11D). Though the intensity of NP staining did not increase, MFN1-knockdown significantly inhibited IFN gene activation. This correlates with prior observations that although IFN production is inhibited by LGP2 overexpression, viral yield does not increase [22]. When control siRNA-treated cells were infected with NDV, a redistribution of IPS-1 was observed (69.3±15.7% of cells positive for NP). In MFN1-knockdown cells, although NDV infection resulted in the formation of NP foci, IPS-1 redistribution did not occur (4.5±1.3% of cells positive for NP). A similar effect was observed when MFN1-knockdown cells were infected with SeV (Fig. 12). These results strongly suggest that MFN1 is critical to the redistribution of IPS-1 triggered by RIG-I mediated sensing of viral RNA.


Discussion
RIG-I mediated antiviral signaling is a critical antiviral response which is initiated when the RIG-I sensor recognizes viral RNA. A signal is relayed to IPS-1, a mitochondrial regulator which delivers the signal downstream. Interestingly, the IPS-1-HeLa clones in this study exhibited very low basal expression of IFN genes, which led us to speculate that IPS-I inhibitory protein(s) is up regulated in these clones. We also examined the expression level of NLRX1, an IPS-1 inhibitor [23], and noted no change in its expression level (not shown). Similarly, levels of MFN1 and MFN2 did not change in the IPS-1-HeLa clones. (Fig. 10, input).
We observed that the IPS-1 level did not change for up to 12 h in virus-infected cells and no specific modification of IPS-1 was identified up to that point. We therefore hypothesize that the activation status of IPS-1 is determined by its localization pattern. We speculate that the mechanism of mitochondrial fusion is mediated by MFN1, and that IPS-1 translocates from some mitochondria and finally accumulates densely on others. On the other hand, forced overexpression of full-length IPS-1 results in constitutive signaling [7], [8], [9], [10]. There is no clear explanation why non-physiological overexpression can by-pass the virus-induced signaling, however, it can be concluded that transient IPS-1 overexpression may quantitatively override the hypothetical inhibitor for IPS-1 (above). Under these conditions, MFN1 is dispensable (Fig. 9G). Consistent with this, artificial aggregation of IPS-1 by cross-linking induced the signaling to activate IFN genes (Tang, E. D. and Wang, C. D. [24] and our unpublished observation). We speculate that under physiological conditions, viral infection induces the local accumulation of IPS-1 (corresponds to “active IPS-1”) on mitochondria. These mitochondria with locally accumulated IPS-1 may function as a platform to recruit downstream molecules.
MFN1 and MFN2 are structurally similar and both occur on the outer membrane of mitochondria. Their functions however are not redundant, as the single knockout of either produces a certain mitochondrial phenotype [19]. Recently two papers were published concerning MFN function in RIG-I-mediated antiviral responses. Yasukawa et al. reported that MFN2 strongly interacts with IPS-1 thereby blocking its function, however MFN1 does not interact with IPS-1 and exhibits no effect [25]. These observations are clearly inconsistent with ours. The report by Castanier et al. however, is consistent with our finding that MFN1, but not MFN2, positively regulates IPS-1 [26]. Most importantly, our knockout results are clearly consistent with their knockdown results (Fig. 9A and B). Castanier et al. observed that a particular variant of SeV (H4) causes elongation of mitochondria, however they did not demonstrate whether this morphological change is common to other viral infections. Unlike Castanier et al. we did not observe mitochondrial elongation by viral infections nor 5′-pppRNA transfection (Fig. 3). The knockdown of a mitochondrial inner membrane protein OPA1 blocked virus-induced signaling (Fig. 9D); thereby indicating that mitochondrial fusion may be necessary for the activation of IPS-1. Castanier et al. observed that knockdown of DRP1 or FIS1 causes mitochondrial elongation and IPS-1's association with STING, an antiviral signaling adaptor. However, neither the mitochondrial elongation nor the IPS-1-STING interaction is sufficient to activate the signaling.
We demonstrated that the redistribution of IPS-1 is induced by various viral infections and 5′ppp-RNA transfection and is dependent on a functional MFN1. The precise mechanism of the IPS-1 redistribution is not known, however we propose a model described in Fig. 13. In uninfected cells, MFN1 and IPS-1 associate constitutively (Fig. 10) and distribute evenly on all mitochondria. When a virus replicates in a specific cytoplasmic compartment, RIG-I is recruited to this area as a result of interaction with viral dsRNA through the C-terminal RNA-binding domain (Fig. 5A). Upon binding with viral RNA, CARD is exposed (activate RIG-I) [27], and then interacts with IPS-1 at the periphery of the viral compartment. At the same time, mitochondria surround the viral compartment because it lacks the ability to penetrate it (Fig. 6). Since mitochondrion is an elastic, movable organelle [28], affinity between RIG-I and IPS-1 may be sufficient for the mitochondrial relocalization. IPS-1/MFN1 complex may facilitate fusion between the surrounding mitochondria. Mitochondrial fission occurs independently to balance the fusion, however since the fusion of IPS-1/MFN1-enriched mitochondria facilitates the redistribution of IPS-1, IPS-1-enriched mitochondria may be generated. Further research is necessary to elucidate if the mitochondrial fusion process is indeed central to IPS-1 redistribution.

In summary, our study provides new insight into why the mitochondrial localization of IPS-1 is essential to its function. We demonstrated that MFN1 regulates the redistribution of IPS-1 in a RIG-I-signal-dependent manner. The mitochondrion provides a platform for the coordination of antiviral signaling by receiving the initial signal from the activated RIG-I. The signal is amplified through the accumulation of IPS-1, and other essential molecules such as tumor necrosis factor receptor-associated factors (TRAFs) and signaling protein kinases are recruited to mobilize active transcriptional regulators. In this regard, another organelle, the late endosome, functions similarly as a platform for antiviral signaling by recruiting different signaling components. This is initiated by Toll-like receptors (TLR3, 7, 8, and 9), another subtype of nucleic acid sensors.
Materials and Methods
Cell culture and transfection
HeLa, SKHep1, MEF, and 293T cells were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and penicillin–streptomycin (100U/ml and 100µg/ml, respectively). L929 cells were maintained in minimum essential medium with 5% fetal bovine serum and penicillin-streptomycin. Immortalized wild-type MEFs and MEFs deficient in Mfn1 or Mfn2 were obtained from Prof. David Chan (Caltech). HeLa, L929, and 293T cells were transfected with Lipofectamine 2000 (Invitrogen). Stable transformants of IPS-1-HeLa cells were established by transfection of a linearized empty plasmid (pEF-Tak) or expression plasmid for FLAG-IPS-1 (pEF-Tak-FLAG-IPS-1), and selected with G418 (1mg/ml). We used HeLa IPS-1#2 clone for most experiments because IPS-1#2 clone showed higher induction of IFN among the stable clones, but we confirmed that other clones also showed the same phenotype. IPS-1/RIG-Iwt and IPS-1/RIG-I K270A-expressing HeLa cells were generated by transduction using a lentivirus system. cDNA for RIG-I wt or RIG-I K270A was cloned into the multi-cloning site of a lentiviral vector, pCSII-CMV-MCS-IRES2-Bsd. The recombinant lentiviruses were generated by co-transfection of the lentiviral vector together with lentivirus constructs, pCMV-VSV-G-RSV-Rev and pCAG-HIVgp, into 293T cells. At 48 h post-transfection, the culture supernatant was collected, and then added to IPS-1#2 HeLa cells. Three days later, the cells were selected with Blasticidin (10µg/ml).
Viral infection and plaque assay
Cells were treated with culture medium or infected with SeV, NDV, Sindbis virus, EMCV, Influenza virus, or VSV at a MOI of 1 or 0.5 for qRT-PCR or immunofluorescence, respectively. The yield of EMCV in the culture supernatant was determined with a standard plaque assay [17].
Plasmid constructs
pEF-Bos-FLAG-RIG-I CARD and pEF-Bos-FLAG-IPS-1 have been described previously [6], [17]. pEF-Tak and pEF-Tak-FLAG-IPS-1 were kindly provided by Dr. M. Gale (University of Washington School of Medicine, USA). pEF-Bos-HA-MFN1 and pEF-Bos-HA-MFN2 were newly constructed. MFN1 and MFN2 cDNA were amplified with a pair of oligonucleotides designed to add an N-terminal HA tag by PCR, and the PCR fragment was inserted into pEF-Bos(+). MFN2 cDNA was purchased from the Biological Resource Center of the National Institute of Technology and Evaluation of Japan. pEF-Bos-HA-MFN1 T109A was constructed with a KOD-Plus-Mutagenesis kit (TOYOBO, Japan). The nucleotide sequences for the constructs were confirmed with the BigDye DNA sequencing kit (Applied Biosystems). Luciferase reporter containing human IFN-β promoter/enhancer (−125-Luc) is described elsewhere [29].
Synthetic RNA
Nucleotide sequences of the synthetic RNA were described previously [14]. 5′ppp-RNA was synthesized by in vitro transcription using the T7 Megascript kit (Ambion). 5′OH-RNA was chemically synthesized (Japan Bio Services Co., Ltd, Japan) and synthetic RNA was transfected by Lipofectamine RNAiMax (20 pmol in a 24-well format).
Immunoblotting, luciferase assay, and antibodies
The preparation of cell extracts, luciferase assay, and immunoblotting have already been described [17], [29]. The antibodies used are: anti-FLAG antibody (SIGMA: F3165 and Affinity Bioreagents: PA1-984B), anti-ERAB antibody (Abcam: ab10260), anti-MFN1 antibody (Santa cruiz: sc-50330), anti-ACTIN antibody (Millipore: MAB150 1R), anti-MFN2 antibody (abcam: ab56889), anti-OPA1 antibody (abcam: ab42364), anti-DRP1 antibody (abcam: ab56788), and anti-BCLXL (Santa cruiz: sc-8392). The anti-NP antibody was produced by Dr. Y. Nagai, and provided by Dr. T. Sakaguchi (Hiroshima University, Japan). The anti-RIG-I antibody was generated by immunizing a rabbit with a synthetic peptide corresponding to a.a. 793–807 of RIG-I. The generation of the anti-IPS-1 guinea pig antibody has already been described [30]. Briefly, anti-IPS-1 rabbit antibody was generated by immunizing a rabbit with a recombinant protein corresponding to a.a. 1–157 of IPS-1.
RNAi
The siRNA negative control and siRNAs targeting MFN1, OPA1, or DRP1 were purchased from Invitrogen, and transfected with RNAi MAX (Invitrogen) according to the manufacturer's recommendation (final concentration of siRNA was 50nM). At 48 or 72 h post-transfection, cells were harvested or infected with NDV or SeV, then subjected to qRT-PCR, immunofluorescence, or SDS-PAGE followed by immunoblotting.
Quantitative real time PCR and microarray analysis
Total RNA was prepared with TRIZOL (Invitrogen) and treated with DNase I (Roche Diagnostics). A High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used for cDNA synthesis, and mRNA level was monitored with the Step One plus Real Time PCR system and TaqMan Fast Universal PCR Master Mix (Applied Biosystems). TaqMan primer-probes for human IFNB1, human OPA1, murine Ifna4, murine Ifnb1, and 18s rRNA were purchased from Applied Biosystems. The RNA copy numbers were normalized to that of internal 18s rRNA. In the microarray analysis, we used the Genopal microarray system according to the manufacturer's instructions (Mitsubishi Rayon). Biotin-labeled RNA was prepared with a MessageAmp II-Biotin Enhanced kit (Ambion).
Immunofluorescence and immunoelectron microscopy
For immunofluorescence analysis, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with an acetone: methanol (1∶1) solution, and blocked with 5mg/ml of BSA in PBST (PBS, 0.04% Tween20) for 1 hour. The cells were next incubated with relevant primary antibodies overnight at 4°C, and then incubated with relevant Alexa Fluor 405, Alexa Fluor 488, or Alexa Fluor 594-conjugated secondary antibodies. Mitochondria were stained with MitoTracker Red CMXRos according to the manufacturer's instructions (Molecular Probes). Nuclei were stained with DAPI (4.6-diamidino-2-phenylinodole). The fluorescence image was quantified by software provided by Leica Microsystems. The percentage of IPS-1 redistribution among NDV-infected cells was scored by 3 persons, and data are shown as means ± s.d. of the three independent scores. Cells were analyzed with a Leica confocal laser-scanning microscope (TCS-SP2). For immunoelectron microscopy, cells were fixed with 4% paraformaldehyde and 0.05% and 0.01% glutaraldehyde for 10 min to detect NP and the FLAG tag, respectively, and then incubated with PBS containing 20% normal goat serum (NGS) and 0.075% Photo-Flo (Kodak) for 30 min at room temperature. The cells were incubated with the anti-NDV NP antibody or anti-FLAG antibody in PBS containing 2% NGS and 0.075% Photo-Flo overnight. After several washes with PBS, the cells were further incubated with a 1.4 nm gold-conjugated anti-mouse IgG goat IgG Fab fragment (Nanoprobes) in PBS containing 2% NGS and 0.075% Photo-Flo overnight. They were then washed with PBS and postfixed with 0.1 M phosphate buffer containing 1% (v/v) glutaraldehyde for 10 min at room temperature. After washing in distilled water, the gold particles were silver-intensified with an HQ silver kit (Nanoprobes) for 10–15 min. Then, the immunostained cells were incubated with 0.5% osmium tetroxide in 0.1M phosphate buffer for 40 min at room temperature. After dehydration with ethanol, cells were embedded in epoxy resin (Luveak 812; Nacalai Tesque, Japan). Once the resin was polymerized, the cells were cut into ultrathin sections on an ultramicrotome, Reichert-Nissei Ultracut S (Leica). The ultrathin sections were mounted on mesh grids, and stained by the Reynolds method. Finally, the ultrathin sections were examined with an electron microscope (H-7650; Hitachi).
Zdroje
1. SamuelCE
2001 Antiviral actions of interferons. Clin Microbiol Rev 14 778 809, table of contents
2. TheofilopoulosAN
BaccalaR
BeutlerB
KonoDH
2005 Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23 307 336
3. KotenkoSV
GallagherG
BaurinVV
Lewis-AntesA
ShenM
2003 IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4 69 77
4. SheppardP
KindsvogelW
XuW
HendersonK
SchlutsmeyerS
2003 IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4 63 68
5. YoneyamaM
FujitaT
2009 RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 227 54 65
6. OnoguchiK
YoneyamaM
TakemuraA
AkiraS
TaniguchiT
2007 Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem 282 7576 7581
7. KawaiT
TakahashiK
SatoS
CobanC
KumarH
2005 IPS-1, an adaptor triggering RIG-I - and Mda5-mediated type I interferon induction. Nat Immunol 6 981 988
8. MeylanE
CurranJ
HofmannK
MoradpourD
BinderM
2005 Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437 1167 1172
9. SethRB
SunL
EaCK
ChenZJ
2005 Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122 669 682
10. XuLG
WangYY
HanKJ
LiLY
ZhaiZ
2005 VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19 727 740
11. ArnoultD
CarneiroL
TattoliI
GirardinSE
2009 The role of mitochondria in cellular defense against microbial infection. Semin Immunol 21 223 232
12. HornungV
EllegastJ
KimS
BrzozkaK
JungA
2006 5′-Triphosphate RNA is the ligand for RIG-I. Science 314 994 997
13. PichlmairA
SchulzO
TanCP
NaslundTI
LiljestromP
2006 RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314 997 1001
14. TakahasiK
YoneyamaM
NishihoriT
HiraiR
KumetaH
2008 Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell 29 428 440
15. BenardG
KarbowskiM
2009 Mitochondrial fusion and division: Regulation and role in cell viability. Semin Cell Dev Biol 20 365 374
16. IwamuraT
YoneyamaM
KoizumiN
OkabeY
NamikiH
2001 PACT, a double-stranded RNA binding protein acts as a positive regulator for type I interferon gene induced by Newcastle disease virus. Biochem Biophys Res Commun 282 515 523
17. YoneyamaM
KikuchiM
NatsukawaT
ShinobuN
ImaizumiT
2004 The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5 730 737
18. SantelA
FrankS
GaumeB
HerrlerM
YouleRJ
2003 Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci 116 2763 2774
19. ChenH
DetmerSA
EwaldAJ
GriffinEE
FraserSE
2003 Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160 189 200
20. CipolatS
Martins de BritoO
Dal ZilioB
ScorranoL
2004 OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101 15927 15932
21. IshiharaN
NomuraM
JofukuA
KatoH
SuzukiSO
2009 Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11 958 966
22. YoneyamaM
KikuchiM
MatsumotoK
ImaizumiT
MiyagishiM
2005 Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175 2851 2858
23. MooreCB
BergstralhDT
DuncanJA
LeiY
MorrisonTE
2008 NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451 573 577
24. TangED
WangCY
2009 MAVS self-association mediates antiviral innate immune signaling. J Virol 83 3420 3428
25. YasukawaK
OshiumiH
TakedaM
IshiharaN
YanagiY
2009 Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2 ra47
26. CastanierC
GarcinD
VazquezA
ArnoultD
2010 Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep 11 133 138
27. FujitaT
2009 A nonself RNA pattern: tri-p to panhandle. Immunity 31 4 5
28. AlbertsB
JohnsonA
LewisJ
RaffM
RobertsK
2008 Energy Conversion: Mitochondria and Chloroplasts. Molecular Biology of the Cell, Fifth Edition New York Garland Science 813 878
29. YoneyamaM
SuharaW
FukuharaY
SatoM
OzatoK
1996 Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3). J Biochem 120 160 169
30. KaukinenP
SillanpaaM
KotenkoS
LinR
HiscottJ
2006 Hepatitis C virus NS2 and NS3/4A proteins are potent inhibitors of host cell cytokine/chemokine gene expression. Virol J 3 66
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 7
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- RNA Virus Replication Complexes
- Functional Genetic Diversity among Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival
- Extreme CD8 T Cell Requirements for Anti-Malarial Liver-Stage Immunity following Immunization with Radiation Attenuated Sporozoites
- A Systems Immunology Approach to Plasmacytoid Dendritic Cell Function in Cytopathic Virus Infections